

Background: eIF2 is a critical point of stress-induced regulation of translation in eukaryotic cells.
Results: eIF2 signaling is activated by bacterial pathogens and regulates two key infection-associated processes.
Conclusion: Regulation of translation in eukaryotic cells is involved in innate immune responses.
Significance: These findings enlarge the possible targets for therapeutic interventions against bacterial pathogens.
Keywords: Bacterial Pathogenesis, Cytokine, Invasion, Stress Response, Virulence Factors
Abstract
In eukaryotic cells, there are two well characterized pathways that regulate translation initiation in response to stress, and each have been shown to be targeted by various viruses. We recently showed in a yeast-based model that the bacterial virulence factor YopJ disrupts one of these pathways, which is centered on the α-subunit of the translation factor eIF2. Here, we show in mammalian cells that induction of the eIF2 signaling pathway occurs following infection with bacterial pathogens and that, consistent with our yeast-based findings, YopJ reduces eIF2 signaling in response to endoplasmic reticulum stress, heavy metal toxicity, dsRNA, and bacterial infection. We demonstrate that the well documented activities of YopJ, inhibition of NF-κB activation and proinflammatory cytokine expression, are both dependent on an intact eIF2 signaling pathway. Unexpectedly, we found that cells with defective eIF2 signaling were more susceptible to bacterial invasion. This was true for pathogenic Yersinia, a facultative intracellular pathogen, as well as for the intracellular pathogens Listeria monocytogenes and Chlamydia trachomatis. Collectively, our data indicate that the highly conserved eIF2 signaling pathway, which is vitally important for antiviral responses, plays a variety of heretofore unrecognized roles in antibacterial responses.
Introduction
Various types of cellular stress result in a transient reduction in general protein synthesis. In eukaryotic cells, one component of this translational inhibitory response is centered on the eIF2 initiation factor, which, together with the initiator methionine tRNA and GTP, forms the ternary complex. Phosphorylation of the α-subunit of eIF2 at Ser-51 by any one of the four so-called eIF2α kinases (GCN2, PERK, protein kinase R (PKR),2 and HRI) negatively affects the GDP-GTP exchange activity of the β-subunit of eIF2, thus reducing the cellular levels of active ternary complexes. Upon the stress-induced activation of the eIF2α kinases and the subsequent phosphorylation of eIF2α, specific mRNAs are selectively translated (by virtue of their 5′-UTRs), and their encoded proteins contribute to cellular recovery (1). The best characterized of these mRNAs is that encoding the basic leucine zipper transcription factor ATF4 (Gcn4 in budding yeast), which, upon its synthesis, translocates to the nucleus, where it transcriptionally activates the expression of a number of stress-adaptive genes, including those encoding ATF3 and CHOP.
Viral infection is one such type of cellular stress that activates eIF2-mediated translation control. The eIF2α kinase PKR becomes activated by virus-derived dsRNA, which has the effect of reducing general protein synthesis and consequently reducing viral replication. Indicative of its importance in antiviral cellular responses, several viruses have independently evolved the means to obstruct the activation of PKR (2). PKR has been shown to be required for LPS-induced activation of STAT1 and the induction of apoptosis in bacterially infected cells (3), suggesting that antiviral and antibacterial response pathways may overlap. There have also been various reports indicating that eIF2 signaling may be active during bacterial infections, the majority of which consist of uncharacterized “hits” from microarray-based screens (4–6). Recently, it has been reported that there is enhanced expression of several endoplasmic reticulum stress proteins (associated with PERK) in lung tissue of mice infected with Mycobacterium tuberculosis (7). It has also been shown that LPS- and Toll-like receptor-mediated signaling is functionally intertwined with HRI- and PERK-mediated stress responses, further suggesting that eIF2 signaling may be involved in antibacterial responses (8–10).
