

Background: Adenosine 5′-phosphosulfate kinase (APSK) catalyzes the synthesis of phosphoadenosine 5′-phosphosulfate, but how APSK coordinates binding of phosphonucleotides is unclear.
Results: Using calorimetry, crystallography, and mutagenesis, this study provides new insight on nucleotide binding in APSK.
Conclusion: The P-loop and a critical aspartate integrate dynamic structural and nucleotide recognition features.
Significance: These results suggest how structural changes guide the order of nucleotide addition for catalysis.
Keywords: Arabidopsis, Calorimetry, Enzyme Catalysis, Nucleotide, Protein Structure
Abstract
Adenosine 5′-phosphosulfate kinase (APSK) catalyzes the ATP-dependent synthesis of adenosine 3′-phosphate 5′-phosphosulfate (PAPS), which is an essential metabolite for sulfur assimilation in prokaryotes and eukaryotes. Using APSK from Arabidopsis thaliana, we examine the energetics of nucleotide binary and ternary complex formation and probe active site features that coordinate the order of ligand addition. Calorimetric analysis shows that binding can occur first at either nucleotide site, but that initial interaction at the ATP/ADP site was favored and enhanced affinity for APS in the second site by 50-fold. The thermodynamics of the two possible binding models (i.e. ATP first versus APS first) differs and implies that active site structural changes guide the order of nucleotide addition. The ligand binding analysis also supports an earlier suggestion of intermolecular interactions in the dimeric APSK structure. Crystallographic, site-directed mutagenesis, and energetic analyses of oxyanion recognition by the P-loop in the ATP/ADP binding site and the role of Asp136, which bridges the ATP/ADP and APS/PAPS binding sites, suggest how the ordered nucleotide binding sequence and structural changes are dynamically coordinated for catalysis.
Introduction
All organisms acquire and assimilate sulfate and/or sulfide for the synthesis of a wide array of metabolites (1–5). Chemical activation of sulfate in organisms that assimilate sulfur from the environment is carried out by ATP sulfurylase and yields adenosine 5′-phosphosulfate (APS)2 (5). In prokaryotes, fungi, and mammals, APS kinase (APSK; also known as adenylyl-sulfate kinase; EC 2.7.1.25) catalyzes the ATP-dependent phosphorylation of APS into PAPS, which is used for sulfur assimilation and as a biosynthetic sulfate donor (1–5). In plants, the sulfur assimilatory pathway branches after formation of APS into primary and secondary pathways. The reductive (i.e. primary) route leads to the production of sulfur-containing amino acids and peptides (2–6). The second metabolic route begins with APS kinase and synthesizes PAPS as a sulfate donor for the sulfonation of brassinosteroids, peptide hormones, and molecules critical for protection against herbivores, such as glucosinolates (8–10). Glucosinolate synthesis also requires metabolites produced by the primary sulfur assimilatory pathway (11–14). PAPS is also a precursor of 3′-phosphoadenosine 5′-phosphate, which is a critical metabolite for stress gene regulation and plant development (15). Control of sulfate flux between the primary (reductive) and secondary metabolic routes in plants helps to maintain the synthesis of the metabolites essential for plant growth and responses to different stresses (11–19).
In Arabidopsis thaliana, APSK is necessary for plant growth and directly connects the sulfur assimilation pathway to the biosynthesis of sulfonated molecules (11–14), but the biochemical regulation of this critical branch point enzyme in plant metabolism is largely unexamined. Initial studies of APSK from A. thaliana (AtAPSK) revealed severe substrate inhibition by APS, as observed in the enzyme from fungi, bacteria, and mammals (7, 20–23); this inhibition presumably results from formation of an E·ADP·APS dead-end complex (22, 24). Steady-state kinetic studies of the Penicillium chrysogenum APSK are consistent with a sequential ordered reaction mechanism in which ATP binds first, followed by APS (20), although later studies of the Escherichia coli APSK suggested that the order of ATP versus APS addition is random and varies with ligand concentrations (21) (Fig. 1A).
FIGURE 1.

Overview of APSK nucleotide binding and structure. A, schematic of ordered and random nucleotide binding proposed for APSK from P. chrysogenum and E. coli, respectively. B, structural overview of AtAPSK and nucleotide binding sites. The ribbon diagram of AtAPSK (24) shows the core α/β-nucleotide binding domain (rose and blue in each monomer of the dimer) and the smaller active site capping domain (gray). APS (green) and ATP (yellow) bound in their respective nucleotide binding sites are shown in each active site of the dimer. Residues of the P-loop are shown as a space-filling model (red). The position of the N-terminal α-helix is indicated by the letter N.
Crystal structures of APSK from Arabidopsis (Fig. 1B) and P. chrysogenum, the APSK domain from human PAPS synthetase, and the bifunctional ATP sulfurylase-APSK from Aquifex aeolicus and Thiobacillus denitrificans (25–30) show that the overall fold of APSK is highly conserved across a variety of organisms. Two nucleotide-binding sites (i.e. ATP/ADP and APS/PAPS sites) span the core α/β-purine nucleotide-binding domain and are capped by a smaller α-helical “lid” domain, which is disordered in the absence of ligands (26). The interface between the ATP/ADP and APS/PAPS binding sites is spanned by a canonical P-loop or Walker A motif (Ser110-Thr116 in AtAPSK), as observed in the structures of all nucleotide kinases (31, 32). Kinetic studies of the P. chrysogenum APSK suggest that APS binds tighter in the presence of ADP and magnesium compared with the unliganded enzyme, which has also been proposed as the basis for substrate inhibition (22); however, these studies relied on enzymatic assays to infer details about the nucleotide binding mechanism. Overall, steady-state kinetic studies and multiple crystal structures of APSK from a variety of species imply that dynamic features around the active site contribute to nucleotide binding.
Using a combination of isothermal titration calorimetry (ITC), x-ray crystallography, and kinetic analysis of point mutants, we examine the formation of AtAPSK nucleotide complexes and probe the role of active site features that coordinate the order of ligand addition. Although energetic analysis of complex formation showed that nucleotides can bind to either the ATP/ADP or APS/PAPS site, initial binding at the ATP/ADP site was favored and enhanced affinity for APS. Interestingly, the thermodynamics of each of the possible binding orders differs, likely reflecting different structural changes associated with nucleotide binding at each site to communicate binding site occupancy. Crystallographic and energetic analysis of oxyanion recognition by the P-loop and a critical aspartate residue that bridges the two nucleotide binding sites of AtAPSK provides insight on how an ordered sequence of binding events and structural changes are coordinated for efficient catalysis.
EXPERIMENTAL PROCEDURES
Reagents
All chemicals and reagents were of analytical grade and purchased from Sigma. The standard experimental buffer condition was 25 mm HEPES, pH 7.5, 200 mm KCl, 5% (v/v) glycerol, and either 1 mm Tris(2-carboxyethyl)phosphine or 5 mm β-mercaptoethanol (βME), unless stated otherwise. Either reducing agent prevented formation of inter-monomer disulfide bonds (25), as judged by nonreducing SDS-PAGE of samples taken before and after ITC titrations.
