

Background: Cholera toxin enters the target cell in a disordered state and must attain a folded, active conformation to modify its G protein target.
Results: Lipid rafts, where G protein is located, shift the disordered toxin to a functional conformation.
Conclusion: Lipids rafts provide a chaperone-like function for cholera toxin.
Significance: Lipid rafts play an important role in regulating toxin function through chaperone-like activity.
Keywords: Cholera Toxin, Cholesterol, Circular Dichroism (CD), Lipid Raft, Protein Folding, Filipin, Lipochaperone, Resonance Energy Transfer
Abstract
Cholera toxin (CT) travels from the cell surface to the endoplasmic reticulum (ER) as an AB holotoxin. ER-specific conditions then promote the dissociation of the catalytic CTA1 subunit from the rest of the toxin. CTA1 is held in a stable conformation by its assembly in the CT holotoxin, but the dissociated CTA1 subunit is an unstable protein that spontaneously assumes a disordered state at physiological temperature. This unfolding event triggers the ER-to-cytosol translocation of CTA1 through the quality control mechanism of ER-associated degradation. The translocated pool of CTA1 must regain a folded, active structure to modify its G protein target which is located in lipid rafts at the cytoplasmic face of the plasma membrane. Here, we report that lipid rafts place disordered CTA1 in a functional conformation. The hydrophobic C-terminal domain of CTA1 is essential for binding to the plasma membrane and lipid rafts. These interactions inhibit the temperature-induced unfolding of CTA1. Moreover, lipid rafts could promote a gain of structure in the disordered, 37 °C conformation of CTA1. This gain of structure corresponded to a gain of function: whereas CTA1 by itself exhibited minimal in vitro activity at 37 °C, exposure to lipid rafts resulted in substantial toxin activity at 37 °C. In vivo, the disruption of lipid rafts with filipin substantially reduced the activity of cytosolic CTA1. Lipid rafts thus exhibit a chaperone-like function that returns disordered CTA1 to an active state and is required for the optimal in vivo activity of CTA1.
Introduction
Cholera toxin (CT)2 exhibits an ADP-ribosyltransferase activity against the stimulatory α subunit of the heterotrimeric G protein (Gsα) (1). ADP-ribosylation of Arg201 in Gsα locks the GTP-bound protein in an active state that promotes the continual stimulation of adenylate cyclase and its production of cAMP. Dysregulation of cAMP-dependent signaling pathways leads to the opening of apical chloride channels in intoxicated enterocytes. The osmotic movement of water to counterbalance chloride accumulation in the lumen of the gut consequently generates the profuse, life-threatening diarrhea of cholera.
CT has an AB5 structural organization that consists of a catalytic A moiety and a homopentameric, cell-binding B moiety (2). For CT to manifest its latent ADP-ribosyltransferase activity, the CTA polypeptide must be proteolytically nicked to generate separate but disulfide-linked CTA1 and CTA2 subunits (3, 4). Nicking can involve Vibrio or other proteases. The disulfide bond linking CTA1 to CTA2 must then be reduced (4), which only occurs after the CT holotoxin moves from the cell surface to the endoplasmic reticulum (ER) through a series of vesicle-mediated trafficking steps collectively termed retrograde transport (5, 6). Reduction of the CTA1/CTA2 disulfide bond permits the chaperone-assisted dissociation of CTA1 from CTA2/CTB5 (7, 8). CTA1 is held in a stable conformation by its interactions with the holotoxin (9, 10), but the isolated CTA1 polypeptide is an unstable protein that will spontaneously unfold at 37 °C upon its separation from CTA2/CTB5 (11). The disordered CTA1 polypeptide is subsequently recognized as a misfolded protein by the host quality control system of ER-associated degradation (ERAD) (12–14). This results in the ER-to-cytosol translocation of CTA1 through one or more protein-conducting channels in the ER membrane (15–18). Most exported ERAD substrates are degraded by the ubiquitin-proteasome system, but the dearth of CTA1 lysine residues for ubiquitin conjugation allows the toxin to evade this fate (19, 20).
The translocated pool of CTA1 must regain a folded, active conformation to modify its Gsα target. This will not occur spontaneously because the isolated CTA1 polypeptide is in a disordered conformation at 37 °C (11). Host ADP-ribosylation factors (ARFs) act as allosteric activators of CTA1 and allow the toxin to exhibit in vitro catalytic activity at 37 °C (21, 22). However, it is possible that other host factors also assist the in vivo gain of structure and gain of function for cytosolic CTA1.
In this paper, we examined the impact of lipid rafts on the structure and function of CTA1. Gsα is found in lipid rafts at the cytosolic face of the plasma membrane (23–26). CTA1 would therefore encounter a lipid raft environment upon contact with its Gsα target. Because two major components of lipid rafts (cholesterol and phosphatidylethanolamine (PE)) can act as lipochaperones (27–29), we hypothesized an interaction with lipid rafts would shift disordered CTA1 to a folded, active conformation. Structural analysis with circular dichroism (CD) and functional analysis with an in vitro ADP-ribosylation assay supported our hypothesis: exposure to lipid rafts but not plasma membrane lipids allowed the disordered CTA1 subunit to attain a folded structure and enzymatic activity at 37 °C. The physiological relevance of these observations was documented with an in vivo intoxication assay which demonstrated intact lipid rafts are required for the ADP-ribosylation of Gsα by cytosolic CTA1. Lipid rafts thus display a chaperone-like activity that modulates the structure and function of CTA1.
EXPERIMENTAL PROCEDURES
Materials
The following were purchased from Avanti Polar Lipids (Alabaster, AL): cholesterol; palmitoyl sphingomyelin; 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC); 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myoinositol) (DOPI); 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myoinositol-4′-phosphate) (PI(4)P); 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myoinositol-4′,5′-bisphosphate) (PI(4,5)P2); 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphoethanolamine (PLPE); 1-palmitoyl-2-linoleoyl-sn-glycero-3-phospho-l-serine (PLPS); 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (SAPC); 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine (SAPE); 1-stearoyl-2-linoleoyl-sn-glycero-3-phosphoethanolamine (SLPE); 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC); and pyrene-labeled PE (pyrene-PE). Filipin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA); CT was purchased from List Biologicals (Campbell, CA); and isoproterenol was purchased from Sigma-Aldrich. CTA1 with a C-terminal His6 tag was purified as described previously (13) and used for all in vitro studies.
Composition and Generation of Large Unilamellar Vesicles (LUVs)
The plasma membrane lipid profile of the rat intestinal epithelium (30–34) was used as the basis for the acyl chain and head group composition of our plasma membrane mimic. Asymmetric lipid distributions between the inner and outer bilayer leaflets (35–37) were further considered to specifically model the composition of the inner leaflet of the plasma membrane (Table 1). To simulate the composition of a lipid raft (38–41), we mixed sphingomyelin and cholesterol with zwitterionic PE and anionic PS (Table 1). The latter two lipids are highly enriched in the inner leaflet of a lipid raft (42–45). Acyl chain selection was based upon the intestinal lipid composition. The inner leaflet of a lipid raft may lack sphingomyelin (46), so we formulated an alternative lipid raft mimic that lacked sphingomyelin (Table 1).
