

Background: NS5A is critical for HCV replication, but its role is poorly understood.
Results: Cysteines Cys-39, Cys-57, Cys-59, and Cys-80 are vital for NS5A dimerization, RNA binding, and viral replication.
Conclusion: NS5A dimerization, RNA binding, and HCV replication are correlated.
Significance: This study addresses an important issue in HCV research with NS5A being a major drug target with inhibitors in advanced stages of clinical development.
Keywords: Hepatitis C Virus, RNA-binding Protein, RNA Viruses, RNA-Protein Interaction, Viral Replication, Dimerization, NS5A, RNA Binding, RNA Replication
Abstract
Hepatitis C virus (HCV) is the main agent of acute and chronic liver diseases leading to cirrhosis and hepatocellular carcinoma. The current standard therapy has limited efficacy and serious side effects. Thus, the development of alternate therapies is of tremendous importance. HCV NS5A (nonstructural 5A protein) is a pleiotropic protein with key roles in HCV replication and cellular signaling pathways. Here we demonstrate that NS5A dimerization occurs through Domain I (amino acids 1–240). This interaction is not mediated by nucleic acids because benzonase, RNase, and DNase treatments do not prevent NS5A-NS5A interactions. Importantly, DTT abrogates NS5A-NS5A interactions but does not affect NS5A-cyclophilin A interactions. Other reducing agents such as tris(2-carboxyethyl)phosphine and 2-mercaptoethanol also abrogate NS5A-NS5A interactions, implying that disulfide bridges may play a role in this interaction. Cyclophilin inhibitors, cyclosporine A, and alisporivir and NS5A inhibitor BMS-790052 do not block NS5A dimerization, suggesting that their antiviral effects do not involve the disruption of NS5A-NS5A interactions. Four cysteines, Cys-39, Cys-57, Cys-59, and Cys-80, are critical for dimerization. Interestingly, the four cysteines have been proposed to form a zinc-binding motif. Supporting this notion, NS5A dimerization is greatly facilitated by Zn2+ but not by Mg2+ or Mn2+. Importantly, the four cysteines are vital not only for viral replication but also critical for NS5A binding to RNA, revealing a correlation between NS5A dimerization, RNA binding, and HCV replication. Altogether our data suggest that NS5A-NS5A dimerization and/or multimerization could represent a novel target for the development of HCV therapies.
Introduction
Hepatitis C virus (HCV)3 is a single-stranded positive sense RNA virus that causes acute and chronic hepatitis, cirrhosis, and hepatocellular carcinoma (1, 2). HCV infects more than 170 million people worldwide and is the most common cause of liver transplantation in the United States (2). There is no vaccine available for HCV and no generally effective therapy for all HCV genotypes. Although the recent approval of HCV protease inhibitors has further increased the treatment response in genotype 1 patients when used in combination with PEGylated interferon α and ribavirin, the utility of the current standard therapy is limited by the significant side effects associated with each of the three drugs (3, 4). Thus, there is an urgent need for the development of new anti-HCV agents with novel mechanisms of antiviral action.
NS5A has recently emerged as an antiviral target for small molecule design after lagging behind the other components of the viral replication complex. Recent publications describe how small compounds, which apparently target NS5A, inhibit replication with extreme potency, highlighting the validity of NS5A as a target for drug development (5–7). NS5A is a nonstructural protein that has no defined role in the virus life cycle but is absolutely required for RNA replication (8–10) and particle assembly (11–14). It has also been shown to interact with a large number of host proteins including CypA (cyclophilin A) (15–22). NS5A is peripherally anchored to membranes by an N-terminal amphipathic helix (23–27). It is organized into three distinct domains, separated by two repetitive low complexity sequence stretches (28). The N-terminal Domain I contains a conserved zinc-binding site that appears to be a structural metal ion required for role(s) of NS5A in RNA replication (28). The structure of Domain I is dimeric in nature and represents a novel class of protein fold (29). The dimer is oriented in such a way that a large groove exists between the two monomers and has been proposed as an RNA binding site (29). NS5A has RNA binding activity that requires a dimer based on glutaraldehyde-cross-linking experiments (30, 33). Unfortunately, a connection between the NS5A RNA binding activity and a specific aspect of RNA replication or virion assembly remains to be established (30). A recent study showed that both Domains I and II of NS5A exhibit RNA binding activities (20). However, the ability to bind specifically to uridine- and guanosine-rich RNA resides in domain I and the adjacent linker region (33). An alternative structure of Domain I has been determined that is also dimeric in nature, although the interface lacks the large groove or any obvious RNA interaction surface (31). The most advanced inhibitor of NS5A, BMS 790052, seems to bind NS5A at its dimer interface based on current models of NS5A structure and analysis of drug resistance mutants (32). However, the mechanisms that dictate how these NS5A inhibitors suppress HCV replication are currently unknown. Some new and attractive models have been recently proposed (33). We thus sought to examine how these NS5A inhibitors exert their antiviral effects. We hypothesize that NS5A dimers/oligomers bind RNA and that NS5A inhibitors suppress RNA replication by preventing NS5A dimer/oligomer formation and RNA binding. In this study, we specifically asked whether NS5A does indeed form dimers/oligomers and whether the NS5A dimerization/multimerization regulates HCV replication. The ultimate goal of this study is to determine whether NS5A dimerization/multimerization represents a new target for the development of anti-HCV therapies.
EXPERIMENTAL PROCEDURES
DNA Constructs
GST-CypA DNA construct was a generous gift from J. Luban. Modified NS5A coding sequences were constructed using PCR mutagenesis and the Con1 genomic DNA (generous gift from R. Bartenschlager) as a template. Sequences of oligonucleotides used for mutagenesis are listed in Table 1. The PCR products were cloned into BamHI and EcoRI sites in the pGEX-2T vector or SacII and HindIII sites in the pET-UbCHis vector. All restriction enzymes and Phusion Hot Start DNA polymerase were obtained from New England Biolabs. T4 DNA ligase was obtained from Roche Diagnostics. All other reagents were obtained of the highest grade available from Sigma, Fisher, or VWR.
TABLE 1.
