

Abstract
The incidence of chromosome 3p gene alterations is one of the most frequent and earliest documented events in lung cancer. This study aimed to investigate promoter methylation in the deleted in lung and esophageal cancer 1 (DLEC1) gene, as well as the p16 and CDH1 genes in Japanese lung cancer cases. The methylation status of the promoter regions of DLEC1, p16 and CDH1 was investigated using methylation-specific PCR. The findings were compared to the clinicopathological features of lung cancer. Methylation-specific PCR showed that the DLEC1 promoter region was methylated in 65 out of 116 (56%) lung cancers. Patients with DLEC1-methylated cancer were associated with a significantly worse prognosis than those with unmethylated cancer (p=0.0368; hazard ratio=1.83). The p16 methylation status correlated with squamous histology (p=0.03) and smoking status (never smoker vs. smoker; p=0.0122). Patients with p16 ummethylated cancer harbored more EGFR mutations (p=0.0071). The CDH1 promoter region was hypermethylated in 65 out of 118 (55.1%) lung cancer cases. However, the CDH1 methylation status was not associated with the clinicopathological characteristics of the lung cancer types. p16 and CDH1 methylation status did not correlate with survival in the lung cancer patients. Thus, in our Japanese cohort, the methylation status of the DLEC1 gene was a marker of poor prognosis independent of stage.
Keywords: methylation, DLEC1 gene, lung cancer
Introduction
Alleic loss of chromosome 3p is one of the most frequent and earliest documented events in lung cancer with a wide range of 3p mutations (12–26), suggesting the presence of multiple tumor-suppressor genes on 3p (1–3). The chromosome locus 3p22.3 is a ‘hot spot’ for chromosomal aberrations and loss of heterozygosity in cancers, including lung cancer (4,5). Further analysis led to the identification of the DLC1 gene (6), which was later renamed deleted in lung and esophageal cancer 1 (DLEC1) on 3p22.3. Loss of DLEC1 expression has been observed in lung, esophageal, renal, ovarian and nasopharyngeal carcinoma cell lines and primary tumors. Moreover, functional analyses strongly suggest that DLEC1 is a tumor-suppressor gene (6–8). Promoter hypermethylation has been shown to be responsible for the silencing of DLEC1 in ovarian cancer and nasopharyngeal carcinoma (7,8). Furthermore, there have been methylation analyses reported for hepatocellular carcinoma (4), gastric (9) and lung cancers (5).
This study investigated whether the promoter hyper-methylation of DLEC1 plays a role in lung cancer in Japanese patients, and whether it has any prognostic significance. The methylation status of the promoter regions of DLEC1 was investigated using methylation-specific PCR. The findings were compared to the clinicopathological features of lung cancer.
Patients and methods
Patients
The study group included lung cancer patients who had undergone surgery at the Nagoya City University Hospital. Written informed consent was obtained. Approval was granted by the Institutional Ethics Committee of the Nagoya City University Graduate School of Medical Sciences. The lung tumors were classified according to the general criteria for the clinical and pathological record of lung cancer in Japan (10). Tumor samples were immediately frozen and stored at −80°C until assayed. The clinical and pathological characteristics of the 116 lung cancer patients for DLEC1 sequencing analysis follow. Eighty-nine patients (76.7%) were male, 27 were female. Thirty-eight patients (32.8%) had squamous cell carcinomas; 65, adenocarcinomas and 10 had adenosquamous cell carcinomas. Eighty-five (73.8%) were smokers and 31 were non-smokers. The clinical and pathological characteristics of the lung cancer patients for the methylation analyses are documented in Tables I, II and III, respectively. The samples from these patients were previously sequenced for EGFR (11–14).
Table I.