TABLE 1.
Lipid composition of LUVs mimicking the plasma membrane or lipid rafts
| Lipid | Percentage of composition |
||
|---|---|---|---|
| Plasma membrane | Lipid raft | Variant lipid raft | |
| Cholesterol | 0 | 30 | 30 |
| PLPS | 13 | 15 | 15 |
| Palmitoyl sphingomyelin | 4 | 30 | 0 |
| PLPE | 27 | 25 | 45 |
| SLPE | 14 | 0 | 0 |
| SAPE | 5 | 0 | 0 |
| SOPC | 20 | 0 | 0 |
| SAPC | 14 | 0 | 0 |
| PI(4,5)P2 | 1 | 0 | 0 |
| PI(4)P | 1 | 0 | 0 |
| DOPI | 1 | 0 | 0 |
| PLPC | 0 | 0 | 10 |
Individual lipids were dissolved in chloroform and mixed according to the percentages listed in Table 1. The total lipid concentration was 8 mm. The solvent was evaporated under a slow stream of nitrogen gas and vacuum dried for 4 h. The dried mixture was suspended in 10 mm borate buffer (pH 7.0) containing 100 mm NaCl and vortexed thoroughly. To generate LUVs, the lipid mixture was passed 30 times through a Liposofast extruder (Avestin, Ottawa, ON, Canada) with 100-nm pore size polycarbonate membranes.
Membrane-binding Experiments
To detect CTA1 interaction with LUVs mimicking the composition of the plasma membrane or lipid rafts, we added 1% pyrene-PE to the vesicles. CTA1 was titrated with pyrene-PE-labeled vesicles in a 4 × 4-mm2 path length quartz cuvette, and membrane binding of CTA1 was measured based on fluorescence resonance energy transfer (FRET) from the three tryptophan (Trp) residues of CTA1 to pyrene-PE using a Jasco 810 spectrofluoropolarimeter (Tokyo, Japan). This is a spectropolarimeter equipped with a Jasco PFD-425S Peltier temperature controller and an additional photomultiplier tube mounted at a right angle, which permits fluorescence measurements in addition to CD. Experiments were performed using a final sample volume of 220 μl containing 15 μm CTA1 in the presence or absence of LUVs, with excitation wavelength of 290 nm. The FRET data were analyzed as described previously (47). Briefly, changes in Trp fluorescence at each addition of pyrene-PE-containing vesicles (ΔF) were measured and plotted against ΔF/[L], where [L] is the total lipid concentration. These Scatchard plots were used to determine the limiting value of ΔF, i.e. ΔFmax. Protein binding isotherms were constructed by plotting ΔF/ΔFmax against [L], after correction of ΔF values for the sample dilution effects and for the changes in ΔF in negative control experiments where the protein was titrated with vesicles without pyrene-PE. Using a Langmuir-type binding model (the membrane surface contains protein binding sites that bind the protein with a dissociation constant of KD and a lipid-to-protein stoichiometry of N lipids/protein), values of KD and N were determined from best fits of simulated and experimental binding isotherms.
CD and Fluorescence Spectroscopy
A Jasco 810 spectrofluoropolarimeter was used for CD measurements of samples placed in a 4-mm path length quartz cuvette. Experiments were performed using a final sample volume of 220 μl containing 15 μm CTA1 in the presence or absence of 800 μm LUVs. Light scattering effects became significant only below 205 nm in experiments with CTA1 and below 210–215 nm in experiments utilizing LUVs at 800 μm lipid concentration (e.g. see Fig. 2). The characteristic spectral minima of CD spectra which were used for Tm determination (see below) were not affected by light scattering effects at this lipid concentration. To monitor the potential stabilizing effect of plasma membrane or lipid raft LUVs on CTA1 structure, near-simultaneous measurements of far-UV CD, near-UV CD, and fluorescence were taken as the temperature was increased stepwise from 20 °C to 60 °C. The temperature was ramped from 20 °C to 34 °C in 2 °C increments; from 34 °C to 40 °C in 1 °C increments, and from 40 °C to 60 °C in 2 °C increments. At each temperature, the sample was equilibrated for 4 min before measurement. The experimentally measured ellipticities (θobs) were converted to mean residue molar ellipticity, [θ], by using the following formula: [θ] = θobs/cnl, where n is the number of amino acid residues in the protein, c is the molar concentration of the protein, and l is the path length of the cuvette in millimeters.
FIGURE 2.

CTA1 is stabilized by its interaction with plasma membrane and lipid raft LUVs. The temperature-induced unfolding of CTA1 (A–C), CTA1 in the presence of plasma membrane LUVs (D–F), and CTA1 in the presence of lipid raft LUVs (G–I) was recorded by far-UV CD (A, D, and G), near-UV CD (B, E, and H), and fluorescence spectroscopy (C, F, and I). The change in color from blue to red denotes the increase in temperature from 20 °C to 60 °C. J–L, thermal unfolding profiles for CTA1 (red), CTA1 in the presence of plasma membrane LUVs (blue), and CTA1 in the presence of lipid raft LUVs (orange) were derived from the data presented in A–I. J, for far-UV CD analysis, the mean residue molar ellipticities at 220 nm ([θ]220) were plotted as a function of temperature. K, for near-UV CD analysis, the mean residue molar ellipticities at 280 nm ([θ]280) were plotted as a function of temperature. L, for fluorescence spectroscopy, the maximum emission wavelength (λmax) was plotted as a function of temperature. One of two independent experiments is shown for each condition.
Calculations of transition temperatures (Tm; the midpoint between folded and final disordered states) were performed as described previously (11). In brief, data were analyzed using the following equation (48):
where X is the measured temperature-dependent variable (i.e. the ellipticity or peak fluorescence emission wavelength), fL is the fraction of amino acids representing the native conformation at low temperature, XL is the limiting value of X at low temperature, and XH is the limiting value of X at high temperature. The parameter, fL, is given by:
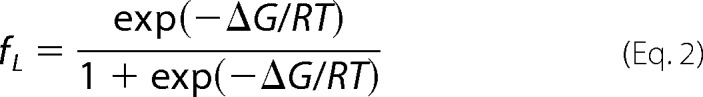 |
The free energy of unfolding (ΔG) is given by
Here, ΔH is the apparent enthalpy of unfolding and ΔC (0.39 kcal mol−1 K−1) is the heat capacity of unfolding.
To monitor the potential renaturation of disordered CTA1 by plasma membrane or lipid raft LUVs, far- and near-UV CD measurements of CTA1 structure were taken at 18 °C. Plasma membrane or lipid raft LUVs were added to a final lipid concentration of 800 μm after heating the toxin to 37 °C. The temperature was maintained at 37 °C, and CD spectra were recorded 5 or 30 min after exposure to the LUVs.