Oligonucleotide sequences
| Name | Sequence |
|---|---|
| GST Domain 1 NS5A | 5′-gctacagttcggatcctccggctcgtggctaagaga-3′ |
| 5′-acgtatgcagaatcctcacgtctccgccgtaatgtggga-3′ | |
| Wild-type NS5A-His | 5′-gcgggtaccccgcggtggatccggctcgtggctaa-3′ |
| 5′-gcgggtaccaagcttctattaatggtggtgatggtggtgaccagaggatccgcagcagacgacgtcctcact-3′ | |
| C13A NS5A-His | 5′-gattggatagccacggtgttg-3′ |
| 5′-caacaccgtggctatccaatc-3′ | |
| C39A NS5A-His | 5′-ttcttctcagcccaacgtggg-3′ |
| 5′-cccacgttgggctgagaagaa-3′ | |
| C57A NS5A-His | 5′-caaaccaccgccccatgtgga-3′ |
| 5′-tccacatggggcggtggtttg-3′ | |
| C59A NS5A-His | 5′-acctgcccagccggagcacag-3′ |
| 5′-ctgtgctccggctgggcaggt-3′ | |
| C80A NS5A-His | 5′-cctaggaccgccagtaacacg-3′ |
| 5′-cgtgttactggcggtcctagg-3′ | |
| C98A NS5A-His | 5′-acgggccccgccacgccctcc-3′ |
| 5′-ggagggcgtggcggggcccgt-3′ | |
| C140A NS5A-His | 5′-aacgtaaaggccccgtgtcag-3′ |
| 5′-ctgacacggggcctttacgtt-3′ | |
| C142A NS5A-His | 5′-aagtgcccggcccaggttccg-3′ |
| 5′-cggaacctgggccgggcactt-3′ | |
| C165A NS5A-His | 5′-gctccagcggccaaacccctc-3′ |
| 5′-gaggggtttggccgctggagc-3′ | |
| C190A NS5A-His | 5′-cagctcccagccgagcccgaa-3′ |
| 5′-ttcgggctcggctgggagctg-3′ | |
| C243A NS5A-His | 5′-aaggcaacagccactacccgt-3′ |
| 5′-acgggtagtggctgttgcctt-3′ | |
| Domain 1 with linker (1–249) | 5′-gcgggtaccccgcggtggatccggctcgtggctaa-3′ |
| 5′-tacgtatgcaaagcttagtgtcatgacgggtagtgcatgt-3′ | |
| Truncated Domain 1 (1–245) | 5′-gcgggtaccccgcggtggatccggctcgtggctaa-3′ |
| 5′-tacgtatgcaaagcttagtggtagtgcatgttgccttcaa-3′ | |
| Truncated Domain 1 (1–240) | 5′-gcgggtaccccgcggtggatccggctcgtggctaa-3′ |
| 5′-tacgtatgcaaagcttagtcttcaaggaaggcgcagacag-3′ | |
| Truncated Domain 1 (1–235) | 5′-gcgggtaccccgcggtggatccggctcgtggctaa-3′ |
| 5′-tacgtatgcaaagcttcagctggctagctgatgagc-3′ | |
| Truncated Domain 1 (1–226) | 5′-gcgggtaccccgcggtggatccggctcgtggctaa-3′ |
| 5′-tacgtatgcaaagcttcaaggaggggggagatcccc-3′ | |
| Truncated Domain 1 (1–223) | 5′-gcgggtaccccgcggtggatccggctcgtggctaa-3′ |
| 5′-tcagtatgcaaagctttcccctggccagcctacgct-3′ | |
| Domain 1 without linker (1–213) | 5′-gcgggtaccccgcggtggatccggctcgtggctaa-3′ |
| 5′-tacgtatgcaaagcttagtcgtctccgccgtaatgtggg-3′ |
Production of His-tagged Recombinant Proteins
Overexpression of NS5A recombinant proteins were performed in the BL21(DE3)pCG1 strain of Escherichia coli (generous gift from C. Cameron) grown overnight in 500 ml of Circlegrow (MP Biomedicals) supplemented with 100 μg/μl kanamycin and 20 μg/μl chloramphenicol at 37 °C. The cells were grown to an A600 of 0.8–1.0 before isopropyl β-d-thiogalactopyranoside was added to a final concentration of 0.5 mm. The cells were grown for an additional 3 h at 30 °C. The cells were harvested by centrifugation in a Fiberlite F10–6 × 500y rotor at 6000 rpm for 20 min at 4 °C. The cell pellets were resuspended in 25 ml of lysis buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10% glycerol, complete mini EDTA-free protease inhibitor mixture (Roche Applied Science) per 50 ml of buffer). Lysozyme (125 μg/μl), 1× Bug Buster (Novagen), DNase I (40 μg/μl), and RNase A (40 μg/μl) were added for complete lysis. The extracts were centrifuged in a Fiberlite F21–8 × 50y rotor at 16,000 × g for 20 min at 4 °C. The supernatants were loaded to a His Bind resin (Novagen) and washed with 10 resin volumes of wash buffer (20 mm Tris-HCl, pH 7.5, 0.5 m NaCl, 60 mm imidazole, 10% glycerol, 10 mm DTT, complete mini EDTA-free protease inhibitor mixture (Roche Applied Science) per 10 ml of buffer). The purified His-tagged proteins were eluted with 1.5 ml elution buffer (20 mm Tris-HCl, pH 7.5, 0.5 m NaCl, 1 m imidazole, 10% glycerol, 1 mm DTT, complete mini EDTA-free protease inhibitor mixture (Roche Applied Science) per 10 ml of buffer), and the eluates were dialyzed against dialysis buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5 mm MgCl2, 10% glycerol, 0.5% Igepal CA-630). The purified proteins were aliquoted and stored at −80 °C.
Production of GST-tagged Recombinant Proteins
Overexpression of GST-tagged recombinant proteins were performed in the BL21(DE3) strain (Novagen) of E. coli grown overnight in 500 ml of Circlegrow (MP Biomedicals) supplemented with 100 μg/μl ampicillin at 37 °C. The cells were grown to an A600 of 0.8–1.0 and 0.6 for GST-NS5BΔ21 (generous gift from G. Luo) before isopropyl β-d-thiogalactopyranoside was added to a final concentration of 0.5 mm and 0.4 mm for GST-NS5BΔ21. The cells were grown for an additional 3 h at 30 °C or 8 h at 25 °C for GST-NS5BΔ21. The cells were harvested by centrifugation in a Fiberlite F10–6 × 500y rotor at 6000 rpm for 20 min at 4 °C. The cell pellets were resuspended in 25 ml of lysis buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10% glycerol, complete mini EDTA-free protease inhibitor mixture (Roche Applied Science) per 50 ml of buffer). Lysozyme (125 μg/μl), 1× Bug Buster (Novagen), DNase I (40 μg/μl), and RNase A (40 μg/μl) were added for complete lysis. For GST-NS5BΔ21, the cells were suspended in 50 ml of 1× PBS containing 1% Triton X-100 and protease inhibitor mixture and frozen at −80 °C; the cell lysates were then sonicated twice on ice for 30 s for complete lysis. The extracts were centrifuged in a Fiberlite F21–8 × 50y rotor at 16,000 × g for 20 min at 4 °C; 12,000 × g for 30 min at 4 °C for GST-NS5BΔ21. The supernatants were loaded to a Glutathione-Sepharose 4B resin (GE Healthcare) and washed with 10 resin volumes of PBS. The purified His-tagged proteins were eluted with 1.5 ml of elution buffer (10 mm reduced glutathione in 50 mm Tris-HCl, pH 8.0), and the eluates were dialyzed against dialysis buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5 mm MgCl2, 10% glycerol, 0.5% Igepal CA-630). The purified proteins were aliquoted and stored at −80 °C.
NS5A Pulldown Studies
Glutathione beads were incubated for 2 h in dialysis buffer (50 mm Tris, pH 7.4, 100 mm NaCl, 5 mm MgCl2, 10% glycerol, 0.5% Nonidet P-40) with 5 mg/ml BSA and washed twice at 4 °C in binding buffer (20 mm Tris, pH 7.9, 0.5 m NaCl, 10% glycerol, and 1% Nonidet P-40). Meanwhile, 5 μg of GST, GST-D1, GST-CypA, or GST-NS5BΔ21 was mixed with 2.5 μg of NS5A-His in a total volume of 100 μl of binding buffer for 3 h at 4 °C on wheel. Glutathione beads (25 μl) were added to the recombinant protein mixture for 30 min at 4 °C and washed three times with 400 μl of binding buffer. The beads were pelleted for 30 s at 2000 × g in a microcentrifuge and bound material was eluted with 25 μl of SDS sample buffer, heated for 5 min, and frozen at −20 °C. Bound material was then analyzed by Western blotting using anti-GST, anti-CypA, and anti-His antibodies.