Protein Expression, Purification, and Site-directed Mutagenesis
The pET-28a-AtAPSKΔ77 bacterial expression construct, which encodes A. thaliana APSK isoform 1 lacking the plastid localization sequence (residues 1–77) and with an N-terminal His6 tag, was previously described (33). Protein overexpression in E. coli BL21(DE3) and purification by nickel affinity and gel-filtration chromatographies were described previously (25), with some modifications. For cell lysis and nickel affinity chromatography, all buffers were supplemented with 5 mm βME. After affinity purification, the protein was dialyzed into 25 mm HEPES, pH 7.5, 200 mm KCl, 5% glycerol, and 5 mm dithiothreitol (DTT), then loaded onto a Superdex-200 26/60 HiLoad FPLC size exclusion column equilibrated in the same buffer. Calibration of the size exclusion column was performed as described previously (34). Purified protein was either flash frozen in liquid nitrogen and stored at −80 °C or dialyzed against 25 mm HEPES, pH 7.5, 200 mm KCl, 5% glycerol, and either 1 mm Tris(2-carboxyethyl)phosphine or 5 mm βME for use in ITC experiments. Site-directed mutants of AtAPSK (G111A, G113A, D136N, and D136A) were generated using the QuikChange PCR method (Agilent) with expression and purification as above.
Calorimetric Measurements
ITC experiments were performed using a VP-ITC calorimeter (Microcal, Inc.). AtAPSK was dialyzed at 4 °C in 25 mm HEPES, pH 7.5, 200 mm KCl, 5% glycerol, and either 1 mm Tris(2-carboxyethyl)phosphine or 5 mm βME. Stock solutions (100 mm) of ATP, ATPγS, AMP-PNP, ADP, and APS were dissolved in NaOH to attain a pH of 7.5 and stored at −20 °C. Prior to ITC experiments, appropriate dilutions were made with dialysis buffer. Protein and nucleotide solutions were degassed at room temperature prior to use. For each titration, 20 to 30 injections of 10 μl of nucleotide were added into sample solutions containing protein (60–100 μm) in the presence or absence of ligands and/or 5 mm Mg2+. AtAPSK complexed with AMP-PNP, ADP, or APS was formed by incubating protein and 2 mm AMP-PNP, 500 μm ADP, or 2 mm APS overnight (4 °C) followed by a 4-h equilibration at 17 °C before titrations. Data were analyzed using either a one-site (i.e. identical sites) binding model (Equation 1) or a two-site binding model (Equation 2), as follows,
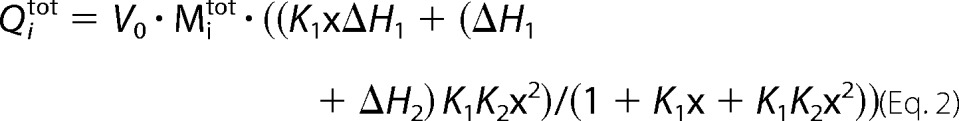 |
where Qitot is the total heat after the ith injection, V0 is the calorimetric cell volume, Mitot is the concentration of protein in the cell after the ith injection, ΔH is the corresponding enthalpy change to AtAPSK·nucleotide binding, n is the number of nucleotide binding sites on the APSK dimer, and K is the equilibrium binding constant. In the latter, k1 and k2 are the observed binding constants for the first and second sites, ΔH1 and ΔH2 are the corresponding enthalpy changes upon nucleotide binding to each site, and p = 1 + k1Xi + k1k2X. Fitting of data were performed using Origin software.
Enzyme Assays
All steady-state kinetic analysis was performed as previously described using an enzyme-coupled spectrophotometric assay (25).
Crystallography
Crystals of the AtAPSK·APS·SO42− complex were formed as follows. Crystals of AtAPSK in complex with APS were first grown at 4 °C in hanging drops with a 1:1 ratio of protein (∼10 mg ml−1) and crystallization buffer (100 mm HEPES, pH 7.25, 200 mm MgCl2, and 15–17.5% PEG-2000) supplemented with 5 mm APS. For x-ray data collection, crystals were transferred to a cryoprotectant solution of mother liquor containing 20% glycerol and 10 mm SO42−. Crystals were frozen in liquid nitrogen after a 2-h incubation period. X-ray diffraction data were collected at SBC beamline 19-ID of the Advanced Photon Source, Argonne National Laboratory. The HKL3000 software suite was used to integrate, merge, and scale diffraction intensities (35). The structure of the AtAPSK·APS·SO42− complex was solved by molecular replacement with PHASER (36) using the previously determined AtAPSK structure (25) with ligands and water molecules removed as a search model. The model was built in COOT (37) and refined using PHENIX (38) until the R-factors converged to those reported in Table 1. Coordinates and structure factors for the AtAPSK·APS·SO42− complex have been deposited in the Protein Data Bank (code 4FXP).
TABLE 1.
Summary of crystallographic statistics for the AtAPSK·APS·SO42− complex
| Crystal | |
|---|---|
| Space group | C2 |
| Cell dimensions | a = 120.9 Å, b = 92.35 Å, c = 73.18 Å; β = 113.5° |
| Data collection | |
| Wavelength (Å) | 0.979 |
| Resolution range (Å) (highest shell resolution) | 33.9–1.95 (2.02–1.95) |
| Reflections (total/unique) | 163,947/46,572 |
| Completeness (highest shell) | 95.5% (100.0%) |
| 〈I/σ〉 (highest shell) | 38.5 (5.0) |
| Rsyma (highest shell) | 5.2% (15.1%) |
| Model and refinement | |
| Rcrystb/Rfreec | 16.1/19.0 |
| No. of protein atoms | 4,738 |
| No. of water molecules | 435 |
| No. of ligand atoms | 96 |
| Root mean square deviation, bond lengths (Å) | 0.006 |
| Root mean square deviation, bond angles (°) | 0.97 |
| Average B-factor (Å2), protein, waters, ligands | 38.7, 46.5, 39.2 |
| Stereochemistry, most favored, allowed, generously allowed | 96.7, 3.1, 0.2% |
a Rsym = Σ|Ih − 〈Ih〉|/ΣIh, where 〈Ih〉 is the average intensity over symmetry.
b Rcryst = Σ|F o − 〈Fc〉|/ΣFo, where summation is over the data used for refinement.
c Rfree is defined the same as Rcryst, but was calculated using 5% of data excluded from refinement.
RESULTS
Calorimetric Analysis of Nucleotide Binding to AtAPSK
To compare nucleotide binding to AtAPSK, ITC experiments were performed using ATP, the ATP analogues AMP-PNP and ATPγS, ADP, and APS in the presence and absence of Mg2+ (Fig. 2, Table 2). All nucleotides showed exothermic binding. Fitting of the ITC data to a one-site binding model failed to adequately describe the observed results, whereas, satisfactory fits were obtained using a two-site binding model with stoichiometries consistent with the binding of two nucleotides per AtAPSK dimer. In all cases, the Kd value of the second site for each nucleotide was 5- to 30-fold higher than the first site, suggesting potential cross-talk between nucleotide binding sites in the homodimer.