In parallel control experiments, CD and fluorescence measurements were recorded at each temperature in the presence of LUVs alone (plasma membrane, lipid raft, or variant lipid raft). CD and fluorescence spectra of the CTA1 samples were then corrected by subtraction of the appropriate temperature-matched spectra of LUVs only. Sphingomyelin produces a strong far-UV CD signal below 200 nm (i.e. between 195 and 197 nm), but around 220 nm its contribution to the far-UV CD signal is only 4–5% of the protein contribution (49). The minimal contribution of sphingomyelin to the 220 nm far-UV CD signal of CTA1 was eliminated by the aforementioned background subtraction procedure before calculating the CTA1 secondary structure Tm at 220 nm.
In Vitro Assay for CTA1 Catalytic Activity
Diethylamino(benzylidine-amino)guanidine (DEA-BAG) is a CTA1 substrate that loses its ability to bind AG-50W-X4 ion exchange resin (Bio-Rad) after ADP-ribosylation (50). DEA-BAG also exhibits an intrinsic fluorescence (361-nm excitation wavelength, 440-nm emission wavelength), so the fluorescent signal remaining after pulldown with AG-50W-X4 resin represents the ADP-ribosylated pool of DEA-BAG. 2-fold serial dilutions of CTA1 were placed in 200 μl of KH2PO4 buffer (pH 7.5) containing 20 mm DTT, 10 mm NAD, and 0.1 mg/ml BSA. In a subset of samples, lipid raft LUVs were also added to a final lipid concentration of 800 μm. LUVs were added at 25 °C or after the toxin had been heated to 37 °C for 30 min. When added after heating, LUVs were incubated with the toxin for 1 h at 37 °C before addition of the DEA-BAG substrate. All toxin samples were incubated with 0.4 mg of DEA-BAG for 2 h at either 25 °C or 37 °C as indicated. After batch removal of unmodified DEA-BAG with 800 μl of a 40% AG-50W-X4 resin slurry, fluorescence intensity in the supernatant was recorded with a Biotek (Winooski, VT) Synergy 2 plate reader.
Cell Culture Studies
HeLa cells seeded to 80% confluence in 6-well plates were transfected with 0.5 μg of pcDNA3.1/mCTA1 (51) using a 5-h exposure to Lipofectamine (Invitrogen) according to the manufacturer's instructions. Transfected cells were chased in the absence or presence of filipin (1 μg/ml) for 2 or 4 h before cAMP levels were quantified using a commercial kit (PerkinElmer Life Sciences). Three samples per condition were run for each experiment. Background levels of cAMP from mock-transfected cells were subtracted from all results before expressing the data as percentages of the maximum cAMP signal obtained for the experiment.
CHO cells stably transfected with a gene encoding the β2-adrenergic receptor (β2AR) were provided by Dr. S. E. Mills (52, 53). Before quantification of cAMP levels, untreated and filipin-treated CHO-β2AR cells were incubated for 4 h with 100 ng/ml CT or 1 μm β2AR agonist isoproterenol. Filipin was co-incubated with CT or isoproterenol and was used at a concentration of 1 μg/ml. In each experiment, three samples were run for each condition. The basal levels of cAMP from untreated cells were subtracted from all results before expressing the data as percentages of the maximum cAMP signal obtained for the experiment.
RESULTS
CTA1 Binds to Lipid Rafts
Toxin-lipid interactions were initially examined by FRET (Fig. 1). CTA1 was incubated at 37 °C with LUVs mimicking the composition of either the plasma membrane or lipid rafts (Table 1). LUVs were prepared using 1% pyrene-PE at the expense of PLPE. Binding of CTA1 to the pyrene-PE-containing membrane results in short range dipolar interactions between the energy donor (Trp) and the acceptor (pyrene-PE), thus decreasing Trp emission intensity between 300 and 360 nm and increasing the 360–450 nm emission intensity of pyrene-PE. This FRET effect was observed when CTA1 was mixed with vesicles mimicking either the plasma membrane (Fig. 1A) or lipid rafts (Fig. 1B). As expected, increasing the LUV concentration produced a concomitant decrease in the Trp emission intensity. No substantial alteration to the CTA1 emission intensity was recorded when the toxin was incubated with plasma membrane (Fig. 1C) or lipid raft (Fig. 1D) LUVs lacking the pyrene-PE acceptor. The loss of CTA1 fluorescence intensity in Fig. 1, A and B, was therefore due to FRET between CTA1 and a membrane containing a pyrene-PE acceptor. Further data analysis was used to calculate a 2.3 μm KD for CTA1 binding to plasma membrane LUVs (Fig. 1E) and a 0.6 μm KD for CTA1 binding to lipid raft LUVs (Fig. 1F). CTA1 thus binds to both the plasma membrane and lipid rafts, but it has a higher affinity for lipid rafts.
FIGURE 1.

CTA1 binds to plasma membrane and lipid raft LUVs. The affinity between CTA1 and the plasma membrane (A, C, and E) or lipid rafts (B, D, and F) was determined by tryptophan FRET using pyrene-labeled LUVs (A and B) or unlabeled LUVs (C and D). For the representative spectra in A–D, a change of color from blue to red signifies an increase in lipid concentration from 100 to 1000 μm. Each measurement of fluorescent intensity for both labeled and unlabeled LUVs was normalized to the CTA1 concentration at the start of the experiment (i.e. corrections were made for the sample dilution effect). In E and F, the solid lines were constructed using a Langmuir-type binding model, and KD values were calculated from curve fitting as described under “Experimental Procedures.”
The C-terminal A13 subdomain of CTA1 was initially thought to activate the ERAD system because this region contains a number of hydrophobic amino acid residues that could have allowed the toxin to masquerade as a misfolded protein (20). Subsequent studies found that the A13 subdomain is not essential for toxin translocation (54), but we hypothesized that this hydrophobic region is instead responsible for CTA1 binding to lipid bilayers. A CTA1 construct lacking the A13 subdomain (CTA11–168) was used to test this prediction. As shown in supplemental Fig. S1, CTA11–168 did not interact with either plasma membrane or lipid raft LUVs. CTA11–168 maintains the same stability and the same general structure as full-length CTA1 (12, 55), so the loss of binding affinity could not be attributed to structural alterations in CTA11–168. Thus, the hydrophobic A13 subdomain of CTA1 appeared to mediate toxin-lipid interactions directly.
