

Abstract
Activating mutations in the cytosolic serine/threonine kinase, BRAF, have been reported in a variety of neoplasms. BRAF activation may contribute to tumor growth via activation of the MAP/ERK kinase pathway, and BRAF represents a possible therapeutic target. Activating BRAF mutations were recently reported in approximately 10% of prostate cancer cases in Asian patients. In the present study, 43 hormone refractory prostate cancers were analyzed for BRAF mutations in order to determine whether anti-BRAF therapy is a suitable approach for advanced prostate cancer patients. In all of the studied tumors, BRAF exons 11 and 15 were PCR-amplified and sequenced, including the backward and forward sequences. BRAF mutations were noted only in the positive control tissues, but were not found in any of the 43 analyzed prostate cancers. We conclude that BRAF mutations occur only rarely in prostate cancers in Caucasian patients and are not associated with tumor progression. The application of anti-BRAF therapies may therefore not be beneficial for prostate cancer.
Keywords: prostate cancer, BRAF, androgen resistance
Introduction
Prostate adenocarcinoma is one of the most commonly diagnosed carcinomas in the Western world and a leading cause of cancer-related death (1). Localized prostate cancer can be cured by various treatment modalities, whereas in advanced stages hormonal ablation represents the most effective systemic treatment option. However, androgen insensitivity invariably develops and is responsible for prostate cancer lethality. Although established, the use of doxetacel chemotherapy is only associated with a 3-month gain in overall survival. Thus, alternative effective systemic therapies are required. An improved insight into the molecular mechanisms of prostate cancer progression, which may result in novel therapeutic options, is therefore crucial.
BRAF, a member of the RAF family of serine/threonine kinases, can be activated by a somatic mutation in many human cancers. BRAF mutations are reported to occur at high frequencies in malignant melanomas and thyroid papillary carcinomas and at lower frequencies in a variety of other human types of cancer (2). Several BRAF inhibitors are clinically available, including the VEGFR/PDGFR/KIT/RAF-inhibitor sorafenib (BAY43-9006) which has been shown to exhibit significant anti-BRAF activity in vitro, suggesting a therapeutic option for tumors carrying BRAF mutations (3,4).
Oncogenic activating BRAF mutations were recently reported in approximately 10% of a series of 204 non-pretreated prostate cancer cases (5). This raises the possibility that BRAF inhibitors are a promising therapeutic option for androgen-independent prostate cancer patients in whom all current treatments have only limited efficacy. Studies involving BRAF mutations in hormone refractory prostate cancer patients are lacking. Therefore, 43 hormone refractory prostate cancer tissue samples were analyzed for BRAF mutations.
Materials and methods
Tissue samples
Paraffin-embedded tissue samples from 43 patients with hormone refractory prostate cancer were collected between 1999 and 2007 at the Institute of Pathology of the University Medical Center Hamburg-Eppendorf. Hormone refractory disease was defined as an increased prostate-specific agent (PSA) during anti-androgen therapy on three consecutive measurements. Tissue samples were obtained by palliative transurethral resections. All patients had high-grade cancer (Table I).
Table I.
Tumor characteristics and the BRAF mutation status in 43 cases of hormone refractory prostate cancer.