FIGURE 2.

ITC analysis of nucleotide binding to AtAPSK. A, titration of AtAPSK with ATP (solid squares), ATPγS (open squares), AMP-PNP (closed circles), and AMP-PNP + 5 mm Mg2+ (open circles). B, titration of AtAPSK with ADP (open squares) and ADP + 5 mm Mg2+ (solid squares). C, titration of AtAPSK with APS (open squares) and APS + 5 mm Mg2+ (solid squares). Representative experimental data for the ATP (A), ADP (B), and APS (C) titrations are plotted as heat signal (μcal s−1) versus time (min) in each upper panel. Each experiment consisted of 20 to 30 injections of 10 μl each of nucleotide into a solution containing AtAPSK dimer. Each lower panel shows the integrated heat responses per injection. The solid line is the linear regression fit using a two-site binding model (see Table 2).
TABLE 2.
Thermodynamic parameters of nucleotide binding to AtAPSK
All titrations were performed at 17 °C with resulting data fit to a two-site binding model as described under “Experimental Procedures.”
| Ligand | K1 | K2 | ΔH1 | ΔH2 |
|---|---|---|---|---|
| μm | kcal mol−1 | |||
| ATP | 1.25 ± 0.09 | 24.7 ± 6.9 | −6.4 ± 0.7 | −5.0 ± 0.7 |
| ATPγS | 0.26 ± 0.05 | 7.9 ± 0.1 | −12.2 ± 0.4 | −5.2 ± 0.3 |
| AMP-PNP | 19.6 ± 1.5 | 361 ± 45 | −4.8 ± 0.5 | −1.2 ± 0.6 |
| AMP-PNP + 5 mm Mg2+ | 16.4 ± 0.2 | 82.6 ± 7.7 | −6.0 ± 0.1 | −3.3 ± 0.4 |
| ADP | 0.18 ± 0.05 | 4.8 ± 0.6 | −16.5 ± 0.2 | −9.4 ± 0.1 |
| ADP + 5 mm Mg2+ | 1.8 ± 0.4 | 38.1 ± 4.2 | −10.7 ± 0.8 | −6.2 ± 1.6 |
| APS | 66.7 ± 10.5 | 325 ± 89 | −5.6 ± 0.9 | −1.9 ± 0.7 |
| APS + 5 mm Mg2+ | 80.4 ± 5.2 | 460 ± 99 | −5.1 ± 2.3 | −2.2 ± 0.8 |
Titration of AtAPSK with ATP, AMP-PNP, and ATPγS showed that the ATP binding site is sensitive to changes in the phosphoanhydride chemical structure (Fig. 2A, Table 2). Substitution of one of the γ-phosphate oxygens with a sulfur in ATPγS increased affinity 5-fold compared with ATP. In contrast, replacement of the oxygen linking the β- and γ-phosphates with a nitrogen (AMP-PNP) decreased affinity 15-fold. As suggested by steady-state kinetic experiments (22), ADP bound with a 7-fold tighter affinity than ATP (Fig. 2B, Table 2). In the presence of Mg2+, AtAPSK hydrolyzed ATP and ATPγS, which prevented accurate binding analysis with these ligands. Addition of Mg2+ had little effect on binding of the first AMP-PNP molecule, but modestly improved affinity for the second molecule by 4-fold (Fig. 2A, Table 2). An opposite effect was observed with ADP and Mg2+, which decreased ligand affinity 10-fold (Fig. 2B, Table 2). To determine whether this change resulted from the ion or increased ionic strength, titrations of AtAPSK with ADP at 0.5 m KCl and NaCl were performed and did not alter the titration profile or the measured parameters, which indicates that the observed decrease in affinity for ADP is a Mg2+-specific effect. Binding of APS was weaker than ATP and was insensitive to Mg2+ (Fig. 2C, Table 2), which is consistent with the location of the ion in the ATP/ADP, not APS/PAPS, binding site of AtAPSK (25).
Calorimetric Analysis of AtAPSK·Nucleotide Ternary Complex Formation
Analysis of single nucleotide interaction with AtAPSK showed that binding at either the ATP/ADP or APS/PAPS site did not require ordered ligand addition. ITC experiments were used to determine whether pre-formed complexes of AtAPSK and either phosphonucleotide (AMP-PNP and ADP) or APS affected the affinity for binding of APS or phosphonucleotides, respectively. First, we compared binding of APS to pre-formed AMP-PNP and ADP complexes, which is the binding order proposed for APSK from P. chrysogenum and the upper path suggested for the E. coli enzyme (Fig. 1A).
Addition of APS to AtAPSK·AMP-PNP in the presence or absence of Mg2+ yielded similar Kd values (Fig. 3A, Table 3), which were ∼50-fold tighter than APS binding to the unliganded enzyme (Table 2). APS binding to the AtAPSK·ADP complex displayed less than 2-fold differences compared with APS binding to the AtAPSK·AMP-PNP complex; however, the presence of Mg2+ altered the binding model with a 5-fold improvement in Kd for the first binding event and a 4-fold decrease in affinity for binding of at the second site (Fig. 3B, Table 3). These results agree with previous kinetic experiments using the fungal APSK, which found that Mg2+ was required for ADP to increase the affinity for APS (22). In the absence of Mg2+, APS binding to either the AMP-PNP or ADP complex was best fit to a one-site binding model, whereas, a two-site model best described binding of APS in the presence of Mg2+. These results provide strong evidence for unidirectional synergistic binding, which would explain the obligate binding order of ATP followed by APS suggested by earlier steady-state kinetic studies of APSK (24).
FIGURE 3.

ITC analysis of nucleotide binding to AtAPSK·nucleotide complexes. A, titration of AtAPSK·AMP-PNP with APS (open squares) and APS + 5 mm Mg2+ (solid squares). B, titration of AtAPSK·ADP with APS (solid squares) and APS + 5 mm Mg2+ (open squares). C, titration of AtAPSK·APS with AMP-PNP (open squares) and ADP (solid squares). Representative experimental data for the APS (A and B) and AMP-PNP (C) titrations are plotted as heat signal (μcal s−1) versus time (min) in each upper panel. Each experiment consisted of 20 to 30 injections of 10 μl each of nucleotide into a solution containing AtAPSK dimer and either 0 or 5 mm Mg2+. Each lower panel shows the integrated heat responses per injection. The solid line is the linear regression fit using either a one- or two-site binding model (see Table 3).
TABLE 3.