Stabilization and Renaturation of CTA1 by Lipid Rafts
We next examined the structural impact of CTA1 binding to either the plasma membrane or lipid rafts. For this work, the thermal stability of CTA1 in the absence or presence of lipid bilayers was examined by CD and fluorescence spectroscopy (Fig. 2). CTA1 was mixed with LUVs mimicking the composition of the plasma membrane or lipid rafts at 20 °C, and measurements were taken during a stepwise increase in temperature from 20 °C to 60 °C. Additional measurements were taken with a CTA1 sample heated in the absence of LUVs. The collective data demonstrated that interactions with plasma membrane and lipid raft LUVs have moderate and prominent stabilizing effects on CTA1, respectively. In the absence of LUVs, CTA1 exhibited a secondary structure transition temperature Tm of 34 °C (Fig. 2A), a tertiary structure Tm of 31 °C (Fig. 2B) derived from CD measurements, and a 34 °C Tm for the red shift to the maximum emission wavelength (λmax) of Trp fluorescence (Fig. 2C). In the presence of plasma membrane LUVs, CTA1 exhibited a secondary structure Tm of 39 °C (Fig. 2D), a tertiary structure Tm of 37 °C (Fig. 2E), and a 37 °C Tm for the λmax of Trp fluorescence (Fig. 2F). These values were similar to the corresponding Tm values recorded for CTA1 in the presence of LUVs mimicking the composition of the ER membrane (56), which suggests that toxin-lipid interactions may have a general stabilizing effect on the structure of CTA1. However, a substantially greater degree of stabilization was observed when CTA1 was exposed to lipid raft LUVs. For this condition, CTA1 exhibited a secondary structure Tm of 47 °C (Fig. 2G), a tertiary structure Tm of 45 °C (Fig. 2H), and a 44 °C Tm for the λmax of Trp fluorescence (Fig. 2I). These Tm values were 10–14 °C higher than the values obtained for untreated CTA1 and 7–8 °C higher than the values recorded for CTA1 in the presence of plasma membrane LUVs, indicating a profound increase in CTA1 thermal stability upon interaction with membranes mimicking lipid rafts. The thermal unfolding profiles for each of our biophysical measurements are shown in Fig. 2, J–L, and the calculated Tm values are summarized in Table 2. Neither plasma membrane nor lipid raft LUVs affected the thermal unfolding of CTA11–168 (supplemental Fig. S2 and Table S1), which exhibits the same conformational stability as full-length CTA1 (12). Lipid-induced alterations to the structure of CTA1 thus required a direct interaction with the toxin.
TABLE 2.
CTA1 stability in the presence of plasma membrane or lipid raft LUVs
Values represent the averages ± ranges from two independent experiments as represented by the data in Fig. 2 and supplemental Fig. S3.
| Toxin condition |
Tm |
||
|---|---|---|---|
| Far-UV CD | Near-UV CD | Fluorescence spectroscopy | |
| °C | °C | °C | |
| Untreated | 34 ± 1 | 31 ± 1 | 34 ± 1 |
| +Plasma membrane | 39 ± 2 | 37 ± 1 | 37 ± 1 |
| +Lipid raft | 47 ± 2 | 45 ± 2 | 44 ± 1 |
| +Variant lipid raft | 45 ± 1 | 41 ± 1 | 41 ± 1 |
CTA1 enters the cytosol in an unfolded state and must therefore regain an active, folded conformation to modify its Gsα target. We hypothesized that an interaction with lipid rafts would facilitate renaturation of the disordered CTA1 polypeptide. Far- and near-UV CD were used to test this prediction (Fig. 3). CTA1 was placed at 18 °C, and measurements of toxin structure were recorded. The toxin was subsequently heated to 37 °C. The disordered CTA1 polypeptide was then left untreated, was mixed with plasma membrane LUVs, or was mixed with lipid raft LUVs. Further measurements of toxin structure were recorded after an additional 5 min and 30 min at 37 °C. In the absence of LUVs, CTA1 progressed to an increasingly disordered state (Fig. 3, A and B). The presence of plasma membrane LUVs prevented the additional time-dependent loss of CTA1 structure but did not promote a gain of secondary or tertiary structure (Fig. 3, C and D). In contrast, CTA1 shifted to a more ordered state in the presence of lipid raft LUVs (Fig. 3, E and F). After 30 min at 37 °C in the presence of lipid rafts, the initially disordered CTA1 polypeptide displayed a microenvironment around its aromatic amino acid residues that was similar to the folded, 18 °C toxin conformation (Fig. 3E). The initial temperature-dependent loss of CTA1 secondary structure was also reversed in the presence of lipid rafts. However, the gain of CTA1 secondary structure was incomplete: the far-UV CD signal for raft-treated toxin was weaker than the initial 18 °C signal for folded CTA1 and was red-shifted by 4 nm from 220 to 224 nm, indicating the movement of amino acid residues into a more hydrophobic environment (Fig. 3F). A further 30-min incubation at 37 °C, for a total of 1 h in the presence of lipid raft LUVs, did not induce a further gain of structure in the CTA1 polypeptide (data not shown). These observations indicated that an interaction with lipid rafts at physiological temperature will promote a partial restoration of structure for the disordered CTA1 polypeptide.
FIGURE 3.

Partial refolding of CTA1 by lipid raft LUVs. CD was used to monitor the structure of CTA1. Measurements of untreated CTA1 were taken at 18 °C (solid lines). The toxin was subsequently heated to 37 °C and then left untreated (A and B), exposed to plasma membrane LUVs (C and D), or exposed to lipid raft LUVs (E and F). Additional near-UV CD (A, C, and E) and far-UV CD (B, D, and F) measurements were taken 5 (dotted lines) and 30 (dashed lines) min after a continued 37 °C incubation in the absence or presence of LUVs. One of three representative experiments is shown.
Sphingomyelin may be largely restricted to the outer leaflet of a lipid raft (46), so we repeated our structural studies with variant lipid raft LUVs devoid of sphingomyelin (Table 1). The alternative, sphingomyelin-depleted lipid rafts also stabilized CTA1 when mixed with the toxin at 20 °C (supplemental Fig. S3) and induced a gain of CTA1 structure when mixed with the disordered toxin at 37 °C (supplemental Fig. S4). The stabilization of CTA1 by sphingomyelin-depleted lipid rafts was less prominent than that observed for sphingomyelin-containing lipid rafts but was still more substantial than the stabilization provided by plasma membrane lipids (Table 2). Moreover, sphingomyelin-depleted lipid rafts but not plasma membrane lipids could promote a gain of structure in disordered CTA1. These collective observations indicated that the presence of sphingomyelin is not critical for the dramatic structural alterations to CTA1 that result from its interaction with lipid rafts.