| Sample no. | BRAF-11 | BRAF-15 | Age (years) | Gleason score |
|---|---|---|---|---|
| Ha 1 | wt | wt | 80 | 4+5 |
| Ha 2 | wt | wt | 79 | 4+5 |
| Ha 3 | wt | wt | 55 | 5+4 |
| Ha 4 | wt | wt | 79 | 5+4 |
| Ha 5 | wt | wt | 84 | 4+4 |
| Ha 6 | wt | wt | 84 | 5+4 |
| Ha 7 | wt | wt | 70 | 4+4 |
| Ha 8 | wt | wt | 76 | 4+5 |
| Ha 9 | wt | wt | 86 | 4+5 |
| Ha 10 | wt | wt | 59 | 4+5 |
| Ha 11 | wt | wt | 77 | 4+5 |
| Ha 12 | wt | wt | 76 | 4+3 |
| Ha 13 | wt | wt | 85 | 4+5 |
| Ha 14 | wt | wt | 83 | 4+5 |
| Ha 15 | wt | wt | 70 | 4+5 |
| Ha 16 | wt | wt | 65 | 4+5 |
| Ha 17 | wt | wt | 79 | 5+5 |
| Ha 18 | wt | wt | 85 | 4+5 |
| Ha 19 | wt | wt | 73 | 5+5 |
| Ha 20 | wt | wt | 78 | 4+5 |
| Ha 21 | wt | wt | 61 | 4+5 |
| Ha 22 | wt | wt | 61 | 4+4 |
| Ha 23 | wt | wt | 64 | 4+3 |
| Ha 24 | wt | wt | 75 | 4+5 |
| Ha 25 | wt | wt | 74 | 5+4 |
| Ha 26 | wt | wt | 80 | 4+5 |
| Ha 27 | wt | wt | 86 | 3+4 |
| Ha 28 | wt | wt | 64 | 5+4 |
| Ha 29 | wt | wt | 87 | 4+5 |
| Ha 30 | wt | wt | 73 | 4+5 |
| Ha 31 | wt | wt | 83 | 4+5 |
| Ha 32 | wt | wt | 88 | 4+5 |
| Ha 33 | wt | wt | 84 | 4+5 |
| Ha 34 | wt | wt | 63 | 4+5 |
| Ha 35 | wt | wt | 80 | 4+4 |
| Ha 36 | wt | wt | 86 | 4+5 |
| Ha 37 | wt | wt | 77 | 5+4 |
| Ha 38 | wt | wt | 70 | 4+5 |
| Ha 39 | wt | wt | 67 | 4+5 |
| Ha 40 | wt | wt | 76 | 4+5 |
| Ha 41 | wt | wt | 89 | 3+4 |
| Ha 42 | wt | wt | 72 | 4+5 |
| Ha 43 | wt | wt | 67 | 5+5 |
| Control | wt | V600E |
wt, wild-type.
BRAF mutation analysis
Representative tumor areas of the paraffin tissue block containing at least 70% tumor cells were selected. Two tissue cylinders (0.6 mm in diameter, ~5-mm long) were punched from each block using a hollow needle and were subjected to DNA isolation using a commercial kit (QIAmp DNA mini kit; Qiagen, Hilden, Germany). Isolated DNA (70 ng) was used for PCR to generate BRAF exons 11- and 15-specific DNA fragments. Primers were: BRAF-11: forward, 5′-tccctctcaggcataaggtaa-3′ and reverse 5′-cgaacagtg aatatttcctttgat-3′; and BRAF-15, forward 5′-tcataatgcttgctctg atagga-3′ and reverse 5′-ggccaaaaatttaatcagtgga-3′. PCR was performed on a thermal cycler with the following steps: i) initial template DNA denaturation at 95˚C for 10 min; ii) 34 cycles of 30 sec at 95˚C, 120 sec at 58˚C and 75 sec at 72˚C and iii) a 7-min extension at 72˚C. For sequencing, 5 μl of the BRAF exons 11- and 15-specific PCR product were treated with 2 μl ExoSAP (USB Europe GmbH, Staufen, Germany) for 15 min at 37˚C and 15 min at 80˚C to remove unincorporated primers. The treated PCR products were subjected to sequencing using the primers outlined above. The sequencing reaction was performed on a thermal cycler with the following steps: i) 10 min at 95˚C (polymerase activation); ii) 10 sec at 95˚C (denaturation) and iii) 35 cycles of 10 sec at 55˚C followed by 2 min at 60˚C. The sequencing products were ethanol-precipitated to remove excess primers and analyzed on an ABI PRISM 3100 genetic analyzer. The samples were processed in duplicate starting from the PCR. Forward and reverse sequences were compared manually to the BRAF reference sequence (NG_007873.1). A mutation was assumed only when it was detected in both analyses. A case of serrated adenoma of the caecum with a known V600E mutation was analyzed in parallel as a positive control.