Thermodynamic parameters of nucleotide binding to AtAPSK·nucleotide complexes
All titrations were performed at 17 °C as described under “Experimental Procedures.” AtAPSK·nucleotide complexes were pre-formed either in the absence or presence of 5 mm Mg2+. ITC data were fit to either a one-site binding model (n = number of sites) or a two-site binding model.
| Pre-bound nucleotide | Titrant | K1 | K2 | ΔH1 | ΔH2 |
|---|---|---|---|---|---|
| μm | kcal mol−1 | ||||
| AMP-PNP | APS | 1.50 ± 0.10 n = 2.00 ± 0.10 | −3.3 ± 0.1 | ||
| AMP-PNP·Mg2+ | APS | 1.50 ± 0.60 | 3.60 ± 0.40 | −4.4 ± 0.1 | −2.3 ± 0.1 |
| ADP | APS | 3.30 ± 0.70 n = 1.93 ± 0.05 | −7.2 ± 0.1 | ||
| ADP·Mg2+ | APS | 0.60 ± 0.20 | 12.9 ± 3.8 | −3.1 ± 0.4 | −2.4 ± 0.5 |
| APS | AMP-PNP | 3.10 ± 0.40 | 18.6 ± 0.9 | 4.3 ± 0.6 | 3.0 ± 0.2 |
| APS | ADP | 0.65 ± 0.04 n = 2.00 ± 0.10 | −3.3 ± 0.4 | ||
In the other possible binding sequence (i.e. APS first; Fig. 1A), the effect of forming the AtAPSK·APS complex on either AMP-PNP or ADP binding was less than 6-fold different compared with the unliganded enzyme titration (Fig. 3C, Table 3). Analysis of the interaction between the AtAPSK·APS complex and AMP-PNP required a two-site binding model. In contrast, the isotherm resulting from ADP titration could be described by a one-site binding model with a 2:1 stoichiometry of ligand to dimer, suggesting that APS does not bind with significant affinity to the ATP/ADP binding site. The affinity of AtAPSK for AMP-PNP improved an order of magnitude in the presence of APS, whereas the affinity for ADP was not significantly changed. The increased affinity for AMP-PNP, but not ADP, may result from interactions formed between the γ-phosphate of AMP-PNP and active site residues.
Surprisingly, binding of AMP-PNP to the AtAPSK·APS complex showed an endothermic interaction (Fig. 3C, Table 3), which markedly contrasts with the exothermic binding displayed by nucleotides to either unliganded or liganded enzyme. Although binding of nucleotides to AtAPSK can occur in either order (Fig. 1A), the two possible routes leading to formation of the AtAPSK·ATP·APS ternary complex are not thermodynamically equal. This difference in binding energetics may reflect structural and/or dynamic differences in the active site that correspond to the order of ligand addition.
Calorimetric, Crystallographic, and Mutagenesis Analysis of P-loop Recognition of Oxyanions
The above results, along with crystal structures of APSK from a variety of sources (25–30), suggests that the P-loop (Fig. 1B) plays a role in nucleotide binding specificity at the ATP/ADP site of AtAPSK. We tested the ability of the P-loop to discern ligands based on anhydride size and charge density by using a series of oxyanions: SO42−, PO42−, NO3−, CO3−, and ClO3−. Titrations of AtAPSK with SO42− resulted in an exothermic heat change (Fig. 4A), but weak binding did not allow for accurate curve fitting. No heat change was observed in titrations with other oxyanions (Fig. 4A). To probe the location of SO42− binding, AtAPSK preincubated with 10 mm SO42− was titrated with ATP, ADP, and APS. Preincubation abrogated binding of ATPγS and ADP (Fig. 4B). In contrast, pre-formation of the AtAPSK·SO42− complex resulted in 10- to 100-fold improvement in binding affinity for APS (Fig. 4C, Table 4), analogous to the effect observed with formation of the AtAPSK·AMP-PNP and AtAPSK·ADP complexes. These results suggest that the sulfate-binding site is likely located within the ATP/ADP site, which was confirmed by x-ray crystallography.
FIGURE 4.

ITC analysis of oxyanion binding to AtAPSK and nucleotide binding to the AtAPSK·SO42− complex. A, titration of AtAPSK with SO42− (closed squares), ClO3− (open squares), and PO42− (closed circles). B, titration of AtAPSK (closed squares) and AtAPSK·SO42− (open squares) with ATPγS. C, titration of AtAPSK (open squares) and AtAPSK·SO42− (closed squares) with APS. D, titration of AtAPSK G111A with ATPγS (open squares) and APS (closed squares). Titrations of the G113A mutant yielded similar results. Representative experimental data for the SO42− (A), AtAPSK/ATPγS (B), AtAPSK·SO42−/APS (C), and G111A/APS (D) titrations are plotted as heat signal (μcal s−1) versus time (min) in each upper panel. Each experiment consisted of 20 to 30 injections of 10 μl each of nucleotide or oxyanion into a solution containing protein. Each lower panel shows the integrated heat responses per injection. The solid line is the linear regression fit using either a one- or two-site binding model (see Table 4).
TABLE 4.
Thermodynamic parameters of APS binding to the AtAPSK·SO42− complex, G111A, and G113A proteins
All titrations were performed at 17 °C as described under “Experimental Procedures.” ITC data were fit to either a one-site binding model (n = number of sites) or a two-site binding model.
| Protein | Titrant | K1 | K2 | ΔH1 | ΔH2 |
|---|---|---|---|---|---|
| μm | kcal mol−1 | ||||
| AtAPSK·SO42− | APS | 3.78 ± 2.55 n = 1.98 ± 0.04 | −8.0 ± 0.6 | ||
| AtAPSK G111A | APS | 102.7 ± 30.1 | 439.0 ± 131.8 | −6.4 ± 2.2 | −2.7 ± 1.9 |
| AtAPSK G113A | APS | 130.0 ± 45.6 | 365.9 ± 93.7 | −9.1 ± 4.6 | −5.4 ± 2.5 |
The 1.95-Å resolution x-ray crystal structure of the AtAPSK·APS·SO42− complex was solved by molecular replacement (Table 1, Fig. 5). The overall structure is similar to that of the AtAPSK·AMP-PNP·APS complex (25) with the root mean square deviation of Cα-atoms at 0.37 Å2. Clear electron density for bound APS was observed in the active site with additional discontinuous density near the P-loop of the ATP/ADP binding site (Fig. 5A), which was modeled as SO42−. Multiple interactions were formed between the SO42− and residues in the P-loop (Fig. 5B), including main chain contacts from Gly111, Ser112, Gly113, Lys114, and Ser115 and hydrogen bonds from the hydroxyl group of Ser115 and amine of Lys114 (Fig. 5B). Compared with the AtAPSK·AMP-PNP·APS complex (Fig. 5, C and D), the position of the SO42− corresponds to that of the β-phosphate of AMP-PNP with a nearly identical set of interactions formed in each structure.
FIGURE 5.