CTA1 Activity at Physiological Temperature Is Enhanced by Lipid Rafts
The isolated CTA1 polypeptide has little to no enzymatic activity at 37 °C (22), which is consistent with the disordered conformation it assumes at physiological temperature (11). We hypothesized the stabilization and renaturation of CTA1 by lipid rafts would allow the toxin to manifest its enzymatic function at 37 °C. This prediction was tested using DEA-BAG, a synthetic substrate for the ADP-ribosyltransferase activity of CTA1 (50). At 25 °C, we documented the dose-dependent modification of DEA-BAG by CTA1 (Fig. 4). A minimal signal was recorded at 37 °C, and this signal most likely represented a background reading because it did not increase with increasing toxin concentration. However, a substantial signal was detected when CTA1 was mixed with lipid raft LUVs before warming to 37 °C. The stabilizing effect of lipid rafts thus allowed CTA1 to maintain enough of its native structure, and hence activity, to effectively modify the DEA-BAG substrate. Moreover, CTA1 exhibited high levels of ADP-ribosylation activity when lipid raft LUVs were added after the toxin had been warmed to 37 °C. An interaction with lipid rafts thus shifted the disordered, 37 °C conformation of CTA1 to an active state. However, lipid rafts did not enhance the activity of folded CTA1 at 25 °C. The raft-induced gain of CTA1 function thus appeared to result from a gain of structure in disordered CTA1 rather than from allosteric activation of the toxin: if lipid rafts acted as an allosteric activator for CTA1, we would have also detected raft-stimulated toxin activity at 25 °C. These results were obtained with both sphingomyelin-containing lipid raft LUVs (Fig. 4) and sphingomyelin-depleted lipid raft LUVs (supplemental Fig. S5). Thus, sphingomyelin was not required for the raft-induced gain of CTA1 function at 37 °C.
FIGURE 4.

CTA1 activity in the presence of lipid raft LUVs. 2-fold dilutions of CTA1 were mixed with lipid raft LUVs at 25 °C for toxicity assays performed at 25 °C (open squares) or 37 °C (filled squares). For a third condition, LUVs were added after the toxin had been heated to 37 °C for 30 min (filled triangles). This toxin-LUV mixture was incubated at 37 °C for an additional hour before initiating the toxicity assay. Toxin samples incubated in the absence of LUVs at 25 °C (open circles) or 37 °C (filled circles) were also used for the assay. Four samples were used for each condition; results are presented as means ± S.E. (error bars). One of three representative experiments is shown.
In Vivo Contribution of Lipid Rafts to G Protein and CTA1 Function
Gsα is found in lipid rafts on the cytoplasmic face of the plasma membrane (23–26). CTA1 would thus encounter a lipid raft environment upon contact with its Gsα target. This suggests that lipid rafts could contribute to CT intoxication by facilitating the return of disordered CTA1 to a folded, active conformation. To examine this possibility, we monitored the enzymatic activity of CTA1 in filipin-treated cells. Filipin blocks CT intoxication by disrupting the lipid rafts which are required for endocytosis and intracellular trafficking of CT (57). To circumvent the upstream roles of lipid rafts in CT intoxication, we used a plasmid-based system (51) for direct expression of CTA1 in the cytosol of cells with intact or filipin-disrupted lipid rafts. At 2 and 4 h after transfection, filipin-treated cells exhibited substantially lower levels of cAMP than the untreated control cells (Fig. 5). Preliminary control experiments ensured that filipin did not alter the level of protein synthesis in drug-treated cells (data not shown), so the low levels of cAMP in filipin-treated cells could not be attributed to the lack of plasmid-borne CTA1 expression. Additional control experiments found that filipin treatment had a moderate, direct effect on Gsα signaling events: when treated with isoproterenol, a ligand for the G-coupled β2AR (52, 53), CHO-β2AR cells produced a robust cAMP response that was reduced by 20% in the presence of filipin. In contrast, filipin reduced the cAMP response from CTA1-expressing cells by 50% at 2 h after transfection and 65% at 4 h after transfection (Fig. 5). The greater extent of inhibition for CTA1-generated cAMP versus isoproterenol-generated cAMP indicated a direct inhibitory effect of filipin on the cytosolic activity of CTA1. Thus, lipid rafts appear to be essential for the optimal in vivo ADP-ribosyltransferase activity of CTA1. Our in vitro biophysical and biochemical studies strongly suggest that the essential function of lipid rafts is linked to their chaperone-like ability to place disordered CTA1 in a folded, active conformation at 37 °C.
FIGURE 5.

Lipid rafts are required for optimal in vivo activity of cytosolic CTA1. HeLa cells were transfected with a plasmid encoding a CTA1 construct that is expressed directly in the cytosol. Intracellular cAMP levels at 2 and 4 h after transfection were then quantified for untreated and filipin-treated cells. The averages ± S.D. (error bars) of 3–4 independent experiments with triplicate samples are shown.
DISCUSSION
To reach its G protein target, CTA1 undergoes what has been termed an order-disorder-order transition (55). The ordered CTA1 polypeptide travels from the cell surface to the ER as part of an intact holotoxin (6). Separation of CTA1 from CTA2/CTB5 in the ER releases the structural constraints on CTA1 unfolding and allows the dissociated CTA1 subunit to spontaneously assume a disordered conformation at physiological temperature (11). This unfolding event identifies CTA1 as a substrate for ERAD-mediated translocation to the cytosol (12–14, 58, 59). The translocated, cytosolic pool of CTA1 must then interact with one or more host factors to regain an ordered conformation. We have previously focused on the order-to-disorder transition that occurs in the ER. Here, we demonstrate lipid rafts play an active role in the disorder-to-order transition for cytosolic CTA1.
As documented with biophysical and biochemical assays, lipid rafts induced disordered CTA1 to assume an ordered, functional conformation. Lipid rafts thus exhibit a chaperone-like activity for CTA1. In vivo disruption of lipid rafts with filipin further demonstrated the physiological importance of this chaperone-like function, because the enzymatic activity of cytosolic CTA1 was greatly attenuated in filipin-treated cells. PE, a major component of the lipid raft inner membrane and the bacterial plasma membrane, is known to exhibit a “lipochaperone” function in the biogenesis of the polytopic bacterial protein lactose permease (29). Dimyristoylphosphatidylethanolamine can likewise induce denatured horseradish peroxidase to attain a folded, active conformation (27). In addition, cholesterol but not sphingolipid has been linked to the proper folding and maturation of cellular prion protein in the ER (28). Because multiple components of the lipid raft inner membrane can modulate protein structure, the chaperone-like activity observed for raft-CTA1 interactions could be a common phenomenon for raft-associated proteins.
Association of the CTB pentamer with its GM1 receptor results in toxin/ganglioside clustering in lipid rafts and subsequent internalization to the endosomal system. Continued interaction with the raft-associated GM1 then directs CT from the endosomes to the trans-Golgi network en route to the ER translocation site (6). Disruption of lipid rafts with filipin or methyl-β-cyclodextrin thus protects cultured cells from intoxication by inhibiting the endocytosis and intracellular transport of CT (57, 60). Given that CT delivery to the ER is dependent upon lipid rafts, the other important function of lipid rafts (i.e. modulation of cytosolic CTA1 structure/function) has gone undetected. We were able to document the in vivo chaperone-like function of lipid rafts toward CTA1 by directly producing CTA1 in the cytosol of cells transfected with the pcDNA3.1/mCTA1 expression vector. This bypassed the upstream raft-dependent CT trafficking events and allowed us to demonstrate an additional role for lipid rafts in the intoxication process. However, the cytosolic pool of transfected CTA1 still exhibited some activity in filipin-treated cells. This suggests that other components of the host cytosol, such as ARF proteins, could also contribute to the in vivo renaturation of disordered CTA1.