In Vitro Transcription of HCV Subgenomic RNA and Electroporation
The in vitro transcription of RNA was accomplished using the T7 MEGAscript kit (Ambion) following the manufacturer's instructions. In vitro transcribed RNAs were introduced into Huh7.5.1 cells by electroporation. Trypsinized cells were washed twice with and resuspended in PBS (calcium-free, magnesium-free) at 1 × 107 cells/ml. Ten micrograms of RNA for each mutant was mixed with 0.4 ml of cells in a 4-mm cuvette, and a Bio-Rad Gene Pulser system was used to deliver a single pulse at 0.27 kV, 100 ohms, and 960 microfarads, and the cells were then plated in 12-well dishes. RNA transfection efficiency and HCV subgenomic replication were assessed by measuring the Renilla and firefly luciferase activities, respectively, 4 h and 1–7 days post-electroporation using the Dual-Luciferase Reporter Assay System (Promega) following the manufacturer's instructions. Relative light units values were normalized to the 4-h time point values. The data are shown as the means ± S.D.
RNA Binding Assay
The RNA binding experiments were performed as previously described using a Beacon2000 fluorescence polarization system (Invitrogen) (34). 0–2000 nm NS5A proteins were incubated for 30 s with 0.1 nm 3′-fluorescein-labeled rU15-Fl RNA in binding buffer (20 mm HEPES, pH 7.5, 5 mm MgCl2, and 0.5 mm TCEP) at 25 °C. Binding of NS5A was measured by the change in polarization (ΔmP) was measured by fluorescence polarization. The data were fit to a hyperbola using KaleidaGraph (Synergy Software).
RNA Immunoprecipitation-PCR
RNA immunoprecipitation-PCR was carried out by transfecting Huh7-Con1 cells (1 million) with 15 μg of NS5A-His pcDNA3 plasmids. Three days post-transfection, the cells were incubated in hypotonic buffer (10 mm HEPES, pH 7.6, 1.5 mm MgCl2, 10 mm KCl, 0.2 mm phenylmethylsulfonyl fluoride) and incubated on ice for 5 min. The cells were lysed with 0.5% Nonidet P-40, vortexed, incubated on ice for an additional 10 min, vortexed, and centrifuged at 4,000 × g at 4 °C for 15 min, and glycerol was added to a final concentration of 5%. Two hundred micrograms of each cell lysate was incubated with 50 μl of Ni+ beads for 1 h at room temperature. The beads were washed three times with wash buffer (10 mm Tris, pH 7.6, 100 mm KCl, 5 mm MgCl2, and 1 mm DTT), and bound material was eluted with 0.5 m imidazole, treated with 100 μg of proteinase K at 37 °C for 30 min followed by two rounds each of phenol-chloroform-isoamyl alcohol (25:24:1) and chloroform extraction, and precipitated by ethanol with 10 μg of glycogen. RNA was purified by centrifugation for 30 min at 13,000 × g, washed with 70% ethanol, and suspended in 5 mm Tris, pH 8.0. Quantitative real time PCR was carried out as we described (18).
RESULTS
NS5A Forms Dimers Directly through Domain I Interactions
Regardless of the differences between the two proposed structures of the Domain I of NS5A, both models show that NS5A is dimeric in nature. We sought to define whether NS5A dimerization has a functional role in viral replication. First, we demonstrated using a GST pulldown assay that NS5A truly forms dimers. Specifically, glutathione beads were incubated for 2 h in dialysis buffer (50 mm Tris, pH 7.4, 100 mm NaCl, 5 mm MgCl2, 10% glycerol, 0.5% Nonidet P-40) with 5 mg/ml BSA and washed twice at 4 °C in binding buffer (20 mm Tris, pH 7.9, 0.5 m NaCl, 10% glycerol, and 1% Nonidet P-40). Meanwhile, 5 μg of GST or GST-Domain I NS5A containing the linker (1–249) was mixed with 2.5 μg of NS5A-His in a total volume of 100 μl of binding buffer for 3 h at 4 °C on wheel. Glutathione beads (25 μl) were added to the recombinant protein mixture for 30 min at 4 °C and washed three times with 400 μl of binding buffer. The beads were pelleted for 30 s at 2000 × g in a microcentrifuge, and bound material was eluted with 25 μl of SDS sample buffer, heated for 5 min, and frozen at −20 °C. Bound material was then analyzed by Western blotting using anti-GST, anti-CypA, and anti-His antibodies. We found that GST-Domain I NS5A containing the linker, but not GST alone, captures full-length NS5A-His (Fig. 1A, top panels). Importantly, we also found that GST-Domain 1 NS5A captures Domain I (Fig. 1B), suggesting that NS5A dimerization occurs via the Domain I of NS5A. Similar amounts of full-length and Domain I of NS5A were used (Fig. 1A, bottom panel). Note that after several attempts, we were unable to obtain significant amounts of soluble GST full-length NS5A (data not shown). This is the reason why GST-Domain 1 NS5A was mainly used for pulldown experiments.
FIGURE 1.

NS5A dimerization occurs directly through Domain I. A and B, GST or GST-Domain I NS5A containing the linker was used as bait to pull down full-length NS5A-His (A) or Domain I NS5A-His (B). Captured proteins were analyzed by Western blotting using anti-His and anti-GST antibodies. C, deletion mutants of Domain I NS5A-His were generated and used as prey in pulldown assays with GST or GST-Domain I NS5A as bait. Captured proteins were analyzed by Western blotting using anti-His and anti-GST antibodies. D, recombinant proteins were treated with benzonase, RNase, or DNase to remove contaminating nucleic acids before pulldown assays. GST-CypA/NS5A-His were used as controls because this interaction has been shown to be direct. Captured proteins were analyzed by Western blotting using anti-His and anti-GST antibodies.
After demonstrating that Domain I of NS5A indeed forms dimers, we examined which regions of Domain I are critical for dimerization. To address this issue, we generated a panel of deletions in Domain I of NS5A and tested these truncated proteins for their capacities to be captured by GST or GST-Domain I NS5A-His as described above. We first compared the dimerization capacities of Domain I NS5A with (1–249) or without linker (1–213) because this low complexity sequence region has been previously shown to confer functional properties to NS5A (34). Importantly, we found that Domain I interactions are lost in the absence of the linker (Fig. 1C). To further characterize which specific regions of the linker are important for dimerization, we made truncations starting from the end of the low complexity sequence: 1–245, 1–240, 1–235, 1–226, and 1–223, and compared the abilities of these truncated proteins to dimerize. The 1–245 and 1–240 truncations were created by postulating that the two prolines, which reside between the two truncations, may be important for NS5A dimerization. We found that dimerization still occurs for these two truncations; however, the interactions are dramatically diminished (Fig. 1C). Importantly, further truncations of Domain I (1–235, 1–226, and 1–223) totally abolished dimerization (Fig. 1C). Similar amounts of recombinant proteins were used as prey (Fig. 1C, top panel) and bait (Fig. 1C, bottom panel). These results suggest that amino acids 1–240 are absolutely necessary for NS5A dimerization.