Results and Discussion
BRAF is a member of the RAF family of protein kinases that includes the members ARAF, BRAF and CRAF (6). BRAF is the only RAF protein to be mutated in approximately 8% of human tumors, resulting in the intensive study of this gene (7). In our study, the BRAF mutation status and tumor characteristics of the patients are shown in Table I. Mutations of BRAF were reported in approximately 66% of melanomas and in smaller percentages in other types of cancer, including thyroid, colonic and ovarian, as well as in some sarcomas (2,7,8). In patients with germ cell tumors, the BRAF mutation appears to indicate resistance against cisplatin treatment (9). However, over 30 different mutations in BRAF have been identified (10,11), a single base-pair substitution in exon 15 at codon 600 (V600E) is found in over 80% of cases. The majority of mutations identified cause constitutive kinase activation with the V600E mutation demonstrating approximately 500-fold greater kinase activity than wild-type BRAF. Supporting its classification as an oncogene, V600E BRAF stimulates ERK signaling, induces proliferation and in model systems is capable of promoting transformation. As suggested by pre-clinical studies, tumors with a V600E BRAF mutation remain dependent upon BRAF for proliferation and survival (12,13). Given its frequent occurrence in human cancer, efforts are underway to develop targeted inhibitors of BRAF and its downstream effectors (14).
Cho et al (5) suggested that BRAF mutations may be found in a significant proportion of prostate cancers. In a series of 206 prostate cancers, these authors reported a BRAF mutation rate of approximately 10%. The BRAF-mutated tumors in this study tended to have a higher Gleason score, a higher pre-operative serum PSA level, as well as a more advanced tumor stage [median PSA, 10.3 ng/ml (5.5–98); Gleason score, ≥7b in 14/21 (67%) patients; stage, ≥pT3a in 12/21 (57.1%) patients]. A role of BRAF activation in prostate cancer development and progression is also consistent with data from Gao et al (15), who demonstrated activation of the Akt and BRAF/Erk MAP kinase signaling pathway in an androgen-independent prostate cancer model established in mice.
However, in our cohort of 43 hormone refractory prostate cancer specimens no BRAF mutations were found, while the control case (serrated adenoma of the caecum) showed the known mutation, V600E (Fig. 1). Since the 204 cancer cases analyzed by Cho et al (5) were untreated primary tumors, one explanation for the different results observed may be that BRAF mutations are involved only in the early stages of prostate cancer development and that tumor maintenance and progression towards advanced and/or hormone-insensitive cancers are independent of the BRAF pathway, in order that BRAF wild-type cells eventually overgrow the BRAF-mutated cells. This assumption is consistent with experimental findings from Jeong et al (16). These authors developed a genetically engineered mouse (GEM) model of invasive prostate cancers, whereby an activating mutation of BRAF V600E was targeted to the epithelial compartment of the prostate gland on the background of Ink4a/Arf deficiency. These GEM mice developed prostate gland hyperplasia with progression to rapidly growing invasive adenocarcinoma without evidence of AKT activation, providing genetic proof that the activation of MAP kinase signaling is sufficient to drive prostate tumorigenesis. Notably, the genetic extinction of BRAF V600E in established prostate tumors did not lead to tumor regression, indicating that, while sufficient to initiate development of invasive prostate adenocarcinoma, BRAF V600E is not required for its maintenance. Based on these findings it can be speculated that BRAF-positive tumor cell clones are eliminated by other tumor cell clones during tumor progression towards hormone resistance, which could explain the lack of BRAF mutations in our series of hormone-resistant prostate cancers.
Figure 1.