In previous work, filipin was applied to Caco-2 cells at various postintoxication time intervals (57). The inhibitory effect of filipin diminished with longer postexposure delays in drug treatment, which suggested that filipin only affected early steps in the intoxication process. However, this procedure could not determine how much CTA1 had already reach Gsα at the time of drug treatment. The use of a cAMP phosphodiesterase inhibitor would further stabilize any cAMP that had already been produced at the time of drug treatment. Our plasmid-based system provided an alternative method to disrupt lipid rafts before CTA1 appearance/expression in the cytosol. Data generated from this approach indicated that the optimal activity of cytosolic CTA1 requires intact lipid rafts, which was consistent with our in vitro biophysical and biochemical analysis of CTA1-raft interactions.
LUVs mimicking the composition of the ER (56) or plasma membrane provide a stabilizing environment for the labile structure of CTA1. However, lipid raft LUVs provide a substantially greater stabilizing environment than either ER or plasma membrane LUVs. Moreover, the gain of structure for disordered CTA1 is a raft-specific event. Because Gsα is present in lipid rafts (23–26), CTA1 would benefit from the chaperone-like function of lipid rafts upon contact with its G protein target. The resulting gain of structure confers a basal level of activity to CTA1 without acting through allostery because no gain of function was recorded for CTA1 treated with lipid rafts at 25 °C. Host ARF proteins instead act as allosteric activators of CTA1 by inducing a conformational change in CTA1 which allows NAD (the donor molecule for the ADP-ribosylation reaction) to readily access the toxin active site (21, 61). Ongoing studies are examining the relative contributions of lipid rafts and ARF proteins to CTA1 activity.
CTA1 exhibited a 2.3 μm affinity for the plasma membrane and a stronger 0.6 μm affinity for lipid rafts. This differential affinity suggests a possible mechanism for CTA1 delivery to raft-localized Gsα. The translocated pool of CTA1 appears to reach its Gsα target by diffusion through the cytosol (51). Binding to the plasma membrane would capture cytosolic CTA1 and place it in the general vicinity of Gsα. The membrane-associated CTA1 subunit would then undergo lateral diffusion along the inner leaflet of the plasma membrane and eventually accumulate in the much smaller surface area of lipid rafts, as it has a higher affinity for lipid rafts than the bulk plasma membrane. A two-stage diffusion trap involving an initial binding to the bulk plasma membrane and a subsequent partitioning into lipid rafts would improve the probability of toxin-target interactions compared with a Gsα contact mechanism involving random toxin diffusion through the cytosol. Sequential interactions with the plasma membrane and lipid rafts would also serve to first stabilize and then refold the disordered, cytosolic CTA1 polypeptide.
The hydrophobic A13 subdomain at the C terminus of CTA1 is responsible for toxin binding to lipid bilayers. Thus, although it is not required for ERAD-mediated toxin translocation (54), the A13 subdomain still plays a functional role in host-toxin interactions. C-terminal domains from the catalytic subunits of Shiga toxin and ricin, two other ER-translocating toxins, also interact with lipid bilayers and play important roles in intoxication (62–65). For ricin A chain, the C terminus has been shown to mediate an interaction with the negatively charged phospholipids of the ER membrane which results in toxin unfolding (62, 63). This would place ricin A chain in a translocation-competent conformation for ERAD-mediated export to the cytosol. The available data suggest a similar mechanism for translocation of the Shiga toxin A1 subunit (64–66), although this model has yet to be directly addressed. An early model of ERAD-mediated toxin translocation noted that multiple ER-translocating toxins contain A chains with hydrophobic C termini and suggested that this could allow the toxin A chains to masquerade as ERAD substrates (20). It now appears that the C termini of toxin A chains, at least for CTA1, ricin A chain, and Shiga toxin A1 chain, play a common role in intoxication. However, rather than serving as a direct trigger for ERAD activation, the C termini seem to facilitate toxin-lipid interactions that promote either toxin unfolding in the ER or toxin refolding in the cytosol.
Supplementary Material
Acknowledgment
We thank Dr. S. E. Mills (Purdue University, West Lafayette, IN) for providing the CHO-β2AR cells.
This work was supported by National Institutes of Health Grant R01 AI073783 (to K. T.).

This article contains supplemental Figs. S1–S5 and Table S1.
- CT
- cholera toxin
- ARF
- ADP-ribosylation factor
- β2AR
- β2-adrenergic receptor
- DEA-BAG
- diethylamino(benzylidine-amino)guanidine
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- LUV
- large unilamellar vesicle
- PE
- phosphatidylethanolamine
- PS
- phosphatidylserine.