Structure of the AtAPSK·APS·SO42− complex. A, active site view showing the |2Fo − Fc| omit map (1.5 σ) for APS and SO42−. In addition, the density for Ser112 and Lys114 in the P-loop are shown. B, schematic of interactions made by P-loop residues and SO42− in the AtAPSK·APS·SO42− complex. C, active site view of the AtAPSK·AMP-PNP·APS complex (25, PDB 3UIE) showing positions of bound ligands. D, schematic of interactions made by P-loop residues and the β-phosphate of AMP-PNP in the AtAPSK (25); the γ-phosphate is not shown for clarity.
To further examine the role of the P-loop in AtAPSK, two site-directed mutants (G111A and G113A) were generated to disrupt the ATP/ADP binding site. Each mutant protein was catalytically inactive. ITC analysis of ATPγS binding to the G111A mutant showed a negligible enthalpy change, although the binding affinity for APS was comparable with binding to the unliganded wild-type enzyme (Fig. 4D, Table 4). Similar results were observed for the G113A mutant (Table 4). Each mutant also disrupted SO42− binding, as there was no heat signature detected by ITC. These results underscore the importance of the P-loop for ATP/ADP binding.
Asp136 as a Critical Determinant of APS Substrate Inhibition and Nucleotide Binding Order
The increased affinity of the plant and fungal APSK (22) for APS, when in complex with ADP, support a model in which APS-dependent substrate inhibition results from formation of a dead-end complex. We examined the structure of AtAPSK (25) to identify residues with the potential to mediate communication between the ATP/ADP and APS/PAPS binding sites with the aim of using mutagenesis to alter the effect of ADP on APS binding and to reduce substrate inhibition. At the interface of the two nucleotide-binding sites in AtAPSK (Fig. 6) and the enzyme from other species (25–30), the P-loop (Ser110–Thr116) is essential for ATP/ADP binding and residues from the α3 helix provide critical contacts with APS. In particular, Arg141 forms charge-charge interactions with the phosphosulfate group of APS and Asp138 provides a bidentate interaction with the hydroxyl groups of the APS ribose and serves as a general base in the reaction mechanism (25, 29). Asp136 anchors an extensive network of contacts with Ser115 in the P-loop, waters coordinated to Mg2+, and the main chain nitrogen of Asp138 in the α3 helix; no other residue in the active site connects multiple structural features.
FIGURE 6.

Structural view of the AtAPSK active site. The positions of APS (green) and AMP-PNP (yellow) are shown in relationship to residues from the P-loop (Ser115) and residues on or near the α3 helix (Asp136, Asp138, and Arg141) that interact with either APS or the water network (red spheres) that coordinates the Mg2+ (green sphere) are shown. Asp136 is highlighted in red to emphasize its location between the P-loop and α3 helix. The figure was generated using the AtAPSK·AMP-PNP·APS complex structure (25).
To examine the role of Asp136, the D136N and D136A mutants were generated, purified, and kinetically characterized. The AtAPSK D136N and D136A mutants displayed 9- and 18-fold reductions in catalytic efficiencies (kcat/Km), respectively, that primarily resulted from reduced turnover rates (Table 5). The D136A mutant also abolished the effect of substrate inhibition by APS, which prompted us to examine nucleotide binding in this protein. ITC analysis of nucleotide binding to D136A showed that the mutation had little effect on ATP, ATPγS, AMP-PNP, and ADP binding either in the presence or absence of Mg2+, as all these ligands displayed exothermic heat signatures and affinities comparable with wild-type protein (Fig. 7A, Table 6). Although Asp136 does not directly contact APS, the D136A mutant displayed drastically reduced affinity for APS compared with the wild-type enzyme (Fig. 7B). Changes in enthalpy were observed upon titrating the D136A mutant with APS, but the binding isotherms were essentially linear and precluded an accurate fitting to determine binding affinity. In contrast to the addition of single nucleotides, preincubation of the D136A mutant with either AMP-PNP or ADP followed by addition of APS showed that the affinity for the second ligand was comparable with that observed for wild-type (Fig. 7C, Table 6). These results implicate Asp136 as required for AtAPSK to bind APS with significant affinity in the absence of ADP or AMP-PNP, as this mutation specifically decreases the affinity for APS in the absence of additional nucleotides without affecting the energetics and affinity for binding in the ATP/ADP site. The AtAPSK D136A mutant exhibits a strict binding order due to the diminished affinity of the enzyme for APS, but not ATP, and suggests that APS binding before ATP is inhibitory to the wild-type enzyme.
TABLE 5.
Steady-state kinetic parameters of wild-type and mutant AtAPSK
Average values ± S.E. (n = 3) are shown.
| Protein | kcat | KmAPS | KiAPS | kcat/Km |
|---|---|---|---|---|
| s−1 | μm | m−1 s−1 × 108 | ||
| AtAPSK | 272 ± 39 | 0.48 ± 0.41 | 37.5 ± 6.9 | 5.67 |
| AtAPSK D136N | 45.0 ± 3.5 | 0.70 ± 0.29 | 32.8 ± 7.7 | 0.64 |
| AtAPSK D136A | 29.0 ± 5.1 | 0.92 ± 0.63 | 0.31 | |
FIGURE 7.

ITC analysis of nucleotide binding to AtAPSK D136A. A, titration of AtAPSK D136A with ADP (solid squares), ATPγS (open squares), ATP (closed circles), and AMP-PNP (open circles). B, titration of AtAPSK D136A with APS (solid squares), APS + 5 mm Mg2+ (open squares), and AtAPSK with APS (open circles). C, titration of AtAPSK D136A·ADP with APS (open squares), APS + 5 mm Mg2+ (solid squares), and AtAPSK D136A·AMP-PNP (open circles) with APS + 5 mm Mg2+. Representative experimental data for the ADP (A) and APS (B and C) titrations are plotted as heat signal (μcal s−1) versus time (min) in each upper panel. Each experiment consisted of 20 to 30 injections of 10 μl each of nucleotide into a solution containing AtAPSK dimer. Each lower panel shows the integrated heat responses per injection. The solid line is the linear regression fit using a two-site binding model (see Table 6).
TABLE 6.