Clinicopathological data of 116 lung cancer patients.
| DLEC1 gene status | |||
|---|---|---|---|
|
|
|||
| Factors | Methylated cases | Unmethylated cases | p-value |
| Mean age (years) | 64.5±8.9 | 66.3±13.3 | 0.2211 |
| Stage | 0.7028 | ||
| I | 24 (36.9%) | 21 (41.2%) | |
| II–IV | 41 (63.1%) | 30 (58.8%) | |
| Lymph node metastasis | 0.5734 | ||
| N0 | 34 (52.3%) | 30 (58.8%) | |
| N+ | 31 (47.7%) | 21 (41.2%) | |
| Smoking | 0.2910 | ||
| Never smoker | 20 (30.8%) | 11 (21.6%) | |
| Smoker | 45 (69.2%) | 40 (79.4%) | |
| EGFR status | 0.1657 | ||
| Wild-type | 49 (75.4%) | 44 (86.3%) | |
| Mutation | 16 (24.6%) | 7 (13.7%) | |
| Pathological subtypes | 0.4269 | ||
| SCC | 19 (29.2%) | 19 (37.3%) | |
| Non-SCC | 46 (70.8%) | 32 (62.7%) | |
| Age | 0.3478 | ||
| ≤65 | 32 (49.2%) | 20 (39.2%) | |
| >65 | 33 (50.8%) | 31 (60.8%) | |
| Gender | 0.9999 | ||
| Male | 50 (76.9%) | 39 (76.5%) | |
| Female | 15 (23.1%) | 12 (23.5%) | |
N+, lymph node metastasis-positive; SCC, squamous cell carcinoma.
Table II.
Clinicopathological data of 221 lung cancer patients.
| p16 gene status | |||
|---|---|---|---|
|
|
|||
| Factors | Methylated cases | Unmethylated cases | p-value |
| Mean age (years) | 66.1±8.9 | 64.3±10.9 | 0.4076 |
| Stage | 0.2726 | ||
| I | 39 (43.3%) | 67 (51.5%) | |
| II–IV | 51 (56.7%) | 63 (48.5%) | |
| Lymph node metastasis | 0.1207 | ||
| N0 | 51 (56.0%) | 87 (66.9%) | |
| N+ | 40 (44.0%) | 43 (33.1%) | |
| Smoking | 0.0122 | ||
| Never smoker | 15 (16.5%) | 41 (31.5%) | |
| Smoker | 76 (83.5%) | 89 (68.5%) | |
| EGFR status | 0.0071 | ||
| Wild-type | 80 (87.9%) | 94 (72.3%) | |
| Mutation | 11 (12.1%) | 36 (27.7%) | |
| Pathological subtype | 0.030 | ||
| SCC | 49 (53.8%) | 54 (38.6%) | |
| Non-SCC | 42 (46.2%) | 86 (61.4%) | |
| Age | 0.2198 | ||
| ≤65 | 37 (40.7%) | 64 (49.2%) | |
| >65 | 54 (59.3%) | 66 (50.8%) | |
| Gender | 0.6141 | ||
| Male | 74 (81.3%) | 101 (77.7%) | |
| Female | 17 (18.7%) | 29 (22.3%) | |
N+, lymph node metastasis positive; SCC, squamous cell carcinoma.
Table III.
Clinicopathological data of 118 lung cancer patients.
| CDH1 gene status | |||
|---|---|---|---|
|
|
|||
| Factors | Methylated cases | Unmethylated cases | p-value |
| Mean age (years) | 66.6±8.9 | 62.7±11.2 | 0.0491 |
| Stage | 0.5669 | ||
| I | 22 (33.8%) | 21 (39.6%) | |
| II–IV | 43 (66.2%) | 32 (60.4%) | |
| Lymph node metastasis | 0.5780 | ||
| N0 | 34 (52.3%) | 31 (58.5%) | |
| N+ | 31 (47.7%) | 22 (41.5%) | |
| Smoking | 0.4073 | ||
| Never smoker | 15 (23.1%) | 16 (30.2%) | |
| Smoker | 50 (76.9%) | 37 (69.8%) | |
| EGFR status | 0.2594 | ||
| Wild-type | 54 (83.1%) | 39 (73.6%) | |
| Mutation | 11 (16.9%) | 14 (26.4%) | |
| Pathological subtypes | 0.0514 | ||
| SCC | 37 (56.9%) | 40 (75.5%) | |
| Non-SCC | 28 (43.1%) | 13 (24.5%) | |
| Age | 0.0275 | ||
| ≤65 | 24 (36.9%) | 29 (54.7%) | |
| >65 | 41 (63.1%) | 24 (43.3%) | |
| Gender | 0.5096 | ||
| Male | 52 (80.0%) | 39 (73.6%) | |
| Female | 13 (20.0%) | 14 (26.4%) | |
N+, lymph node metastasis positive; SCC, squamous cell carcinoma.