To determine whether NS5A dimerization is mediated by nucleic acids that may serve as a bridge for NS5A-NS5A contacts, we treated recombinant proteins with benzonase, RNase, or DNase to eliminate any contaminating nucleic acids prior to conducting pulldown experiments. As a control, we used GST-CypA as bait to capture full-length NS5A-His because this interaction has been shown to be direct (15–22). As expected, both GST-CypA and GST-Domain I, but not GST, capture full-length NS5A-His (Fig. 1D). Importantly, benzonase, RNase, or DNase treatments do not affect these interactions (Fig. 1D), demonstrating that NS5A-NS5A contacts are not mediated by nucleic acids. Note that NS5A-NS5A interactions are apparently weaker than NS5A-CypA interactions (Fig. 1D, top panel) and that similar amounts of bait (Fig. 1D, middle panel) and prey proteins (Fig. 1D, bottom panel) were used.
Cyclophilin and NS5A Inhibitors Do Not Affect NS5A Dimerization
After demonstrating direct contacts between Domain I of NS5A, we tested our initial hypothesis that NS5A inhibitors block HCV replication by disrupting NS5A dimer formation. To test this hypothesis, we examined the inhibitory effect of the NS5A inhibitor BMS-790052 on NS5A dimerization. We also examined the effect of another class of anti-HCV agents, the cyclophilin inhibitors CsA and alisporivir, which have been shown to disrupt NS5A-CypA interactions (35–37). We first found that CsA (Fig. 2A) or alisporivir (Fig. 2B) do not interfere with the capture of either full-length (Fig. 2, A and B, top panels) or Domain I NS5A (Fig. 2, A and B, middle panel) by GST-Domain I NS5A. This is in accordance with the fact that cyclophilin inhibitors target cyclophilin A and not NS5A. Interestingly, we found that the NS5A inhibitor BMS-790052 fails to prevent NS5A dimerization (Fig. 2C), suggesting that the mechanism of antiviral action of NS5A inhibitors do not involve the rupture of NS5A-NS5A interactions. While our studies were in progress, a report showed that NS5A inhibitors such as BMS-790052 impair hyperphosphorylation of NS5A and that this activity is mediated by sequences within Domain 1 (38).
FIGURE 2.

Cyclophilin inhibitors and NS5A inhibitors do not affect NS5A dimerization. Pulldown assays using GST or GST-Domain I NS5A as bait to capture full-length NS5A-His or Domain I NS5A-His were performed in the presence of 2 μm CsA (A), alisporivir (B), or BMS-790052 (C). Captured proteins were analyzed by Western blotting using anti-His and anti-GST antibodies.
Reducing Agents and EDTA Reduce NS5A Dimerization, whereas Zn2+, but Not Mg2+ or Mn2+, Increases Dimerization
In light of these somewhat unexpected results, we decided to further dissect the mechanism of action of NS5A dimerization. Because the Domain I of NS5A is enriched in cysteines, we postulated that NS5A-NS5A contacts occur through disulfide bridges via these cysteines. To test this hypothesis, we asked whether reducing agents inhibit NS5A dimerization. Remarkably, the addition of DTT prevents the capture of full-length NS5A-His (Fig. 3A, top panel) or Domain I NS5A-His (Fig. 3A, middle panel) by GST-Domain I NS5A. This finding suggests that NS5A dimerization occurs through disulfide bridge formation between cysteines, which reside in Domain I of NS5A. To further confirm these findings, we tested other reducing agents such as 2-mercaptoethanol and TCEP. Importantly, these reducing agents also block NS5A dimerization (Fig. 3, B and C). More importantly, the block is specific because reducing agents such as DTT do not prevent NS5A-CypA interactions (Fig. 3D).
FIGURE 3.
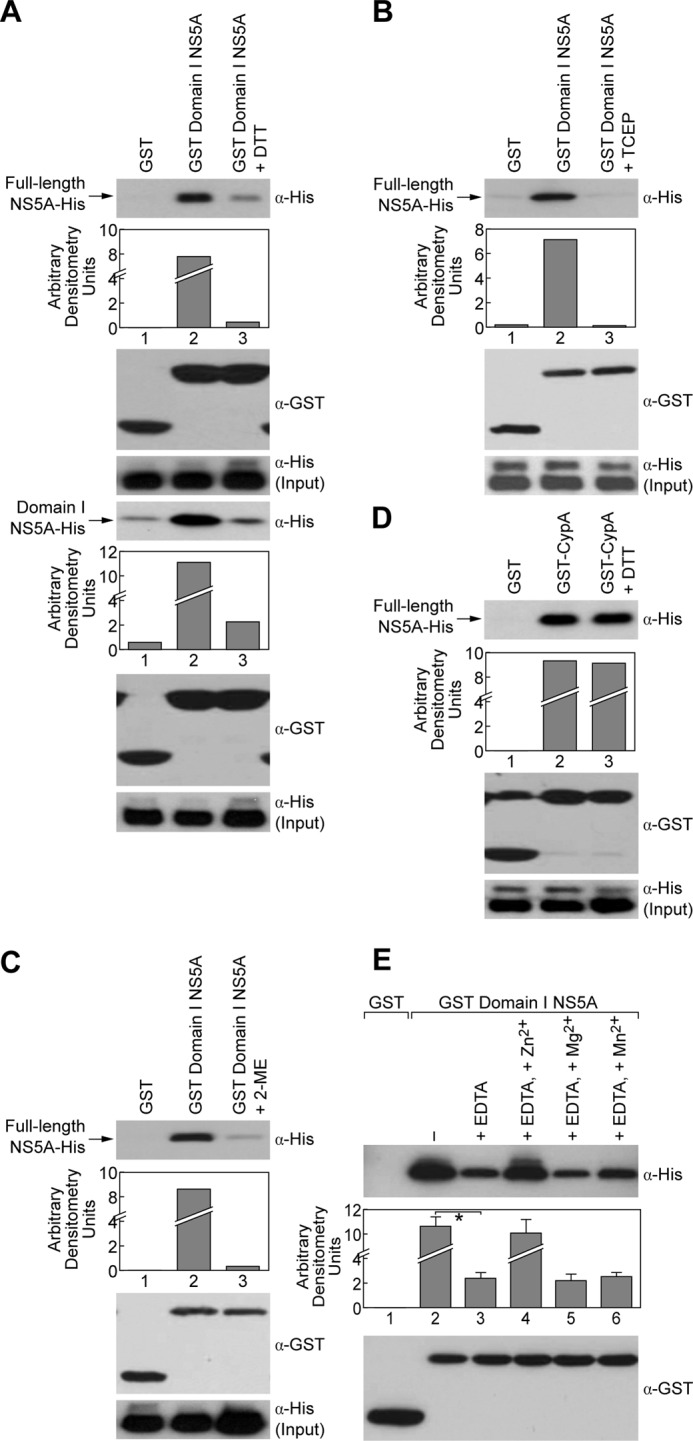
Reducing agents and EDTA reduce NS5A dimerization, whereas Zn2+, but not Mg2+ or Mn2+, increases dimerization. A, pulldown assays using GST or GST-Domain I NS5A as bait to capture full-length NS5A-His or Domain I NS5A-His were performed in the presence of DTT (5 μm). B, pulldown assays using GST or GST-CypA as bait to capture full-length NS5A-His were performed in the presence of DTT (5 μm). C and D, pulldown assays using GST or GST-Domain I NS5A as bait to capture full-length NS5A-His were performed in the presence of TCEP (C, 5 μm) and 2-mercaptoethanol (D, 50 μm). E, pulldown assays were performed using GST or GST-Domain 1 NS5A to capture full-length NS5A-His in the presence of EDTA (20 mm), EDTA (5 mm) + Zn2+(10 mm), EDTA (5 mm) + Mg2+ (10 mm), or EDTA (5 mm) + Mn2+ (10 mm). Captured proteins were analyzed by Western blotting using anti-His and anti-GST antibodies. In three independent experiments, we measured the relative intensity of specific bands using a GS-800 densitometer and performed statistical analysis using the Student's t test. A p value <0.05 (*) denotes a statistically significant difference.