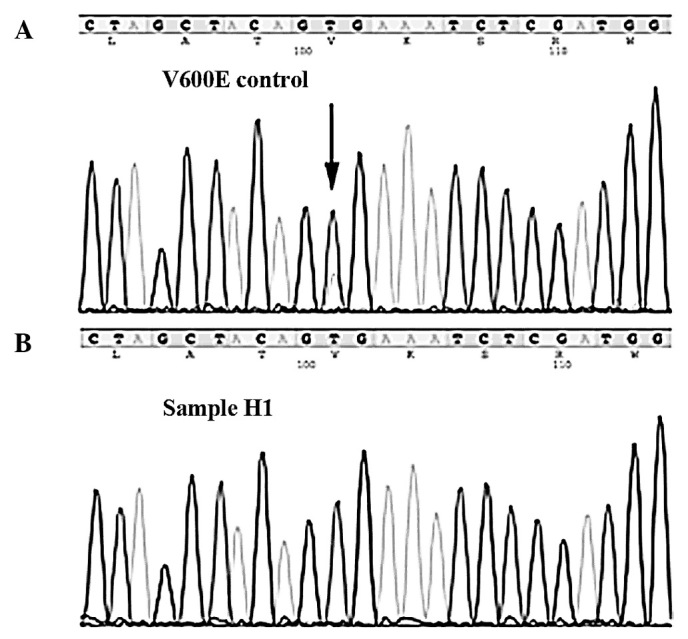
Examples of BRAF sequencing results. (A) Control cases of a serrated adenoma of the caecum showing a V600E mutation (arrow) in exon 15. (B) Case H1 showing the wild-type sequence.
However, other groups studying less advanced prostate cancers failed to show BRAF mutations. For example, Liu et al did not find any BRAF mutations in 93 non-pretreated prostate cancer tissues (17). The same holds true for a smaller study by Cohen et al who also failed to show mutations in a small set of 17 untreated prostate cancer samples (2). An ethnic bias towards the prevalence of prostate BRAF mutations may represent an alternative explanation for the difference in results. In contrast to Cho et al (5) who exclusively analyzed Korean patients, our study as well as the aforementioned studies by Liu et al (17) and Cohen et al (2) examined Caucasian patients. The prevalence for an ethnic relationship between tumor molecular abnormalities and ethnic variations is well documented from the study of epidermal growth factor receptor mutations in non-small cell lung cancer. Patients from Asian descent are more likely to have non-small cell lung cancer with epidermal growth factor receptor-activating mutations than patients from the US (18). Moreover, HER2 amplification is more frequent in breast cancers of Korean and Saudi Arabian patients than in those of Caucasian patients (19,20).
Technical errors are another possible explanation for conflicting results in molecular analyses. The majority of BRAF mutations reported by Cho et al (5) were V600A or V600M. These mutations are transitions uncommon in cancers in Caucasian individuals (21). It is known that such transitions can be artificially incorporated into DNA which has been fixed in formalin (22). However, even in the event that all V600A or V600M mutations were caused by technical errors in the study of Cho et al (5), the BRAF mutation rate would not have dropped to zero. The large scale of their study at least allowed for the detection of 2 of 206 cases with V600E mutations, which is not a typical technical artifact.
In conclusion, BRAF mutations are rare in early- and late-stage prostate cancer. Collectively, approximately 1% of prostate cancers harbor BRAF mutations. Since more than 11,000 men die from hormone refractory prostate cancer in Germany alone, testing a cohort of prostate cancers for BRAF mutations must be considered in a clinical trial when potent BRAF-inhibiting drugs become available.
Acknowledgements
A grant support was obtained from the German Federation Ministry of Education and Science in the framework of the program for medical genome research (FKZ:01GS08189).