Thermodynamic comparison of nucleotide binding to AtAPSK D136A and AtAPSK D136A·nucleotide complexes
All titrations were performed at 17 °C as described under “Experimental Procedures.” AtAPSK D136A·nucleotide complexes were pre-formed either in the absence or presence of 5 mm Mg2+. ITC data were fit to either a one-site binding model (n = number of sites) or a two-site binding model.
| Pre-bound nucleotide | titrant | K1 | K2 | ΔH1 | ΔH2 |
|---|---|---|---|---|---|
| μm | kcal mol−1 | ||||
| ATP | 1.60 ± 0.50 | 23.6 ± 3.1 | −9.0 ± 0.6 | −4.2 ± 0.8 | |
| ATPγS | 0.80 ± 0.50 | 10.9 ± 5.8 | −11.6 ± 2.7 | −8.6 ± 1.4 | |
| AMP-PNP | 36.9 ± 6.2 | 48.0 ± 10.4 | −5.0 ± 1.6 | −2.2 ± 0.9 | |
| ADP | 0.17 ± 0.02 | 3.40 ± 1.10 | −13.2 ± 0.9 | −6.3 ± 0.5 | |
| ADP + 5 mm Mg2+ | 0.41 ± 0.02 | 6.0 ± 1.3 | −12.8 ± 1.4 | −9.7 ± 2.2 | |
| AMP-PNP | APS | 5.3 ± 0.5 n = 1.95 ± 0.04 | −2.7 ± 0.1 | ||
| AMP-PNP + 5 mm Mg2+ | APS | 6.0 ± 0.3 n = 2.08 ± 0.07 | −2.3 ± 0.1 | ||
| ADP | APS | 4.0 ± 0.5 n = 2.10 ± 0.10 | −4.1 ± 0.4 | ||
| ADP + 5 mm Mg2+ | APS | 1.80 ± 0.20 | 11.1 ± 0.9 | −3.5 ± 0.4 | −2.6 ± 0.5 |
DISCUSSION
Substrate inhibition by APS is a hallmark feature of APSK from bacteria, fungi, plants, and mammals and has been examined using kinetic and structural approaches (7, 20–24, 30), which suggest different models for nucleotide binding (Fig. 1A). To date, the thermodynamic basis for the order of ligand binding and how the ATP/ADP and APS/PAPS binding sites interact remains unclear. The combined calorimetric, crystallographic, and kinetic analyses presented here reveal a highly integrated system of dynamic structural and nucleotide recognition features in APSK.
Calorimetric analysis of ligand binding to form AtAPSK binary and ternary nucleotide complexes provides new insight on the interaction between the ATP/ADP and APS/PAPS binding sites within the monomer and helps to define the molecular basis for the proposed ordered sequential mechanism of binding. Steady-state kinetic analysis of APSK from P. chrysogenum and E. coli suggest alternative models for the order of ligand addition (Fig. 1A) (20, 21). As shown here, ATP, AMP-PNP, ATPγS, ADP, and APS all bind to AtAPSK with APS binding displaying the weakest affinity (Fig. 2, Table 2). Analysis of ternary complex formation shows that ordered addition of AMP-PNP enhances the affinity for APS by 50-fold (Fig. 3, Table 3). In contrast, addition of AMP-PNP to the AtAPSK·APS complex does not significantly change affinity. Moreover, the tighter binding of ADP compared with ATP and its analogs to either unliganded AtAPSK or the AtAPSK·APS complex confirms that substrate inhibition results from facile formation of a dead-end form of the enzyme (22, 24, 30).
Although binding of nucleotides to AtAPSK can occur in either order, the two possible routes leading to formation of the AtAPSK·ATP·APS ternary complex are not thermodynamically equivalent. For example, addition of ATP first followed by APS showed exothermic binding interactions (Kd = 1.50 μm; ΔH = −3.3 kcal mol−1; ΔG = +7.73 kcal mol−1; −TΔS = 11.0 kcal mol−1), whereas addition of APS followed by AMP-PNP was endothermic (Kd = 3.10 μm; ΔH = +4.3 kcal mol−1; ΔG = +7.31 kcal mol−1; −TΔS = 3.0 kcal mol−1) (Fig. 3, Table 3). The enthalpic changes are offset by concomitant entropic changes that maintain binding affinity. This difference in the binding energetics of the two reaction sequences likely reflects structural changes in the active site that correspond to the order of ligand addition, which are discussed below. Overall, these results are consistent with a preferred order of ligand binding in APSK, in which ATP binds first followed by APS, and the proposed ordered sequential mechanism determined by steady-state kinetics (20).
The ITC data presented here also supports a previous suggestion of cross-talk between active sites within the APSK homodimer based on crystallographic studies (25, 39). Single nucleotide titrations of AtAPSK yielded binding isotherms best fit by a two-site model in which the Kd values for the second site were 5–30-fold higher than the first site for each nucleotide (Fig. 2, Table 2). Moreover, pre-ordering of the active site by formation of a binary complex before nucleotide addition either reduces or eliminates the effectiveness of potential communication within the dimer (Fig. 3, Table 3). The lack of binding asymmetry for formation of the ternary complexes suggests that preincubation with ligands occupies the binding site and organizes the active sites in the dimer for a more symmetric interaction with the second nucleotide. Although the molecular basis of this effect needs to be further examined, the N-terminal region of APSK has been implicated as a possible control feature for this behavior. Earlier crystallographic studies of the APSK domain from human PAPS synthetase revealed asymmetry in substrate binding between monomers of the protein (30, 39). This led to the suggestion that nucleotide binding in one active site alters the position of the N-terminal domain adjacent to the monomer, which entwines the other monomer (Fig. 1B), to provide a “coupling function” that communicates nucleotide occupancy between active sites (30). A similar asymmetry was observed in the disulfide linkages of the N-terminal domain of the AtAPSK·AMP-PNP·APS complex (25). The dynamic nature of structural features of APSK is also underscored by the nucleotide binding analyses presented above and by the effects of mutations in the P-loop and Asp136 in the active site.
As in other ATP-dependent enzymes, the P-loop of APSK is critical for enzymatic function (40, 41). Binding AMP-PNP, ADP, or SO42− in the ATP/ADP site results in increased affinity of AtAPSK for APS (Tables 3 and 4). In multiple crystal structures, AMP-PNP and ADP interact with residues in the P-loop (Fig. 5), which provides a driving force for nucleotide recognition as it forms nine hydrogen bonds with the β-phosphate compared with two hydrogen bonds made between the rest of the nucleotide and protein (23–30). Comparable interactions are observed for SO42− bound in the AtAPSK·APS·SO42− complex (Fig. 5). To examine the role of the P-loop in determining binding order, nucleotide binding to two AtAPSK mutants (G111A and G113A) was examined (Fig. 4D, Table 4). The resultant isotherms from titrations with either ATPγS or ADP could not be fit to obtain binding constants, but APS binding was comparable that of the wild-type apoenzyme (Table 1). Based on the energetic analysis of wild-type and mutant AtAPSK, interactions made between ATP and the P-loop help order the active site and position the γ-phosphate in proximity to APS for catalysis.
This work also identifies Asp136 as a critical determinant for determining the order of nucleotide binding and for APS substrate inhibition. The position of Asp136 in the AtAPSK active site anchors an extensive set of interactions that connect the P-loop and the α3-helix in the APS binding site (Fig. 6). Steady-state kinetic characterization of the D136N and D136A mutants reveals a modest change in catalytic efficiency, but a loss of substrate inhibition by APS in the D136A mutant (Table 5). Thus, kinetic analysis would suggest a minor role for this residue; however, examination of nucleotide binding in the D136A mutant by ITC reveals a critical role for guiding ligand binding. Mutation of Asp136 to an alanine drastically reduced the Kd for APS without altering binding at the ATP/ADP site (Fig. 7, Table 6), even though this residue does not directly contact APS. In contrast, the D136A mutant showed a slightly decreased affinity for APS after formation of the AtAPSK-D136A·AMP-PNP complex. This demonstrates that the D136A mutation affects ligand addition to effectively allow only an ordered sequential binding of ATP and then APS, not vice versa. These results suggest a structural model for distinguishing the order of nucleotide binding to APSK (Fig. 8).