Methylation-specific polymerase chain reaction analysis
DNA was prepared from tissue samples using standard methods. Bisulfite modification of genomic DNA was performed using the MethylCode Bisulfite Conversion Kit (Invitrogen, CA, USA). Briefly, 500 ng of genomic DNA was denatured by incubation with CT Conversion Reagent at 98°C for 10 min and 68°C for 2.5 h and at 4°C for several min. Modified DNA was purified by the Spin Column and then eluted with Elution Buffer.
The primer sequences for the DLEC1 gene for methylated (M) sequences were: forward, 5-GTTTCGTAGTTCGGTT TCGT C-3 and reverse, 5-CGAAATATCTTAAATACGCA ACG-3 (107 bp). The primer sequences for the DLEC1 gene for unmethylated (U) sequences were: forward, 5-TAGTTTT GTAGTTTGGTTTTGTT-3 and reverse, 5-ACAAAATATCT TAAATACACACAACA-3. The primer sequences for the CDH1 gene for methylated (M) sequences were: forward, 5-GGTGAATTTTTAGTTAATTAGCGGTAC-3 and reverse, 5-CATAACTAACCGAAAACGCCG-3. The primer sequences for the CDH1 gene for unmethylated (U) sequences were: forward, 5-GGTAGGTGAATTTTTAGTTAATTAGTGGTA-3 and reverse, 5-ACCCATAACTAACCAAAAACACCA-3. The primer sequences for the p16 gene for methylated (M) sequences were: forward, 5-TTATTAGAGGGTGGGGTG GATTGT-3 and reverse, 5-G ACCCCGAACCGCGACCG TAA-3. The primer sequences for the p16 gene for unmethylated (U) sequences were: forward, 5-TTATTAGAGGGT GGGGTGGATTGT-3 and reverse, 5-CAACCCCAAACC ACAACCATA-3. The cycling conditions were: initial denaturation at 94°C for 5 min, followed by 40 cycles at 94°C for 45 sec, 65°C (p16, M), 60°C (p16, U), 58°C (DLEC1, M), 57°C (CDH1) or 55°C (DLEC1, U) for 45 sec, and 72°C for 45 sec.
Statistical analysis
Statistical analyses were carried out using the Mann-Whitney U test for unpaired samples and the Wilcoxon signed-rank test for paired samples. Linear relationships between variables were determined by means of simple linear regression. Correlation coefficients were determined by rank correlation using Spearman’s and the χ2-test. Survival of the lung cancer patients was examined by the Kaplan-Meier method, and differences were examined by the log-rank test. Analyses were carried out using the Stat-View software package (Abacus Concepts Inc., Berkeley, CA, USA), and differences were considered significant at p<0.05.
Results
DLEC1 gene methylation status in Japanese lung cancer patients
Methylation-specific PCR showed that the DLEC1 promoter region was methylated in 65 out of 116 (79.8%) lung cancer types. The methylation status of DLEC1 was not associated with squamous histology (squamous cell carcinoma 29.2% vs. non-squamous cell carcinoma 37.3%; p=0.4269) and smoking status (never smoker 30.8% vs. smoker 21.6%; p=0.291). In addition, the DLEC1 methylation status did not correlate with gender (p=0.9999), age (p=0.3478), lymph node metastasis (p=0.5734) and pathological stages (I vs. II–IV; p=0.7028). DLEC1 methylation independently existed with EGFR mutations (p=0.1657).
The p16 promoter region was methylated in 91 out of 221 (78.8%) lung cancer types (Table II). The methylation status was correlated with squamous histology (p=0.03), smoking status (never smoker vs. smoker; p=0.0122) and EGFR wild-type (p=0.0071). However, the p16 methylation status did not correlate with survival (p=0.6215).