By performing a yeast-based mutagenesis screen for eukaryotic factors that are responsive to the bacterial virulence factor YpkA (Yersinia protein kinase A) (11), we discovered that eIF2 signaling is specifically activated by the kinase activity of YpkA and that this response reduces the cellular toxicity of YpkA (12). Unexpectedly, we found that the cellular activity of an additional Yersinia virulence factor, YopJ, which is encoded on the same transcript as YpkA, is entirely dependent on a properly functioning eIF2 signaling pathway (12). YopJ has no apparent phenotype in unstressed yeast cells. However, YopJ-expressing yeast cells are extremely sensitive to various types of stresses (e.g. osmotic, oxidative, and nutritional). Interestingly, YopJ also altered the eIF2-mediated cellular stress response induced by the kinase activity of YpkA.
Although our yeast-based findings indicate that the genetic interactions between YopJ and eIF2 signaling can shape the stress response, it was not immediately obvious how these interactions would affect the infection process. Here, we examined eIF2 responses in mammalian cells following infection with bacterial pathogens and determined whether those responses are sensitive to bacterially encoded virulence factors. We describe two separate findings that were not foreseen from our prior yeast-based studies: that eIF2 signaling is linked to infection-induced expression of cytokines as well as to bacterial invasion.
EXPERIMENTAL PROCEDURES
Infection-based Experiments
Macrophage-like RAW 264.7 cells (or wild-type and eIF2α(S51A) mouse embryonic fibroblasts (MEFs) (13)) were seeded in 24-well plates at 2 × 105 cells/well and infected the next day. For the Yersinia infections, the Yersinia pseudotuberculosis wild-type (YPIII/pIB102) (14) and ΔyopJ (YPIII/pIB232) (15) strains were grown in tissue culture medium, diluted to A595 = 0.1, and propagated for 2 h at 26 °C and then for 1 h at 37 °C. The bacterial strains were further diluted to achieve the desired titer and added to cultured cells.
For the translocation assay, a plasmid encoding a hybrid protein consisting of YopE (residues 1–130) and a 40-residue Elk tag (16) was transformed into a multiple yop mutant strain of Y. pseudotuberculosis (17); the resulting strain was then used to infect wild-type and eIF2α(S51A) MEFs. After a 2-h infection period, cells were lysed, and the resulting lysates were analyzed using anti-phospho-Elk (Ser-383) and anti-Elk antibodies (Cell Signaling) to detect phosphorylated and total Elk, respectively.
For gene expression assays, at the indicated times, total RNA was extracted using a ZR RNA MiniPrep kit (Zymo Research), and cDNA was prepared using an Omniscript RT kit (Qiagen). Real-time quantitative PCR (qPCR) was performed on a ABI PRISM 7700 sequence detection system (Applied Biosystems) using TaqMan probes (Applied Biosystems). Signals of specific mRNAs were normalized with mouse GAPDH as the housekeeping gene, and the relative -fold changes were determined by calculating 2−ΔΔCt.
For the Yersinia internalization assay, infection was initiated as described in the figure legends, and the fraction of internalized bacteria was determined by calculating the ratio of the number of bacteria recovered from the gentamycin-containing wells at 1 h to the number of bacteria recovered from the untreated wells. Each data point is the average of three independent wells, and the results are representative of three separate experiments.
Listeria monocytogenes was obtained from American Type Culture Collection (DMX 09-082) and grown to stationary phase in brain-heart infusion broth (BD Biosciences) at 37 °C. Bacteria were diluted in tissue culture medium and added to cells. The infection proceeded as described in the figure legends and was terminated by removing the medium from the wells and lysing the cells with water, and the resulting lysates were plated on standard LB medium to determine their titers. The Chlamydia trachomatis infection assays (18) were performed as described in the figure legends.