FIGURE 8.

Model for ligand binding to AtAPSK. The structure of AtAPSK is shown as a schematic with the core domain of each monomer colored rose and blue, respectively, and the lid domain colored white. The P-loop is shown as a red loop with Asp136 and the α3-helix in green. Ligands are indicated by colored rectangles in each binding site as follows: ATP (yellow rectangle) and APS (green rectangle). Thicker arrows indicate the preferred order of ligand binding.
Crystallographic studies of the unliganded and nucleotide bound forms of APSK indicate that the mobile active site capping or lid domain binds to the P-loop and provides interactions with APS to lock substrates into the active site (23, 26). It is unclear what coordinates the movement of this domain with nucleotide binding, but loop and ligand interactions likely alter the flexibility of this loop. Based on the ITC analysis presented here, binding of ATP·Mg2+ to AtAPSK first pre-organizes the active site for subsequent addition of APS (Fig. 8, top). ATP binding establishes extensive interactions between the β-phosphate and P-loop and places the γ-phosphate in proximity to the acceptor hydroxyl group of APS once bound, and helps position Asp136 to bridge Ser115 in the P-loop, the water molecules coordinating the Mg2+ ion, and the α3 helix to bring Asp138 and Arg141 into proximity for APS binding and phosphoryl-group transfer. These results imply highly interconnected roles for the P-loop and Asp136 that allow for energetically efficient formation of the AtAPSK·ATP·APS ternary complex and ensuing catalysis. In contrast, initial APS binding is possible (Fig. 8, bottom), but alters movement of the lid domain and P-loop for subsequent ATP binding, as suggested by the thermodynamic differences associated with the AMP-PNP binding to the AtAPSK·APS complex (Fig. 3C, Table 3). Thus, APS binding first has an overall inhibitory effect on the reaction sequence and changes the energetics that allow optimal active site conformation. These studies of AtAPSK suggest an important role for the dynamic motions of active site features in response to ligand binding order. As recently shown for adenylate kinase (42, 43), movement in localized structures can preferentially create active site configurations that favor efficient catalysis. A similar series of events can now be proposed for the APSK reaction mechanism.
Acknowledgments
Portions of this research were carried out at the Argonne National Laboratory Structural Biology Center of the Advanced Photon Source, a national user facility operated by the University of Chicago for the Dept. of Energy Office of Biological and Environmental Research (DE-AC02–06CH11357).
This work was supported by National Science Foundation Grant MCB-0904215.
The atomic coordinates and structure factors (code 4FXP) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- APS
- adenosine 5′-phosphosulfate
- AMP-PNP
- β,γ-imidoadenosine-5-triphosphate
- APSK
- adenosine-5′-phosphosulfate kinase, also known as adenylyl-sulfate kinase
- AtAPSK
- Arabidopsis thaliana APSK
- ATPγS
- adenosine 5′-(γ-thio)triphosphate
- βME
- β-mercaptoethanol
- ITC
- isothermal titration calorimetry
- PAPS
- adenosine 3′-phosphate 5′-phosphosulfate.
REFERENCES
- 1. Schelle M. W., Bertozzi C. R. (2006) Sulfate metabolism in mycobacteria. ChemBioChem 7, 1516–1524 [DOI] [PubMed] [Google Scholar]
- 2. Patron N. J., Durnford D. G., Kopriva S. (2008) Sulfate assimilation in eukaryotes. Fusions, relocations, and lateral transfers. BMC Evol. Biol. 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yi H., Galant A., Ravilious G. E., Preuss M. L., Jez J. M. (2010) Sensing sulfur conditions. Simple to complex protein regulatory mechanisms in plant thiol metabolism. Mol. Plant 3, 269–279 [DOI] [PubMed] [Google Scholar]
- 4. Yi H., Ravilious G. E., Galant A., Krishnan H. B., Jez J. M. (2010) From sulfur to homoglutathione. Thiol metabolism in soybean. Amino Acids 39, 963–978 [DOI] [PubMed] [Google Scholar]
- 5. Takahashi H., Kopriva S., Giordano M., Saito K., Hell R. (2011) Sulfur assimilation in photosynthetic organisms. Molecular functions and regulations of transporters and assimilatory enzymes. Annu. Rev. Plant Biol. 62, 157–184 [DOI] [PubMed] [Google Scholar]
- 6. Ravilious G. E., Jez J. M. (2012) Structural biology of plant sulfur metabolism. From assimilation to biosynthesis. Nat. Prod. Rep., in press [DOI] [PubMed] [Google Scholar]
- 7. Lillig C. H., Schiffmann S., Berndt C., Berken A., Tischka R., Schwenn J. D. (2001) Molecular and catalytic properties of Arabidopsis thaliana adenylyl sulfate (APS) kinase. Arch. Biochem. Biophys. 392, 303–310 [DOI] [PubMed] [Google Scholar]
- 8. Klein M., Papenbrock J. (2004) The multiprotein family of Arabidopsis sulfotransferases and their relatives in other plant species. J. Exp. Bot. 55, 1809–1820 [DOI] [PubMed] [Google Scholar]
- 9. Halkier B. A., Gershenzon J. (2006) Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 57, 303–333 [DOI] [PubMed] [Google Scholar]
- 10. Amano Y., Tsubouchi H., Shinohara H., Ogawa M., Matsubayashi Y. (2007) Tyrosine-sulfated glycopeptide involved in cellular proliferastion and expansion in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 104, 18333–18338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mugford S. G., Yoshimoto N., Reichelt M., Wirtz M., Hill L., Mugford S. T., Nakazato Y., Noji M., Takahashi H., Kramell R., Gigolashvili T., Flügge U. I., Wasternack C., Gershenzon J., Hell R., Saito K., Kopriva S. (2009) Disruption of adenosine 5′-phosphosulfate kinase in Arabidopsis reduces levels of sulfated secondary metabolites. Plant Cell 21, 910–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kopriva S., Mugford S. G., Matthewman C., Koprivova A. (2009) Arabidopsis root growth dependence on glutathione is linked to auxin transport. Plant Cell Rep. 28, 1769–1780 [DOI] [PubMed] [Google Scholar]
- 13. Mugford S. G., Matthewman C. A., Hill L., Kopriva S. (2010) Adenosine 5′-phosphosulfate kinase is essential for Arabidopsis viability. FEBS Lett. 584, 119–123 [DOI] [PubMed] [Google Scholar]
- 14. Yatusevich R., Mugford S. G., Matthewman C., Gigolashvili T., Frerigmann H., Delaney S., Koprivova A., Flügge U. I., Kopriva S. (2010) Genes of primary sulfate assimilation are part of the glucosinolate biosynthetic network in Arabidopsis thaliana. Plant J. 62, 1–11 [DOI] [PubMed] [Google Scholar]