The CDH promoter region was methylated in 65 out of 118 (78.8%) lung cancer types (Table III). A higher frequency of methylation cases of the CDH1 promoter region was associated with younger patients (p=0.0491). However, the methylation status did not correlate with survival (p=0.8011).
DLEC1 gene methylation status and survival in lung cancer patients
Of the 65 DLEC1 methylated cases, 37 patients succumbed to the disease, while of the 51 unmethylated cases, 18 patientts succumbed to the disease. Thus, patients with DLEC1 methylated cancer were significantly associated with poor survival (log-rank test, p=0.0407) (Fig. 1). In the multivariate analysis, pathological stage [stage I vs. stage III–IV, p=0.0038, relative risk, 2.495 (1.342–4.636)] and DLEC1 methylation status were independent prognostic factors [p=0.0348, relative risk, 1.865 (1.046–3.362)].
Figure 1.
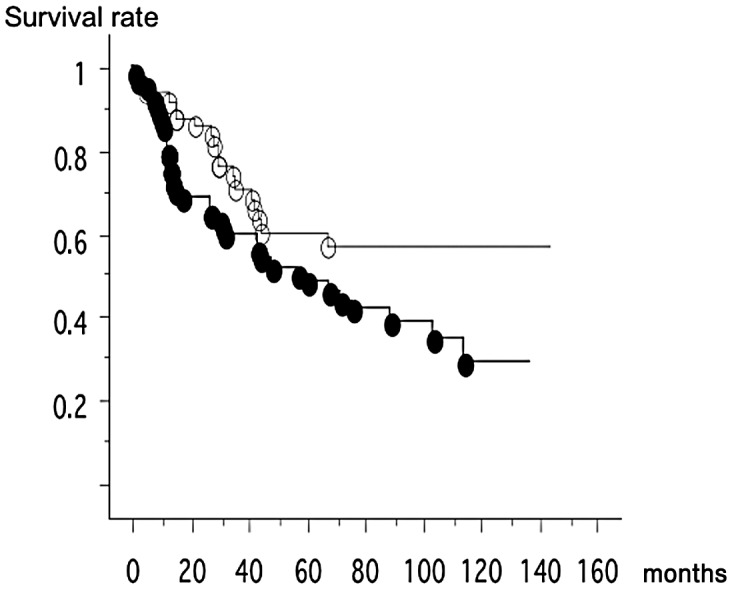
Of the 65 DLEC1 methylated cases, 37 patients succumbed to the disease (mean survival, 62.4 months). Of the 51 unmethylated cases, 18 patients succumbed to the disease (mean survival, 51.1 months). Thus, patients with DLEC1 methylated cancer were significantly associated with poor survival (log-rank test, p=0.0407). Black ovals, unmethylated cases; white ovals, methylated cases.
Discussion
We demonstrated that the DLEC1 gene was hypermethylated in Japanese lung cancer patients. We did not find any correlations between methylation status and gender, pathological stage and smoking status in Japanese NSCLC patients. However, the DLEC1 methylation status was correlated with survival in Japanese lung cancer patients.
DLEC1 is a candidate tumor-suppressor gene found in multiple cancer types (3–5,7,15,16). DLEC1 suppresses tumor growth or reduces the invasiveness of cancer cells. In ovarian cancer cell lines, the number of colonies formed in DLEC1 transfectants was significantly lower than that in mock transfectants (8), which showed that DLEC1 suppressed the growth of ovarian cancer cells and/or inactivation of CDC2 kinase, thereby blocking cells at the G2/M phase and preventing tumor development in nude mice (4,5). The demethylating agent 5-aza caused the loss of mRNA expression in lung cancer cell lines (16). DLEC1 methylation is cancer-specific, as it was only rarely detected in matched normal lung tissue (5). As there is no antibody available for DLEC1, we were unable to determine whether methylated tumors show a loss or reduced DLEC1 protein expression. However, a loss of DLEC1 RNA expression was previously shown in 8 out of 30 primary lung cancer types, although this loss was not due to gene mutations (6). No correlation between DELC1 methylation and clinical parameters of gastric cancer was found (17), similar to our investigation.