Because a previous study elegantly showed that the N-terminal Domain I of NS5A contains a set of conserved cysteines that serve as binding site for Zn2+, a structural metal ion apparently required for NS5A function in HCV RNA replication (28), we asked whether chelating agents such as EDTA affect NS5A dimerization. We found that EDTA significantly reduced NS5A dimerization (Fig. 3E), but more importantly, we found that the addition of Zn2+, but not Mg2+ or Mn2+, totally restored NS5A dimerization in the presence of EDTA. Together, these results suggest that Zn2+ and/or the Zn2+-binding site of NS5A regulate NS5A dimerization.
Cysteines at Positions 39, 57, 59 and 80 Are Critical for Dimerization
Our two independent observations that reducing agents inhibit NS5A dimerization and that Zn2+ increases NS5A dimerization led us to postulate that cysteines in the Domain I of NS5A are responsible for NS5A-NS5A interactions. To test this hypothesis, we replaced each cysteine in Domain I of NS5A, including the linker region, with an alanine resulting in a panel of cysteine mutants. We first verified that similar amounts of NS5A proteins were used as input (Fig. 4A, top panel). We then conducted pulldown assays as above using GST or GST-Domain I NS5A as bait. We found that C13A, C98A, C140A, C142A, C165A, C190A, and C243A NS5A mutant proteins behave like wild-type NS5A in terms of dimerization (Fig. 4A). In sharp contrast, we found that the C39A, C57A, C59A, and C80A NS5A mutant proteins fail to dimerize (Fig. 4, bottom panels), suggesting that these four cysteines are crucial for NS5A dimerization. To exclude the possibility that the introduced mutations profoundly altered the structure or folding of NS5A, we asked whether these NS5A mutant proteins are still able to bind CypA. Importantly, all cysteine mutants retained their abilities to bind CypA, and these interactions are inhibited by CsA (Fig. 4B). Together these results suggest that the four cysteines: Cys-39, Cys-57, Cys-59, and Cys-80, are specific key residues for NS5A-NS5A interactions.
FIGURE 4.

Cysteines at positions 39, 57, 59, and 80 of Domain I are critical for NS5A dimerization. A, full-length NS5A-His with single mutations at each cysteine residue in Domain I were generated and used as prey in pulldown assays with GST or GST-Domain I NS5A as bait. These data are representative of three independent experiments using three different batches of recombinant NS5A proteins. B, cysteine mutants in A were tested for their ability to bind cyclophilin A with or without the presence of CsA (2 μm).
Cysteines Critical for Dimerization Are Also Important for RNA Binding and Replication
Because NS5A has been shown to bind RNA (34, 39), we then examined whether dimerization has any functional aspect related to RNA binding. To address this issue, we measured the RNA binding capacity of each NS5A cysteine mutant as described previously (34). We obtained the following Kd values: 215 nm for wild type NS5A, 830 nm for C13A, 840 nm for C98A, 790 nm for C140A, 600 nm for C142A, 502 nm for C165A, 260 nm for C190A, and 430 nm for C243A (Table 2 and Fig. 5A). Importantly, we obtained significantly higher Kd values for the four cysteine NS5A mutants, which fail to dimerize −1300 nm for C39A, 1304 nm for C57A, 1213 nm for C59A, and 1006 nm for C80A (Table 2 and Fig. 5A). This represents an average 5.6-fold increase for the cysteine mutants that fail to dimerize compared with an average of 2.8-fold increase for the rest of the cysteine mutants relative to wild-type NS5A. We thus identified four conserved cysteines in the Domain I of NS5A that are important for both NS5A dimerization and NS5A RNA binding. This is the first link between NS5A dimerization and NS5A binding to RNA.
TABLE 2.
NS5A cysteine mutant that fail to dimerize/multimerize bind weakly RNA
| FL-NS5A-His | Kd |
|---|---|
| WT | 215 ± 60 nM |
| C39A | 1300 ± 544 nM |
| C57A | 1304 ± 595 nM |
| C59A | 1213 ± 550 nM |
| C80A | 1006 ± 430 nM |
| C13A | 830 ± 400 nM |
| C98A | 840 ± 340 nM |
| C140A | 790 ± 416 nM |
| C142A | 600 ± 143 nM |
| C165A | 502 ± 153 nM |
| C190A | 260 ± 84 nM |
| C243A | 430 ± 75 nM |
FIGURE 5.

Cysteines critical for dimerization are also important for RNA-binding and replication. A, NS5A (0–2000 nm) and 0.1 nm rU15-FI were gently mixed in binding reaction buffer (20 mm HEPES, pH 7.5, 100 mm NaCI, 5 mm MgCl2, and 0.5 mm TCEP) and incubated for 30 s at 25 °C. Binding of NS5A was measured by the change in polarization (mP). The data were fit to a hyperbola using KaleidaGraph (Synergy Software). B, full-length NS5A-His with alanine substitutions at Cys-39, Cys-57, Cys-59, and Cys-80 were tested for their ability to bind GST-NS5BΔ21 relative to wild-type NS5A. C, Huh7.5.1 cells were electroporated with luciferase reporter subgenomic RNA transcripts containing cysteine mutations in NS5A. The cells were lysed at the given time points and post-electroporation, and the replication fitness was measured using a luciferase assay. The results (triplicates) are representative of three independent experiments. D, Huh7-Con1 cells were transfected with wild-type or cysteine NS5A-His plasmids for 3 days. The cell lysates were incubated with Ni+ beads for 1 h, bound material was eluted, RNA was purified, and quantitative real time PCR was executed as described (18). The amount of HCV RNA precipitated by wild-type NS5A-His was arbitrary fixed at 100. The results (triplicates) are representative of two independent experiments.
Because a previous study showed that the Domain I of NS5A contains an NS5B-binding site (40), we asked whether the four cysteine mutations: C39A, C57A, C59A and C80A, influence the contact between NS5A and the HCV NS5B polymerase. Importantly, we found that all four cysteine NS5A mutants bind to NS5B at wild-type levels (Fig. 5B). Thus, NS5A-CypA and NS5A-NS5B contacts are preserved for the four cysteine NS5A mutants.
Because NS5A is a key component of the HCV replication machinery, we investigated whether the four cysteine NS5A mutants identified above also play a role in HCV RNA replication. Specifically, HCV subgenomic RNAs containing cysteine mutations in the Domain I of NS5A were created and electroporated into Huh-7.5 cells. RNA replication was monitored by determination of luciferase activity in cell lysates at 4, 24, 48, and 72 h post-electroporation. The 4-h time point value was used for normalization and for correction for different transfection efficiencies. We found that wild-type virus and C13A mutant virus replicate well, whereas the four cysteine C39A, C57A, C59A, and C80A NS5A mutant viruses fail to replicate (Fig. 5C). It is important to note that cysteine mutant viruses other than the C39A, C57A, C59A, and C80A NS5A mutant viruses also fail to replicate (Fig. 5C). These replication defects are likely unrelated to NS5A dimerization because these cysteine NS5A mutants dimerize like wild-type NS5A (Fig. 4A).