- 15. Chen H., Zhang B., Hicks L. M., Xiong L. (2011) A nucleotide metabolite controls stress-responsive gene expression and plant development. PLoS One 6, e26661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bick J. A., Setterdahl A. T., Knaff D. B., Chen Y., Pitcher L. H., Zilinskas B. A., Leustek T. (2001) Regulation of plant-type 5′-adenylyl sulfate reductase by oxidative stress. Biochemistry 40, 9040–9048 [DOI] [PubMed] [Google Scholar]
- 17. Martin M. N., Tarczynski M. C., Shen B., Leustek T. (2005) The role of 5′-adenylylsulfate reductase in controlling sulfate reduction in plants. Photosynth. Res. 86, 309–323 [DOI] [PubMed] [Google Scholar]
- 18. Loudet O., Saliba-Colombani V., Camilleri C., Calenge F., Gaudon V., Koprivova A., North K. A., Kopriva S., Daniel-Vedele F. (2007) Natural variation for sulfate content in Arabidopsis thaliana is highly controlled by APR2. Nat. Genet. 39, 896–900 [DOI] [PubMed] [Google Scholar]
- 19. Scheerer U., Haensch R., Mendel R. R., Kopriva S., Rennenberg H., Herschbach C. (2010) Sulfur flux through the sulfate assimilation pathway is differently controlled by adenosine 5′-phosphosulphate reductase under stress and in transgenic poplar plants overexpressing γECS, SO, or APR. J. Exp. Bot. 61, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Renosto F., Seubert P. A., Knudson P., Segel I. H. (1985) Adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Determining ligand dissociation constants of binary and ternary complexes from the kinetics of enzyme inactivation. J. Biol. Chem. 260, 11903–11913 [PubMed] [Google Scholar]
- 21. Satishchandran C., Markham G. D. (2000) Mechanistic studies of Escherichia coli adenosine 5′-phosphosulfate kinase. Arch. Biochem. Biophys. 378, 210–215 [DOI] [PubMed] [Google Scholar]
- 22. Renosto F., Martin R. L., Segel I. H. (1991) Adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Ligand binding properties and the mechanism of substrate inhibition. Arch. Biochem. Biophys. 284, 30–34 [DOI] [PubMed] [Google Scholar]
- 23. Lansdon E. B., Segel I. H., Fisher A. J. (2002) Ligand-induced structural changes in adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry 41, 13672–13680 [DOI] [PubMed] [Google Scholar]
- 24. MacRae I. J., Segel I. H. (1999) Adenosine 5′-phosphosulfate (APS) kinase, diagnosing the mechanism of substrate inhibition. Arch. Biochem. Biophys. 361, 277–282 [DOI] [PubMed] [Google Scholar]
- 25. Ravilious G. E., Nguyen A., Francois J. A., Jez J. M. (2012) Structural basis and evolution of redox regulation in plant adenosine 5′-phosphosulfate kinase. Proc. Natl. Acad. Sci. U.S.A. 109, 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacRae I. J., Segel I. H., Fisher A. J. (2000) Crystal structure of adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry 39, 1613–1621 [DOI] [PubMed] [Google Scholar]
- 27. Yu Z., Lansdon E. B., Segel I. H., Fisher A. J. (2007) Crystal structure of the bifunctional ATP sulfurylase-APS kinase from the chemolithotrophic thermophile Aquifex aelicus. J. Mol. Biol. 365, 732–743 [DOI] [PubMed] [Google Scholar]
- 28. Gay S. C., Segel I. H., Fisher A. J. (2009) Structure of the two-domain hexameric APS kinase from Thiobacillus denitrificans. Structural basis for the absence of ATP sulfurylase activity. Acta Crystallogr. D Biol. Crystallogr. 65, 1021–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sekulic N., Dietrich K., Paarmann I., Ort S., Konrad M., Lavie A. (2007) Elucidation of the active conformation of the APS-kinase domain of human PAPS synthetase 1. J. Mol. Biol. 367, 488–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sekulic N., Konrad M., Lavie A. (2007) Structural mechanism for substrate inhibition of the adenosine 5′-phosphosulfate kinase domain of human 3′-phosphoadenosine-5′-phosphosulfate synthetase 1 and its ramifications for enzyme regulation. J. Biol. Chem. 282, 22112–22121 [DOI] [PubMed] [Google Scholar]
- 31. Walker J. E., Saraste M., Runswick M. J., Gay N. J. (1982) Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases, and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saraste M., Sibbald P. R., Wittinghofer A. (1990) The P-loop, a common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 15, 430–434 [DOI] [PubMed] [Google Scholar]
- 33. Phartiyal P., Kim W. S., Cahoon R. E., Jez J. M. (2006) Soybean ATP sulfurylase, a homodimeric enzyme involved in sulfur assimilation, is abundantly expressed in roots and induced by cold treatment. Arch. Biochem. Biophys. 450, 20–29 [DOI] [PubMed] [Google Scholar]
- 34. Kumaran S., Yi H., Krishnan H. B., Jez J. M. (2009) Assembly of the cysteine synthase complex and the regulatory role of protein-protein interactions. J. Biol. Chem. 284, 10268–10275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Minor W., Cymborowski M., Otwinowski Z., Chruszcz H. (2006) HKL-3000, the integration of data reduction and structure solution. From diffraction images to an initials model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 36. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and developmen of COOT. Acta Cryststallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX, a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harjes S., Bayer P., Scheidig A. J. (2005) The crystal structure of human PAPS synthetase 1 reveals asymmetry in substrate binding. J. Mol. Biol. 347, 623–635 [DOI] [PubMed] [Google Scholar]
- 40. Deyrup A. T., Krishnan S., Cockburn B. N., Schwartz N. B. (1998) Deletion and site-directed mutagenesis of the ATP-binding motif (P-loop) in the bifunctional murine ATP-sulfurylase/adenosine 5′-phosphosulfate kinase enzyme. J. Biol. Chem. 273, 9450–9456 [DOI] [PubMed] [Google Scholar]
- 41. MacRae I. J., Rose A. B., Segel I. H. (1998) Adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Site-directed mutagenesis at putative phosphoryl-accepting and ATP P-loop residues. J. Biol. Chem. 273, 28583–28589 [DOI] [PubMed] [Google Scholar]
- 42. Henzler-Wildman K. A., Thai V., Lei M., Ott M., Wolf-Watz M., Fenn T., Pozharski E., Wilson M. A., Petsko G. A., Karplus M., Hübner C. G., Kern D. (2007) Intrinsic motions along an enzymatic reaction trajectory. Nature 450, 838–844 [DOI] [PubMed] [Google Scholar]
- 43. Henzler-Wildman K., Kern D. (2007) Dynamic personalities of proteins. Nature 450, 964–972 [DOI] [PubMed] [Google Scholar]