REFERENCES
- 1. De Haan L., Hirst T. R. (2004) Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms (review). Mol. Membr. Biol. 21, 77–92 [DOI] [PubMed] [Google Scholar]
- 2. Spangler B. D. (1992) Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol. Rev. 56, 622–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kassis S., Hagmann J., Fishman P. H., Chang P. P., Moss J. (1982) Mechanism of action of cholera toxin on intact cells: generation of A1 peptide and activation of adenylate cyclase. J. Biol. Chem. 257, 12148–12152 [PubMed] [Google Scholar]
- 4. Mekalanos J. J., Collier R. J., Romig W. R. (1979) Enzymic activity of cholera toxin. II. Relationships to proteolytic processing, disulfide bond reduction, and subunit composition. J. Biol. Chem. 254, 5855–5861 [PubMed] [Google Scholar]
- 5. Majoul I., Ferrari D., Söling H. D. (1997) Reduction of protein disulfide bonds in an oxidizing environment: the disulfide bridge of cholera toxin A-subunit is reduced in the endoplasmic reticulum. FEBS Lett. 401, 104–108 [DOI] [PubMed] [Google Scholar]
- 6. Wernick N. L., Chinnapen D. J., Cho J. A., Lencer W. I. (2010) Cholera toxin: an intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins 2, 310–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor M., Banerjee T., Ray S., Tatulian S. A., Teter K. (2011) Protein disulfide isomerase displaces the cholera toxin A1 subunit from the holotoxin without unfolding the A1 subunit. J. Biol. Chem. 286, 22090–22100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsai B., Rodighiero C., Lencer W. I., Rapoport T. A. (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104, 937–948 [DOI] [PubMed] [Google Scholar]
- 9. Goins B., Freire E. (1988) Thermal stability and intersubunit interactions of cholera toxin in solution and in association with its cell-surface receptor ganglioside GM1. Biochemistry 27, 2046–2052 [DOI] [PubMed] [Google Scholar]
- 10. Surewicz W. K., Leddy J. J., Mantsch H. H. (1990) Structure, stability, and receptor interaction of cholera toxin as studied by Fourier-transform infrared spectroscopy. Biochemistry 29, 8106–8111 [DOI] [PubMed] [Google Scholar]
- 11. Pande A. H., Scaglione P., Taylor M., Nemec K. N., Tuthill S., Moe D., Holmes R. K., Tatulian S. A., Teter K. (2007) Conformational instability of the cholera toxin A1 polypeptide. J. Mol. Biol. 374, 1114–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Banerjee T., Pande A., Jobling M. G., Taylor M., Massey S., Holmes R. K., Tatulian S. A., Teter K. (2010) Contribution of subdomain structure to the thermal stability of the cholera toxin A1 subunit. Biochemistry 49, 8839–8846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Massey S., Banerjee T., Pande A. H., Taylor M., Tatulian S. A., Teter K. (2009) Stabilization of the tertiary structure of the cholera toxin A1 subunit inhibits toxin dislocation and cellular intoxication. J. Mol. Biol. 393, 1083–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taylor M., Banerjee T., Navarro-Garcia F., Huerta J., Massey S., Burlingame M., Pande A. H., Tatulian S. A., Teter K. (2011) A therapeutic chemical chaperone inhibits cholera intoxication and unfolding/translocation of the cholera toxin A1 subunit. PLoS ONE 6, e18825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schmitz A., Herrgen H., Winkeler A., Herzog V. (2000) Cholera toxin is exported from microsomes by the Sec61p complex. J. Cell Biol. 148, 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bernardi K. M., Forster M. L., Lencer W. I., Tsai B. (2008) Derlin-1 facilitates the retro-translocation of cholera toxin. Mol. Biol. Cell 19, 877–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saslowsky D. E., Cho J. A., Chinnapen H., Massol R. H., Chinnapen D. J., Wagner J. S., De Luca H. E., Kam W., Paw B. H., Lencer W. I. (2010) Intoxication of zebrafish and mammalian cells by cholera toxin depends on the flotillin/reggie proteins but not Derlin-1 or -2. J. Clin. Invest. 120, 4399–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dixit G., Mikoryak C., Hayslett T., Bhat A., Draper R. K. (2008) Cholera toxin up-regulates endoplasmic reticulum proteins that correlate with sensitivity to the toxin. Exp. Biol. Med. 233, 163–175 [DOI] [PubMed] [Google Scholar]
- 19. Rodighiero C., Tsai B., Rapoport T. A., Lencer W. I. (2002) Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 3, 1222–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hazes B., Read R. J. (1997) Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 36, 11051–11054 [DOI] [PubMed] [Google Scholar]
- 21. Welsh C. F., Moss J., Vaughan M. (1994) ADP-ribosylation factors: a family of approximately 20-kDa guanine nucleotide-binding proteins that activate cholera toxin. Mol. Cell. Biochem. 138, 157–166 [DOI] [PubMed] [Google Scholar]
- 22. Murayama T., Tsai S. C., Adamik R., Moss J., Vaughan M. (1993) Effects of temperature on ADP-ribosylation factor stimulation of cholera toxin activity. Biochemistry 32, 561–566 [DOI] [PubMed] [Google Scholar]
- 23. Oh P., Schnitzer J. E. (2001) Segregation of heterotrimeric G proteins in cell surface microdomains. Gq binds caveolin to concentrate in caveolae, whereas Gi and Gs target lipid rafts by default. Mol. Biol. Cell 12, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ostrom R. S., Insel P. A. (2004) The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br. J. Pharmacol. 143, 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Allen J. A., Halverson-Tamboli R. A., Rasenick M. M. (2007) Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 8, 128–140 [DOI] [PubMed] [Google Scholar]
- 26. Kamata K., Manno S., Ozaki M., Takakuwa Y. (2008) Functional evidence for presence of lipid rafts in erythrocyte membranes: Gsα in rafts is essential for signal transduction. Am. J. Hematol. 83, 371–375 [DOI] [PubMed] [Google Scholar]
- 27. Debnath D., Bhattacharya S., Chakrabarti A. (2003) Phospholipid assisted folding of a denatured heme protein: effect of phosphatidylethanolamine. Biochem. Biophys. Res. Commun. 301, 979–984 [DOI] [PubMed] [Google Scholar]
- 28. Sarnataro D., Campana V., Paladino S., Stornaiuolo M., Nitsch L., Zurzolo C. (2004) PrP(C) association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol. Biol. Cell 15, 4031–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bogdanov M., Dowhan W. (1999) Lipid-assisted protein folding. J. Biol. Chem. 274, 36827–36830 [DOI] [PubMed] [Google Scholar]
- 30. Lencer W. I., Chu S. H., Walker W. A. (1987) Differential binding kinetics of cholera toxin to intestinal microvillus membrane during development. Infect. Immun. 55, 3126–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Proulx P. (1991) Structure-function relationships in intestinal brush-border membranes. Biochim. Biophys. Acta 1071, 255–271 [DOI] [PubMed] [Google Scholar]
- 32. Forstner G. G., Tanaka K., Isselbacher K. J. (1968) Lipid composition of the isolated rat intestinal microvillus membrane. Biochem. J. 109, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Waheed A. A., Yasuzumi F., Gupta P. D. (1998) Lipid and fatty acid composition of brush-border membrane of rat intestine during starvation. Lipids 33, 1093–1097 [DOI] [PubMed] [Google Scholar]
- 34. Forstner G. G., Sabesin S. M., Isselbacher K. J. (1968) Rat intestinal microvillus membranes: purification and biochemical characterization. Biochem. J. 106, 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Meer G., Voelker D. R., Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boon J. M., Smith B. D. (2002) Chemical control of phospholipid distribution across bilayer membranes. Med. Res. Rev. 22, 251–281 [DOI] [PubMed] [Google Scholar]