We then asked whether NS5A dimerization influences the contact between NS5A and HCV RNA in a cellular context. To address this issue, Huh7-Con1 cells were transfected with His-tagged wild type or cysteine NS5A mutants. The cells were lysed, and NS5A proteins were precipitated with Ni+ beads. Bound viral RNA was eluted, purified, and quantified by quantitative real time PCR. Importantly, the four cysteine mutants, C39A, C57A, C59A, and C80A, that poorly dimerize (Fig. 4A) and that bind less efficiently synthetic RNA than wild-type NS5A (Fig. 5A), precipitate HCV RNA less efficiently than wild-type NS5A or a cysteine mutant that dimerizes well (C190A) (Fig. 5D). Altogether our results reveal a correlation between NS5A dimerization, NS5A RNA binding, and HCV replication.
DISCUSSION
NS5A has long been overlooked as a drug target because of its lack of enzymatic activity and poorly characterized role in HCV RNA replication. Clinical validation, however, has recently been achieved with NS5A inhibitors, indicating that small molecules targeting the viral protein without any known enzymatic activity can have profound antiviral effects and have the potential to be part of a therapeutic regimen based on combinations of HCV inhibitors (5–7). Although NS5A has been shown to be an attractive target for inhibition of HCV RNA replication, the exact mechanisms by which these NS5A inhibitors suppress viral replication is unknown. In this study, we sought to determine how NS5A inhibitors exert their antiviral effects. We hypothesize that HCV RNA replication is impeded by the prevention of NS5A dimer formation. The N-terminal Domain I of NS5A has been shown in crystallography structural models to be dimeric in nature (29, 31). In addition, this domain has been shown to be most conserved among genotypes with Zn2+ binding and RNA binding activities (28–30). In this study, we demonstrated that Domain I of NS5A is sufficient to mediate NS5A-NS5A interactions, verifying the published structural and functional data (29, 31). Interestingly, we found that deleting the linker located on the C terminus of Domain I abrogates NS5A dimerization. It is attractive to postulate that this linker, which is critical for NS5A dimerization, has a direct implication on HCV RNA replication. Importantly, a large number of NS5A adaptive mutations map exactly to this linker region (8, 41). Furthermore, this linker region along with Domain I has also been shown to be the minimal RNA-binding domain of NS5A (34). One can envision that deleting the linker of NS5A triggers a conformational change, rendering NS5A less able to form dimers. We also demonstrated that NS5A dimerization occurs via direct contacts between NS5A proteins and not via RNA or DNA. Indeed, we showed that nucleic acid removal by enzymatic treatment did not affect NS5A dimerization. It is important to mention that published crystallographic data showed two distinct conformations of NS5A dimers that are both based on constructs of Domain I without the linker. One possibility to explain this apparent discrepancy is that in our study we used a few micrograms of NS5A to observe dimerization, whereas crystallization studies used larger amounts of proteins (mg range). One thus cannot exclude the possibility that the use of lower protein concentrations of NS5A for crystallization would have resulted in monomeric rather than dimeric forms. Another possibility is that crystallization of NS5A Domain I with the linker would facilitate dimer or even oligomer formation.
Note that we conducted FRET microscopy analyses to confirm NS5A-NS5A contacts in live cells. We observed a major FRET signal in the endoplasmic reticulum using wild-type NS5A, suggesting close proximity between the viral proteins. However, we did not observe a significant decrease in FRET signal using NS5A cysteine mutants (data now shown). We speculate that even if NS5A cysteine mutant proteins do not form dimers in a cell, their close association in the endoplasmic reticulum membrane via their N-terminal anchor preserves the FRET signal.
We observed NS5A dimers in our pulldown experiments when using 10–20 μg of NS5A protein. However, when using NS5A at higher concentrations (>100 μg), we observed NS5A forms of higher molecular weight that could represent trimers and tetramers (data not shown). At this stage, we do not know whether these high molecular forms represent nonspecific NS5A aggregation complexes or true higher order NS5A complexes. It is important to note that Love et al. (31) observed an oligomeric state of NS5A and modeled possible NS5A oligomers based on their crystal structure.
Symmetry and dimeric structures are apparently important contributors to the antiviral activities of NS5A inhibitors (5, 7). Biochemical studies have also suggested that these inhibitors directly bind to NS5A (5, 7). In addition, mutations that confer resistance to these inhibitors mapped to the N-terminal region of NS5A (6, 32). Therefore, we expected NS5A-NS5A interactions to be disrupted by the NS5A inhibitor BMS-790052. Surprisingly, it did not affect NS5A dimerization. We thus decided to further characterize the molecular features of NS5A dimerization by determining how NS5A dimerization could be interrupted. Our hypothesis that dimerization occurs through disulfide bridge-forming cysteines in Domain I was proved to be correct because NS5A interactions were reduced upon treatment with reducing agents such as DTT, TCEP, and 2-mercaptoethanol. Importantly, this block is specific because reducing agents such as DTT do not affect NS5A-CypA interactions. This led us to examine which cysteines in the Domain I of NS5A are responsible for NS5A dimerization. Site-directed mutagenesis revealed that four cysteine mutants, C39A, C57A, C59A, and C80A, fail to dimerize, whereas the others behave like wild-type NS5A. It was important to show that all of NS5A cysteine mutants are still able to bind to CypA or NS5B, demonstrating that the cysteine mutations do not affect the overall folding of NS5A. Coincidentally, the four cysteines, Cys-39, Cys-57, Cys-59, and Cys-80, have been shown to serve as the Zinc2+-binding site in NS5A (Fig. 6) (28). NS5A has been shown to coordinate one Zn2+ atom/protein within the N-terminal Domain I (28), and its RNA binding activity requires Zn2+ (34). Supporting the role of Zinc2+ binding in NS5A function, we demonstrated that EDTA reduces dimerization, whereas Zn2+, but not Mg2+ or Mn2+, facilitates NS5A dimerization. To further highlight the importance of these four cysteine residues in NS5A function, we showed through a fluorescence polarization assay that NS5A with any of its Cys-39, Cys-57, Cys-59, and Cys-80 residues substituted exhibits a reduced capacity to bind synthetic or viral RNA. Collectively, these results suggest that NS5A dimerization, through Cys-39, Cys-57, Cys-59, and Cys-80, plays an important role in RNA binding and that this activity requires Zn2+. Zn2+ coordination in proteins has been demonstrated to be involved in a wide range of enzymatic activities, nucleic acid interactions, metabolic regulatory functions, protein-protein interactions, and structural fold maintenance (42). Moreover, Zn2+ appears to function in different capacities in NS5A (28).
FIGURE 6.

Model for Domain I-Domain I contacts displaying Cys-39, Cys-57, Cys-59, and Cys-80 required for NS5A dimerization. The model was prepared using Chem3D Pro software with the Tellinghuisen et al. (29) and Love et al. (31) crystal structures of NS5A Domain I (Protein Data Bank codes 1ZH1 and 3FQM, respectively) (29, 31). The two monomers are cyan and purple. The zinc atoms are represented as white balls. Cys-39, Cys-57, Cys-59, and Cys-80 are highlighted in yellow on both monomers with amino acid side chains displayed. The Tyr-93 residue in Domain I responsible for the NS5A inhibitor BMS 790052 resistance is highlighted in dark pink. For orientation purposes, the two N-terminal amino acids are colored green in each model: Phe-36/Phe-37 for the Tellinghuisen et al. structure and Leu-32/Gly-33 for the Love et al. structure. Three different views of the dimers are presented.