It was demonstrated that p16 methylation occurs more frequently in squamous cell carcinoma (16,18). These previous results are consistent with our present findings. Several studies have demonstrated a significant association between DNA methylation and tobacco smoking (20,21). Methylation of the p16INK4A gene was induced in rats treated with tobacco-specific NKK[4-N-methyl-N-nitrosamino-1-3-prydil 1-1-butanone (NNK), polyaromatic hydrocarbon] (20). Lung tumors that were induced in F344/N rats after exposure to cigarette smoke by inhalation displayed de novo methylation of p16INK4a (21).
The correlation between CDH1 gene methylation and survival is controversial. Although Nakata et al demonstrated a marginal correlation between CDH1 methylation and survival (p=0.0473) (16), Kim et al demonstrated that CDH1 methylation itself did not correlate with either survival or clinicopathological factors (22).
Preclinically, the majority of ovarian cancer cell lines siginificantly up-regulated DLEC1 transcripts after histone deacetylase (HDAC) inhibitor treatment (8). Moreover, exposure to the HDAC inhibitor PXD101 (belinostat) had varying effects on hepatocellular carcinoma cell lines (23). Thus, several HDAC inhibitors were found to exhibit antiproliferative activity and induce apoptosis in human cancer cells (24). Moreover, the restoration of tumor-suppressor genes, such as DLEC1, by HDAC inhibitors may contribute to antitumor effects.
Acknowledgements
The authors would like to thank Mrs. Tomomi Shibata for the excellent technical assistance. This work was supported by the Grand-in-Aid for Research, Nagoya City University (2006) and Grants-in-Aid for Scientific Research, Japan Society for the Promotion of Science (JSPS) (nos. 21591820 and 21390394).
References
- 1.Hung J, Kishimoto Y, Sugio K, et al. Allele-specific chromosome 3p deletions occur at an early stage in the pathogenesis of lung carcinoma. JAMA. 1995;273:558–563. [PubMed] [Google Scholar]
- 2.Wistuba II, Behrens C, Virmani AK, et al. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res. 2000;60:1949–1960. [PubMed] [Google Scholar]
- 3.Zaborovsky ER, Lerman MI, Minna JD. Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene. 2002;21:6915–6935. doi: 10.1038/sj.onc.1205835. [DOI] [PubMed] [Google Scholar]
- 4.Qiu GH, Salto-Tellez M, Ross JA, et al. The tumor suppressor gene DLEC1 is frequently silenced by DNA methylation in hepatocellular carcinoma and induces G1 arrest in cell cycle. J Hepatol. 2008;48:433–441. doi: 10.1016/j.jhep.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 5.Seng TJ, Currey N, Cooper WA, et al. DLEC1 and MLH1 promoter methylation are associated with poor prognosis in non-small cell lung carcinoma. Br J Cancer. 2008;99:375–382. doi: 10.1038/sj.bjc.6604452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daigo Y, Nishiwaki T, Kawasoe T, Tamari M, Tsuchiya E, Nakamura Y. Molecular cloning of a candidate tumor suppressor gene, DLC1, from chromosome 3p. 213. Cancer Res. 1999;59:1966–1972. [PubMed] [Google Scholar]
- 7.Kwong J, Chow LS, Wong WK, et al. Epigenetic inactivation of deleted in lung and esophageal cancer 1 gene in nasopharyngeal carcinoma. Genes Chromosomes Cancer. 2007;46:171–180. doi: 10.1002/gcc.20398. [DOI] [PubMed] [Google Scholar]
- 8.Kwong J, Lee JY, Wong KK, et al. Candidate tumor-suppressor gene DLEC1 is frequently downregulated by promoter hypermethylation and histone hypoacetylation in human epithelial ovarian cancer. Neoplasia. 2006;8:268–278. doi: 10.1593/neo.05502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang GH, Lee S, Cho NY, et al. DNA methylation profiles of gastric carcinoma characterized by quantitative DNA methylation analysis. Lab Invest. 2008;88:161–170. doi: 10.1038/labinvest.3700707. [DOI] [PubMed] [Google Scholar]