Transfection-based Experiments
105 HEK 293T cells were seeded in 24-well plates and transfected the next day. Transfection mixtures contained Lipofectamine 2000 (Invitrogen), the pTK-ATF4-Luc reporter (provided by David Ron, University of Cambridge, Cambridge, United Kingdom), pRL-TK (encoding Renilla luciferase, which serves as an transfection control), and YopJ-encoding sequences cloned into Retro-X Tet-Off (Clontech). YopJ expression in transfected cells was verified by qPCR using YopJ-specific primers as described below. No signals were obtained in non-reverse transcriptase controls, indicating that YopJ-specific cDNAs were derived from mRNAs. In some experiments, the transfected cells were treated the next day with either 10 ng/ml thapsigargin (Calbiochem) or arsenic trioxide (Sigma) at the concentrations indicated in the figure legends, and 4 h later, the cells were lysed, and luciferase activity was measured using the Dual-Luciferase reporter system assay (Promega). Firefly luciferase signals were normalized against the Renilla-derived signals. PKR+/+ and PKR−/− MEFs (gift of J. Durbin) were transfected with the IFN-β-Luc reporter (19), pRL-TK, and the YopJ-encoding expression plasmid. The following day, cells were supertransfected with 6 μg of poly(I·C) (GE Healthcare) in Lipofectamine 2000, and 6 h later, cells were lysed, and luciferase levels were determined as described above. Salubrinal, the NF-κB-Luc reporter plasmid, and recombinant TNFα were purchased from Tocris Bioscience, Stratagene, and R&D Systems, respectively.
RESULTS
eIF2 Signaling in Yersinia-infected Cells
To determine whether eIF2 signaling is activated following exposure to a bacterial pathogen, the levels of phosphorylated eIF2α were evaluated in cultured murine macrophage-like cells infected with the bacterial pathogen Y. pseudotuberculosis. There was a notable increase in eIF2α phosphorylated at Ser-51 in cells following a 60-min infection compared with uninfected cells that was comparable with that observed in cells exposed to arsenite (Fig. 1A). To further evaluate eIF2 signaling in infected cells, we analyzed the transcript levels of endogenous Atf3, which is an immediate downstream target gene of ATF4 and a commonly used readout for eIF2 signaling in mammalian cells (20). In human HEK 293 cells infected with Yersinia, the kinetics of inductive ATF3 expression were comparable with those in cells treated with thapsigargin (see Fig. 2B below) (data not shown). In murine macrophage-like cells, inductive Atf3 expression increased over a 4-h infection period in contrast to TNFα expression, which reached a plateau after 30–60 min of infection (Fig. 1B).
FIGURE 1.
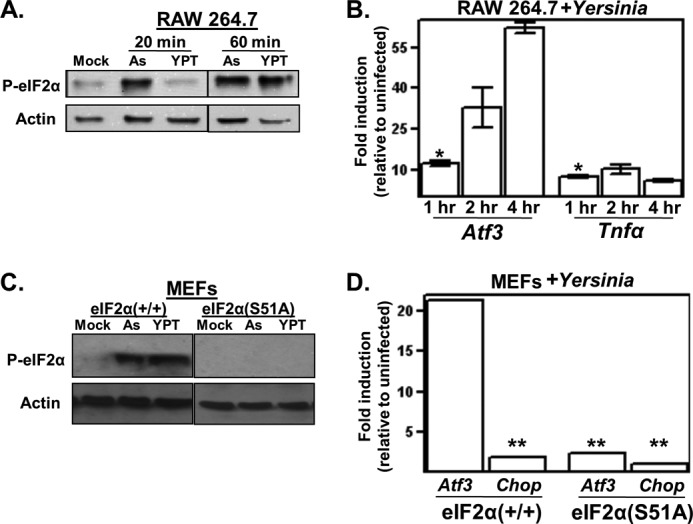
Bacterial pathogen Yersinia activates eIF2 signaling. A, macrophage-like RAW 264.7 cells were left untreated (Mock) or either exposed to 10 μm sodium arsenite (As) or infected with Y. pseudotuberculosis (YPT; multiplicity of infection (m.o.i.) = 50) for 20 or 60 min. The resulting whole cell lysates were analyzed by Western blotting using antibodies specific for eIF2α phosphorylated at Ser-51 (P-eIF2α) and actin. For presentation purposes, the lanes of a single blot were rearranged. B, RAW 264.7 cells were infected with Y. pseudotuberculosis, and at the indicated times, cells were collected, and Atf3 and TNFα transcript levels were determined by qPCR. Shown are the average GAPDH-normalized signals of three separate wells relative to the uninfected controls. C, wild-type and eIF2α(S51A) MEFs were left untreated or infected with Y. pseudotuberculosis at m.o.i. = 10 for 30 min and analyzed for eIF2α phosphorylation. For presentation purposes, the lanes of a single blot were rearranged. D, wild-type and eIF2α(S51A) MEFs were infected with Y. pseudotuberculosis at m.o.i. = 5 for 3 h, followed by analysis of Atf3 and Chop transcript levels by qPCR. All results shown are representative of at least three independent experiments. *, p < 0.05 between the indicated groups and transcript levels in uninfected cells; **, p > 0.05 between the indicated groups and uninfected cells using two-tailed Student's t test.