We are postulating that EDTA chelates Zn2+ from NS5A, rendering it unable to fold properly and form higher order structures. This is consistent with our mutational analysis that suggests changing the Zn2+-binding cysteine residues diminishes dimerization, RNA binding, and replication. Thus, our mechanistic explanation is that the presence of Zn2+ atoms is needed for the structural integrity of NS5A. As previously reported by Tellinghuisen et al. (28), Zn2+ coordination in proteins has been demonstrated to be involved in a wide range of enzymatic activities, nucleic acid interactions, metabolic regulatory functions, protein-protein interactions, and structural fold maintenance (28). The NS5A Zn2+-binding site does not possess the characteristics of any of the classes of enzymatic functions associated with Zn2+-binding proteins (28). Moreover, this site is not similar to known classes of Zn2+-dependent nucleic acid interaction motifs (28). The coordination of the NS5A Zn2+ atom by four cysteines suggests this is a structural metal ion necessary for NS5A folding or stability.
The four Cys-39, Cys-57, Cys-59, and Cys-80 residues are conserved across all HCV genotypes, with the exception of Cys-39 in genotype 4A (28), which led us to hypothesize that they must be essential for the HCV life cycle. We demonstrated here that mutating any of the four Cys-39, Cys-57, Cys-59, and Cys-80 residues led to a lethal phenotype in HCV replication. It is possible that the Cys-39, Cys-57, Cys-59, and Cys-80 residues influence HCV RNA replication by affecting the activity RNA-dependent RNA polymerase NS5B because the Domain I of NS5A contains an NS5B-binding site (40). However, we showed that the C39A, C57A, C59A, and C80A NS5A mutants were still able to bind NS5B. This is in accordance with previous data showing that NS5B binds NS5A via two discontinuous regions, amino acids 105–162 (Domain I) and 277–334 (Domain II) (40), which do not include the four cysteines critical for NS5A dimerization and RNA binding. Our results may suggest that NS5A dimerization plays an important role in replication, likely by enhancing NS5A binding to RNA. Other cysteine mutations also inhibit RNA replication. However, these mutations do not affect NS5A dimerization. It is likely that these other cysteines play a critical role in HCV replication unrelated to NS5A dimerization. Cys-142 and Cys-190 have been shown to form disulfide bonds, resulting in a covalent link between β-strands in one of the NS5A structural models (29). We showed here that C142A and C190A mutants still dimerize but fail to replicate, indicating that these cysteines may be important in intramolecular interactions within NS5A and that these are important for the RNA replicase functions of NS5A.
We showed that HCV RNA replication is greatly impacted when any of the cysteine in the Domain I of NS5A is mutated. It is important to emphasize that NS5A plays multiple roles in HCV replication (replication complex formation, RNA replication, budding, modulating the innate response, etc.) and has multiple viral and host partners. Thus, mutating residues in the Domain I of NS5A can influence multiple properties of NS5A that are critical for HCV replication. It is likely that the best or even a unique way to definitely demonstrate that NS5A dimerization is vital for HCV replication would be to identify specific inhibitors of NS5A dimerization and to test them for their capacities to block viral replication. We are currently screening compound libraries to identify such inhibitors.
This study provides the first correlation between NS5A dimerization without the aid of cross-linking agents and HCV RNA replication. NS5A has been shown to interact with other HCV nonstructural proteins (43), and it has been demonstrated that its N-terminal helix anchors it into the endoplasmic reticulum membrane (23, 25, 26). One can envision that in the context of the viral replication complex, NS5A is present in very high concentrations such that NS5A exists on membranes as dimers and/or multimers and may act as a scaffold for the viral replication complex. Our data provide a molecular perspective on this scaffolding function of NS5A and possible mechanisms for its inhibition. Furthermore, our data provide the first link between NS5A dimerization, NS5A binding to RNA, and HCV replication. This makes NS5A dimerization and/or multimerization an attractive putative target for the development of novel anti-HCV agents.
Acknowledgments
We thank J. Kuhns for secretarial assistance, G. Luo for the GST-NS5BΔ21 plasmid, F. Chisari for the Huh7.5.1. cells, and R. Bartenschlager for Huh7-Luc/Neo ET cells and Con1 DNA plasmid.
This work was supported by U.S. Public Health Service Grants AI087746 (to P. A. G.) and GM089001 (to C. E. C.).
- HCV
- hepatitis C virus
- TCEP
- tris(2-carboxyethyl)phosphine
- DTT
- dithiothreitol
- CsA
- cyclosporine A.
REFERENCES
- 1. Hoofnagle J. H. (2002) Course and outcome of hepatitis C. Hepatology 36, S21–S29 [DOI] [PubMed] [Google Scholar]
- 2. Alter H. J., Seeff L. B. (2000) Recovery, persistence, and sequelae in hepatitis C virus infection. A perspective on long-term outcome. Semin. Liver Dis. 20, 17–35 [DOI] [PubMed] [Google Scholar]
- 3. Di Bisceglie A. M., McHutchison J., Rice C. M. (2002) New therapeutic strategies for hepatitis C. Hepatology 35, 224–231 [DOI] [PubMed] [Google Scholar]
- 4. Tan S. L., Pause A., Shi Y., Sonenberg N. (2002) Hepatitis C therapeutics. Current status and emerging strategies. Nat. Rev. Drug Discov. 1, 867–881 [DOI] [PubMed] [Google Scholar]
- 5. Gao M., Nettles R. E., Belema M., Snyder L. B., Nguyen V. N., Fridell R. A., Serrano-Wu M. H., Langley D. R., Sun J. H., O'Boyle D. R., 2nd, Lemm J. A., Wang C., Knipe J. O., Chien C., Colonno R. J., Grasela D. M., Meanwell N. A., Hamann L. G. (2010) Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465, 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lemm J. A., O'Boyle D., 2nd, Liu M., Nower P. T., Colonno R., Deshpande M. S., Snyder L. B., Martin S. W., St Laurent D. R., Serrano-Wu M. H., Romine J. L., Meanwell N. A., Gao M. (2010) Identification of hepatitis C virus NS5A inhibitors. J. Virol. 84, 482–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lemm J. A., Leet J. E., O'Boyle D. R., 2nd, Romine J. L., Huang X. S., Schroeder D. R., Alberts J., Cantone J. L., Sun J. H., Nower P. T., Martin S. W., Serrano-Wu M. H., Meanwell N. A., Snyder L. B., Gao M. (2011) Discovery of potent hepatitis C virus NS5A inhibitors with dimeric structures. Antimicrob. Agents Chemother. 55, 3795–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blight K. J., Kolykhalov A. A., Rice C. M. (2000) Efficient initiation of HCV RNA replication in cell culture. Science 290, 1972–1974 [DOI] [PubMed] [Google Scholar]
- 9. Pietschmann T., Lohmann V., Rutter G., Kurpanek K., Bartenschlager R. (2001) Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 75, 1252–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krieger N., Lohmann V., Bartenschlager R. (2001) Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75, 4614–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Appel N., Zayas M., Miller S., Krijnse-Locker J., Schaller T., Friebe P., Kallis S., Engel U., Bartenschlager R. (2008) Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathogens 4, e1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masaki T., Suzuki R., Murakami K., Aizaki H., Ishii K., Murayama A., Date T., Matsuura Y., Miyamura T., Wakita T., Suzuki T. (2008) Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82, 7964–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hughes M., Griffin S., Harris M. (2009) Domain III of NS5A contributes to both RNA replication and assembly of hepatitis C virus particles. J. Gen. Virol. 90, 1329–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benga W. J., Krieger S. E., Dimitrova M., Zeisel M. B., Parnot M., Lupberger J., Hildt E., Luo G., McLauchlan J., Baumert T. F., Schuster C. (2010) Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51, 43–53 [DOI] [PubMed] [Google Scholar]