- 37. D'Antuono C., Fernández-Tomé M. C., Sterin-Speziale N., Bernik D. L. (2000) Lipid-protein interactions in rat renal subcellular membranes: a biophysical and biochemical study. Arch. Biochem. Biophys. 382, 39–47 [DOI] [PubMed] [Google Scholar]
- 38. Dietrich C., Bagatolli L. A., Volovyk Z. N., Thompson N. L., Levi M., Jacobson K., Gratton E. (2001) Lipid rafts reconstituted in model membranes. Biophys. J. 80, 1417–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schroeder F., Woodford J. K., Kavecansky J., Wood W. G., Joiner C. (1995) Cholesterol domains in biological membranes. Mol. Membr. Biol. 12, 113–119 [DOI] [PubMed] [Google Scholar]
- 40. Frazier M. L., Wright J. R., Pokorny A., Almeida P. F. (2007) Investigation of domain formation in sphingomyelin/cholesterol/POPC mixtures by fluorescence resonance energy transfer and Monte Carlo simulations. Biophys. J. 92, 2422–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hammond A. T., Heberle F. A., Baumgart T., Holowka D., Baird B., Feigenson G. W. (2005) Cross-linking a lipid raft component triggers liquid ordered-liquid disordered phase separation in model plasma membranes. Proc. Natl. Acad. Sci. U.S.A. 102, 6320–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pike L. J., Han X., Chung K. N., Gross R. W. (2002) Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: a quantitative electrospray ionization/mass spectrometric analysis. Biochemistry 41, 2075–2088 [DOI] [PubMed] [Google Scholar]
- 43. Bakht O., Pathak P., London E. (2007) Effect of the structure of lipids favoring disordered domain formation on the stability of cholesterol-containing ordered domains (lipid rafts): identification of multiple raft-stabilization mechanisms. Biophys. J. 93, 4307–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Delaunay J. L., Breton M., Trugnan G., Maurice M. (2008) Differential solubilization of inner plasma membrane leaflet components by Lubrol WX and Triton X-100. Biochim. Biophys. Acta 1778, 105–112 [DOI] [PubMed] [Google Scholar]
- 45. Grzybek M., Kubiak J., Łach A., Przybyło M., Sikorski A. F. (2009) A raft-associated species of phosphatidylethanolamine interacts with cholesterol comparably to sphingomyelin: a Langmuir-Blodgett monolayer study. PLoS ONE 4, e5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brown D. A., London E. (2000) Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275, 17221–17224 [DOI] [PubMed] [Google Scholar]
- 47. Qin S., Pande A. H., Nemec K. N., Tatulian S. A. (2004) The N-terminal α-helix of pancreatic phospholipase A2 determines productive-mode orientation of the enzyme at the membrane surface. J. Mol. Biol. 344, 71–89 [DOI] [PubMed] [Google Scholar]
- 48. Lavigne P., Crump M. P., Gagné S. M., Hodges R. S., Kay C. M., Sykes B. D. (1998) Insights into the mechanism of heterodimerization from the 1H-NMR solution structure of the c-Myc-Max heterodimeric leucine zipper. J. Mol. Biol. 281, 165–181 [DOI] [PubMed] [Google Scholar]
- 49. Litman B. J., Barenholz Y. (1975) The optical activity of d-erythro-sphingomyelin and its contribution to the circular dichroism of sphingomyelin-containing systems. Biochim. Biophys. Acta 394, 166–172 [DOI] [PubMed] [Google Scholar]
- 50. Soman G., Narayanan J., Martin B. L., Graves D. J. (1986) Use of substituted (benzylidineamino)guanidines in the study of guanidino group specific ADP-ribosyltransferase. Biochemistry 25, 4113–4119 [DOI] [PubMed] [Google Scholar]
- 51. Teter K., Jobling M. G., Holmes R. K. (2004) Vesicular transport is not required for the cytoplasmic pool of cholera toxin to interact with the stimulatory α subunit of the heterotrimeric G protein. Infect. Immun. 72, 6826–6835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang W., Bidwell C. A., Collodi P. R., Mills S. E. (2000) Expression of the porcine β2-adrenergic receptor in Chinese hamster ovary cells. J. Anim. Sci. 78, 2329–2335 [DOI] [PubMed] [Google Scholar]
- 53. Mills S. E., Kissel J., Bidwell C. A., Smith D. J. (2003) Stereoselectivity of porcine β-adrenergic receptors for ractopamine stereoisomers. J. Anim. Sci. 81, 122–129 [DOI] [PubMed] [Google Scholar]
- 54. Teter K., Jobling M. G., Sentz D., Holmes R. K. (2006) The cholera toxin A13 subdomain is essential for interaction with ADP-ribosylation factor 6 and full toxic activity but is not required for translocation from the endoplasmic reticulum to the cytosol. Infect. Immun. 74, 2259–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ampapathi R. S., Creath A. L., Lou D. I., Craft J. W., Jr., Blanke S. R., Legge G. B. (2008) Order-disorder-order transitions mediate the activation of cholera toxin. J. Mol. Biol. 377, 748–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ray S., Taylor M., Burlingame M., Tatulian S. A., Teter K. (2011) Modulation of toxin stability by 4-phenylbutyric acid and negatively charged phospholipids. PLoS ONE 6, e23692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Orlandi P. A., Fishman P. H. (1998) Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 141, 905–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Teter K., Holmes R. K. (2002) Inhibition of endoplasmic reticulum-associated degradation in CHO cells resistant to cholera toxin, Pseudomonas aeruginosa exotoxin A, and ricin. Infect. Immun. 70, 6172–6179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Teter K., Jobling M. G., Holmes R. K. (2003) A class of mutant CHO cells resistant to cholera toxin rapidly degrades the catalytic polypeptide of cholera toxin and exhibits increased endoplasmic reticulum-associated degradation. Traffic 4, 232–242 [DOI] [PubMed] [Google Scholar]
- 60. Wolf A. A., Fujinaga Y., Lencer W. I. (2002) Uncoupling of the cholera toxin-GM1 ganglioside receptor complex from endocytosis, retrograde Golgi trafficking, and downstream signal transduction by depletion of membrane cholesterol. J. Biol. Chem. 277, 16249–16256 [DOI] [PubMed] [Google Scholar]
- 61. O'Neal C. J., Jobling M. G., Holmes R. K., Hol W. G. (2005) Structural basis for the activation of cholera toxin by human ARF6-GTP. Science 309, 1093–1096 [DOI] [PubMed] [Google Scholar]
- 62. Day P. J., Pinheiro T. J., Roberts L. M., Lord J. M. (2002) Binding of ricin A-chain to negatively charged phospholipid vesicles leads to protein structural changes and destabilizes the lipid bilayer. Biochemistry 41, 2836–2843 [DOI] [PubMed] [Google Scholar]
- 63. Mayerhofer P. U., Cook J. P., Wahlman J., Pinheiro T. T., Moore K. A., Lord J. M., Johnson A. E., Roberts L. M. (2009) Ricin A chain insertion into endoplasmic reticulum membranes is triggered by a temperature increase to 37 °C. J. Biol. Chem. 284, 10232–10242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Menikh A., Saleh M. T., Gariépy J., Boggs J. M. (1997) Orientation in lipid bilayers of a synthetic peptide representing the C terminus of the A1 domain of Shiga toxin: a polarized ATR-FTIR study. Biochemistry 36, 15865–15872 [DOI] [PubMed] [Google Scholar]
- 65. Saleh M. T., Ferguson J., Boggs J. M., Gariépy J. (1996) Insertion and orientation of a synthetic peptide representing the C terminus of the A1 domain of Shiga toxin into phospholipid membranes. Biochemistry 35, 9325–9334 [DOI] [PubMed] [Google Scholar]
- 66. LaPointe P., Wei X., Gariépy J. (2005) A role for the protease-sensitive loop region of Shiga-like toxin 1 in the retrotranslocation of its A1 domain from the endoplasmic reticulum lumen. J. Biol. Chem. 280, 23310–23318 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.