References
- 1.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;1:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Cohen Y, Xing M, Mambo E, et al. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst. 2003;8:625–627. doi: 10.1093/jnci/95.8.625. [DOI] [PubMed] [Google Scholar]
- 3.Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;19:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 4.Preto A, Goncalves J, Rebocho AP, et al. Proliferation and survival molecules implicated in the inhibition of BRAF pathway in thyroid cancer cells harbouring different genetic mutations. BMC Cancer. 2009;1:387. doi: 10.1186/1471-2407-9-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho NY, Choi M, Kim BH, Cho YM, Moon KC, Kang GH. BRAF and KRAS mutations in prostatic adenocarcinoma. Int J Cancer. 2006;8:1858–1862. doi: 10.1002/ijc.22071. [DOI] [PubMed] [Google Scholar]
- 6.Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal. 2003;5:463–469. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- 7.Davies H, Dicks E, Stephens P, et al. High throughput DNA sequence variant detection by conformation sensitive capillary electrophoresis and automated peak comparison. Genomics. 2006;3:427–432. doi: 10.1016/j.ygeno.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 8.Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;7:1454–1457. [PubMed] [Google Scholar]
- 9.Honecker F, Wermann H, Mayer F, et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J Clin Oncol. 2009;13:2129–2136. doi: 10.1200/JCO.2008.18.8623. [DOI] [PubMed] [Google Scholar]
- 10.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, Wooster R. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 12.Hoeflich KP, Gray DC, Eby MT, et al. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res. 2006;2:999–1006. doi: 10.1158/0008-5472.CAN-05-2720. [DOI] [PubMed] [Google Scholar]
- 13.Sumimoto H, Miyagishi M, Miyoshi H, et al. Inhibition of growth and invasive ability of melanoma by inactivation of mutated BRAF with lentivirus-mediated RNA interference. Oncogene. 2004;36:6031–6039. doi: 10.1038/sj.onc.1207812. [DOI] [PubMed] [Google Scholar]
- 14.Pratilas CA, Solit DB. Therapeutic strategies for targeting BRAF in human cancer. Rev Recent Clin Trials. 2007;2:121–134. doi: 10.2174/157488707780599393. [DOI] [PubMed] [Google Scholar]
- 15.Gao H, Ouyang X, Banach-Petrosky WA, Gerald WL, Shen MM, Abate-Shen C. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc Natl Acad Sci USA. 2006;39:14477–14482. doi: 10.1073/pnas.0606836103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong JH, Wang Z, Guimaraes AS, et al. BRAF activation initiates but does not maintain invasive prostate adenocarcinoma. PLoS One. 2008;12:e3949. doi: 10.1371/journal.pone.0003949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu T, Willmore-Payne C, Layfield LJ, Holden JA. Lack of BRAF activating mutations in prostate adenocarcinoma: a study of 93 cases. Appl Immunohistochem Mol Morphol. 2009;2:121–125. doi: 10.1097/PAI.0b013e31818816b9. [DOI] [PubMed] [Google Scholar]
- 18.Riely GJ, Politi KA, Miller VA, Pao W. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res. 2006;24:7232–7241. doi: 10.1158/1078-0432.CCR-06-0658. [DOI] [PubMed] [Google Scholar]
- 19.Al-Kuraya K, Schraml P, Sheikh S, et al. Predominance of high-grade pathway in breast cancer development of Middle East women. Mod Pathol. 2005;7:891–897. doi: 10.1038/modpathol.3800408. [DOI] [PubMed] [Google Scholar]
- 20.Choi DH, Shin DB, Lee MH, et al. A comparison of five immunohistochemical biomarkers and HER-2/neu gene amplification by fluorescence in situ hybridization in white and Korean patients with early-onset breast carcinoma. Cancer. 2003;8:1587–1595. doi: 10.1002/cncr.11703. [DOI] [PubMed] [Google Scholar]
- 21.Michaloglou C, Vredeveld LC, Mooi WJ, Peeper DS. BRAF(E600) in benign and malignant human tumours. Oncogene. 2008;7:877–895. doi: 10.1038/sj.onc.1210704. [DOI] [PubMed] [Google Scholar]
- 22.Marchetti A, Felicioni L, Buttitta F. Assessing EGFR mutations. N Engl J Med. 2006;5:526–528. doi: 10.1056/NEJMc052564. [DOI] [PubMed] [Google Scholar]