- 15. Hanoulle X., Badillo A., Wieruszeski J. M., Verdegem D., Landrieu I., Bartenschlager R., Penin F., Lippens G. (2009) Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 284, 13589–13601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Waller H., Chatterji U., Gallay P., Parkinson T., Targett-Adams P. (2010) The use of AlphaLISA technology to detect interaction between hepatitis C virus-encoded NS5A and cyclophilin A. J. Virol. Methods 165, 202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chatterji U., Lim P., Bobardt M. D., Wieland S., Cordek D. G., Vuagniaux G., Chisari F., Cameron C. E., Targett-Adams P., Parkinson T., Gallay P. A. (2010) HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J. Hepatol. 53, 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coelmont L., Hanoulle X., Chatterji U., Berger C., Snoeck J., Bobardt M., Lim P., Vliegen I., Paeshuyse J., Vuagniaux G., Vandamme A. M., Bartenschlager R., Gallay P., Lippens G., Neyts J. (2010) DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 5, e13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fernandes F., Ansari I. U., Striker R. (2010) Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. PLoS One 5, e9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Foster T. L., Gallay P., Stonehouse N. J., Harris M. (2011) Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J. Virol. 85, 7460–7464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Verdegem D., Badillo A., Wieruszeski J. M., Landrieu I., Leroy A., Bartenschlager R., Penin F., Lippens G., Hanoulle X. (2011) Domain 3 of NS5A protein from the hepatitis C virus has intrinsic α-helical propensity and is a substrate of cyclophilin A. J. Biol. Chem. 286, 20441–20454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang F., Robotham J. M., Grise H., Frausto S., Madan V., Zayas M., Bartenschlager R., Robinson M., Greenstein A. E., Nag A., Logan T. M., Bienkiewicz E., Tang H. (2010) A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathogens 6, e1001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brass V., Bieck E., Montserret R., Wölk B., Hellings J. A., Blum H. E., Penin F., Moradpour D. (2002) An amino-terminal amphipathic α-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 277, 8130–8139 [DOI] [PubMed] [Google Scholar]
- 24. Brass V., Pal Z., Sapay N., Deléage G., Blum H. E., Penin F., Moradpour D. (2007) Conserved determinants for membrane association of nonstructural protein 5A from hepatitis C virus and related viruses. J. Virol. 81, 2745–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elazar M., Cheong K. H., Liu P., Greenberg H. B., Rice C. M., Glenn J. S. (2003) Amphipathic helix-dependent localization of NS5A mediates hepatitis C virus RNA replication. J. Virol. 77, 6055–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Penin F., Brass V., Appel N., Ramboarina S., Montserret R., Ficheux D., Blum H. E., Bartenschlager R., Moradpour D. (2004) Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 279, 40835–40843 [DOI] [PubMed] [Google Scholar]
- 27. Sapay N., Montserret R., Chipot C., Brass V., Moradpour D., Deléage G., Penin F. (2006) NMR structure and molecular dynamics of the in-plane membrane anchor of nonstructural protein 5A from bovine viral diarrhea virus. Biochemistry 45, 2221–2233 [DOI] [PubMed] [Google Scholar]
- 28. Tellinghuisen T. L., Marcotrigiano J., Gorbalenya A. E., Rice C. M. (2004) The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 279, 48576–48587 [DOI] [PubMed] [Google Scholar]
- 29. Tellinghuisen T. L., Marcotrigiano J., Rice C. M. (2005) Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435, 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang L., Hwang J., Sharma S. D., Hargittai M. R., Chen Y., Arnold J. J., Raney K. D., Cameron C. E. (2005) Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 280, 36417–36428 [DOI] [PubMed] [Google Scholar]
- 31. Love R. A., Brodsky O., Hickey M. J., Wells P. A., Cronin C. N. (2009) Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 83, 4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fridell R. A., Qiu D., Wang C., Valera L., Gao M. (2010) Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob. Agents Chemother. 54, 3641–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Targett-Adams P., Graham E. J., Middleton J., Palmer A., Shaw S. M., Lavender H., Brain P., Tran T. D., Jones L. H., Wakenhut F., Stammen B., Pryde D., Pickford C., Westby M. (2011) Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target. Insights into compound modes of action. J. Virol. 85, 6353–6368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hwang J., Huang L., Cordek D. G., Vaughan R., Reynolds S. L., Kihara G., Raney K. D., Kao C. C., Cameron C. E. (2010) Hepatitis C virus nonstructural protein 5A. Biochemical characterization of a novel structural class of RNA-binding proteins. J. Virol. 84, 12480–12491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fernandes F., Poole D. S., Hoover S., Middleton R., Andrei A. C., Gerstner J., Striker R. (2007) Sensitivity of hepatitis C virus to cyclosporine A depends on nonstructural proteins NS5A and NS5B. Hepatology 46, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 36. Ishii N., Watashi K., Hishiki T., Goto K., Inoue D., Hijikata M., Wakita T., Kato N., Shimotohno K. (2006) Diverse effects of cyclosporine on hepatitis C virus strain replication. J. Virol. 80, 4510–4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watashi K., Hijikata M., Hosaka M., Yamaji M., Shimotohno K. (2003) Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 38, 1282–1288 [DOI] [PubMed] [Google Scholar]
- 38. Qiu D., Lemm J. A., O'Boyle D. R., 2nd, Sun J. H., Nower P. T., Nguyen V., Hamann L. G., Snyder L. B., Deon D. H., Ruediger E., Meanwell N. A., Belema M., Gao M., Fridell R. A. (2011) The effects of NS5A inhibitors on NS5A phosphorylation, polyprotein processing and localization. J. Gen. Virol. 92, 2502–2511 [DOI] [PubMed] [Google Scholar]
- 39. Foster T. L., Belyaeva T., Stonehouse N. J., Pearson A. R., Harris M. (2010) All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. J. Virol. 84, 9267–9277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shirota Y., Luo H., Qin W., Kaneko S., Yamashita T., Kobayashi K., Murakami S. (2002) Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J. Biol. Chem. 277, 11149–11155 [DOI] [PubMed] [Google Scholar]
- 41. Lohmann V., Korner F., Dobierzewska A., Bartenschlager R. (2001) Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J. Virol. 75, 1437–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berg J. M., Shi Y. (1996) The galvanization of biology. A growing appreciation for the roles of zinc. Science 271, 1081–1085 [DOI] [PubMed] [Google Scholar]
- 43. Dimitrova M., Imbert I., Kieny M. P., Schuster C. (2003) Protein-protein interactions between hepatitis C virus nonstructural proteins. J. Virol. 77, 5401–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]