FIGURE 2.
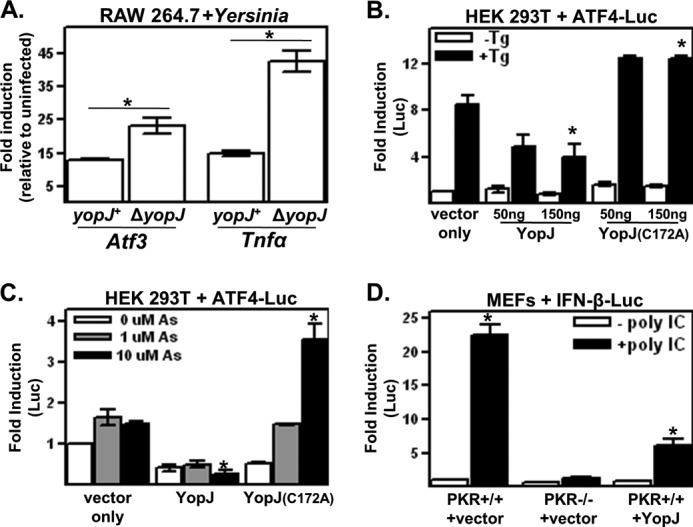
YopJ limits eIF2α signaling in mammalian cells. A, macrophage-like RAW 264.7 cells were infected with the wild-type (yopJ+) or yopJ deletion mutant (ΔyopJ) strain of Y. pseudotuberculosis for 1 h, and Atf3 and TNFα transcript levels were determined as described for Fig. 1B. B and C, HEK 293T cells were cotransfected with the YopJ expression and ATF4-Luc reporter plasmids and, 24 h later, either left untreated or treated with 10 μg/ml thapsigargin (Tg) for 4 h (B) or with the indicated concentrations of sodium arsenite (As) for 3 h (C). Reporter protein signals were normalized to a transfection control and are expressed relative to the untreated vector-only controls. D, wild-type or PKR-deficient MEFs were cotransfected with IFN-β-Luc and either empty vector or YopJ-encoding plasmids. Transfection mixtures were either supplemented or not with 10 ng/ml poly(I·C) (poly IC). Cells were harvested 18 h later, and reporter protein signals are expressed relative to the untreated PKR+/+ vector sample. All results shown are representative of at least three independent experiments. *, p < 0.05 between the indicated groups.
To determine whether infection-induced Atf3 expression is in fact dependent on the phosphorylation of eIF2α, we used MEF cells expressing a non-phosphorylatable mutant, eIF2α(S51A). Like RAW 264.7 cells, wild-type MEFs exhibited both higher levels of eIF2α phosphorylation and Atf3 transcript levels in response to infection with Yersinia; this inductive expression did not occur in eIF2α(S51A) MEFs (Fig. 1, C and D). Interestingly, whereas the ATF4 target gene Chop was significantly induced in response to thapsigargin (data not shown), it was not induced in Yersinia-infected cells (Fig. 1D). The relative preferential expression of Atf3 compared with Chop was also observed in infected murine macrophage-like cells, as well as peritoneal and bone marrow-derived macrophages (data not shown). Infection-induced expression of Atf3 (but not Chop) has been noted in previous microarray-based reports of Yersinia- and Staphylococcus-infected epithelial cells (5, 6). Collectively, these data indicate that eIF2 signaling leading to Atf3 expression occurs in response to bacterial infection.