- 10.Japan Lung Cancer Society. General Rule for Clinical and Pathological Record of Lung Cancer. Jpn Lung Cancer Soc. (5th edition) 1999;5:1–177. [Google Scholar]
- 11.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 12.Endo K, Konishi A, Sasaki H, et al. Epidermal growth factor receptor gene mutation in non-small cell lung cancer using highly sensitive and fast TaqMan PCR assay. Lung Cancer. 2005;50:375–384. doi: 10.1016/j.lungcan.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki H, Shimizu S, Endo K, et al. EGFR and erbB2 mutation status in Japanese lung cancer patients. Int J Cancer. 2006;118:180–184. doi: 10.1002/ijc.21301. [DOI] [PubMed] [Google Scholar]
- 14.Sasaki H, Endo K, Konishi A, et al. EGFR mutation status in Japanese lung cancer patients: genotyping analysis using LighyCycler. Clin Cancer Res. 2005;11:2924–2929. doi: 10.1158/1078-0432.CCR-04-1904. [DOI] [PubMed] [Google Scholar]
- 15.Smith IM, Mithani SK, Liu C, et al. Novel integrative methods for gene discovery associated with head and neck squamous cell carcinoma development. Arch Otolaryngol Head Neck Surg. 2009;135:487–495. doi: 10.1001/archoto.2009.43. [DOI] [PubMed] [Google Scholar]
- 16.Nakata S, Sugio K, Uramoto H, et al. The methylation status and protein expression of CDH1, p16INK4A, and fragile histidine triad in nonsmall cell lung carcinoma: epigenetic silencing, clinical features, and prognostic significance. Cancer. 2006;106:2190–2199. doi: 10.1002/cncr.21870. [DOI] [PubMed] [Google Scholar]
- 17.Ying J, Poon FF, Yu J, et al. DLEC1 is a functional 3p22.3 tumor suppressor silenced by promoter CpG methylation in colon and gastric cancers. Br J Cancer. 2009;100:663–669. doi: 10.1038/sj.bjc.6604888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarmalaite S, Kannio A, Anttila S, et al. Aberrant p16 promoter methylation in smokers and former smokers with non-small cell lung cancer. Int J Cancer. 2003;106:913–918. doi: 10.1002/ijc.11322. [DOI] [PubMed] [Google Scholar]
- 19.Kim DH, Nelson HH, Wiencke JK, et al. p16(INK4a) and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res. 2001;61:3419–3424. [PubMed] [Google Scholar]
- 20.Rom WN, Jay JG, Lee TC, Jiang Y, Tchou-Wong KM. Molecular and genetic aspects of lung cancer. Am J Respir Crit Care Med. 2000;161:1355–1367. doi: 10.1164/ajrccm.161.4.9908012. [DOI] [PubMed] [Google Scholar]
- 21.Swafford DS, Middleton SK, Palmisano WA, et al. Frequent aberrant methylation of p16INK4a in primary rat lung tumors. Mol Cell Biol. 1997;17:1366–1374. doi: 10.1128/mcb.17.3.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim DS, Kim MJ, Lee JY, et al. Aberrant methylation of E-cadherin and H-cadherin genes in non-small cell lung cancer and its relation to clinicopathologic features. Cancer. 2007;110:2785–2792. doi: 10.1002/cncr.23113. [DOI] [PubMed] [Google Scholar]
- 23.Ma BB, Sung F, Tao Q, et al. The preclinical activity of the histone deacetylase inhibitor PXD101 (belinostat) in hepatocellular carcinoma cell lines. Invest New Drugs. 2009 Jan 27; doi: 10.1007/s10637-009-9219-7. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 24.Takai N, Kawamata N, Gui D, Said JW, Miyakawa I, Koeffler HP. Human ovarian carcinoma cells: histone deacetylase inhibitors exhibit antiproliferative activity and potently induce apoptosis. Cancer. 2004;15:2760–2770. doi: 10.1002/cncr.20709. [DOI] [PubMed] [Google Scholar]