Bacterial Virulence Factor YopJ Impacts eIF2 Signaling
In yeast cells, the activity of the Yersinia-encoded virulence factor YopJ is dependent on an intact eIF2 signaling pathway (12). To determine whether YopJ affects eIF2 signaling in mammalian cells, Atf3 expression was assayed in cells infected with either the yopJ+ or ΔyopJ strain of Yersinia. Atf3 expression was greater in cells infected with the ΔyopJ strain compared with the yopJ+ strain (Fig. 2A), showing that YopJ, when delivered into cells by the bacterial type 3 secretion system, negatively affects eIF2 signaling. This inhibitory activity of YopJ toward eIF2 signaling (Fig. 2A) is reminiscent of its previously reported negative effect on the expression of proinflammatory cytokines such as TNFα (21, 22).
To determine whether YopJ affects other previously characterized inducers of eIF2 signaling, cells were cotransfected with YopJ-encoding expression plasmids and a reporter plasmid that constitutively expresses a transcript consisting of the ATF4 5′-UTR control region upstream of sequences encoding firefly luciferase (23). As mentioned in the Introduction, stress-induced activation of eIF2 signaling results in the preferential translation of the ATF4-Luc transcript. HEK 293T cells expressed transfected wild-type YopJ and an inactive variant, YopJ(C172A), at comparable levels (data not shown). Treatment of cells with either thapsigargin or arsenite induce ATF4 expression by activating the eIF2α kinases PERK and HRI, respectively (Fig. 2, B and C) (24, 25). Wild-type YopJ reduced both thapsigargin- and arsenite-induced ATF4 5′-UTR activity in contrast to YopJ(C172A) (Fig. 2, B and C). For unknown reasons, YopJ(C172A) expression caused heighten thapsigargin- and arsenite-induced expression levels of ATF4-Luc. The eIF2α kinase PKR is activated by dsRNA, which results in the enhanced expression of IFN-β (26). Similar to results reported previously by Sweet et al. (27), expression of YopJ greatly reduced the dsRNA-induced expression of an IFN-β-Luc reporter protein (Fig. 2D). The dsRNA-induced activation of IFN-β promoter activity in YopJ-expressing cells, in terms of -fold increase, was similar to that observed in cells lacking PKR. Collectively, these data show that, in addition to infection, YopJ negatively affects stress-induced eIF2 signaling mediated by three of the four mammalian eIF2α kinases.
Infection-induced TNFα Expression Is Regulated by eIF2 Signaling
The inhibitory effect of YopJ on both eIF2 signaling and TNFα expression motivated us to test whether induction of TNFα expression requires an intact eIF2 signaling pathway. TNFα transcript levels were measured in infected wild-type and eIF2α(S51A) MEFs. Similar to macrophages (Fig. 2A), TNFα expression in wild-type MEFs was induced following infection with the yopJ+ strain of Yersinia, and this inductive expression was markedly enhanced in wild-type MEFs infected with the ΔyopJ strain (Fig. 3A). In striking contrast, in eIF2α(S51A) MEFs, there was no increase in TNFα transcript levels following infection (Fig. 3A), indicating that eIF2 signaling regulates the infection-induced expression of this cytokine.
FIGURE 3.
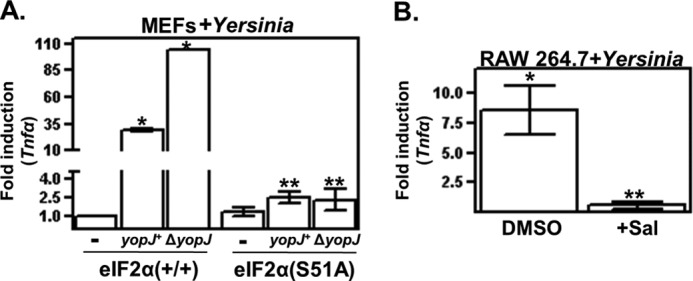
YopJ limits eIF2-dependent expression of TNFα in infected cells. A, wild-type and eIF2α(S51A) MEFs were infected with the indicated strains of Y. pseudotuberculosis for 2 h, and TNFα transcript levels were then determined as described for Fig. 1B. Shown are the average GAPDH-normalized signals of three separate wells relative to the matched uninfected controls. *, p < 0.05 between the indicated groups; **, p > 0.05 between the indicated groups. B, RAW 264.7 cells were treated or not with salubrinal (Sal) for 18 h and then infected with Y. pseudotuberculosis. After 2 h, cells were collected, and TNFα transcript levels were determined. Shown are the average GAPDH-normalized signals of three separate wells relative to the uninfected controls. All results shown are representative of at least three independent experiments. *, p < 0.05 between the indicated group and matched uninfected; **, p > 0.05 between the indicated group and matched uninfected.
We also tested whether pharmacological disruption of eIF2 signaling affects infection-specific TNFα expression by treating cells with the small molecule salubrinal, which specifically inhibits the regulatory subunit PPP1r15ab/GADD34 of the eIF2α phosphatase complex PP1c and results in enhanced levels of phosphorylated eIF2α (28). Salubrinal treatment of culture macrophages resulted in a severalfold reduction in infection-induced expression of TNFα (Fig. 3B). There were no detectable differences in cell viability between untreated and salubrinal-treated cells (data not shown). Salubrinal has similarly been shown to abrogate β-amyloid- and free fatty acid-induced eIF2 signaling (29, 30), although it is not currently known how salubrinal-induced enhancement of phosphorylated eIF2α levels negatively affects eIF2 signaling. However, considered together with the results derived from the eIF2α(S51A)-expressing cells, these data indicate that perturbing eIF2α-mediated signaling negatively affects infection-induced cytokine expression.
A number of experiments were performed to corroborate the unexpected linkage of infection-induced eIF2 signaling and cytokine expression. Because inductive cytokine expression is regulated by NF-κB, we determined whether NF-κB activation is sensitive to genetic or pharmacological disruption of eIF2 signaling. Wild-type and eIF2α(S51A) MEFs were transfected with a NF-κB-Luc reporter plasmid and subsequently stimulated with recombinant TNFα (rTNFα), a potent activator of NF-κB. In striking contrast to wild-type MEFs, rTNFα-induced NF-κB activation was completely abrogated in eIF2α(S51A) MEFs (Fig. 4A).
FIGURE 4.
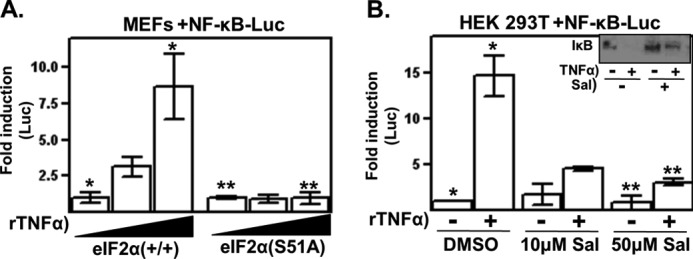
Genetic and pharmacological disruption of eIF2 signaling inhibits NF-κB activation. A, wild-type and eIF2α(S51A) MEF cells were transfected with a NF-κB-Luc reporter plasmid and then treated with 0, 1, and 10 ng/ml rTNFα for 5 h, at which time, reporter gene expression was determined. B, cells were cotransfected with a NF-κB-Luc reporter plasmid and treated 6 h later with either vehicle or the indicated doses of salubrinal (Sal) for 18 h. Cells were then treated or not with rTNFα. Cells were harvested 3 h later, and reporter gene expression was determined and normalized to the untreated sample. Inset, HEK 293T cells were treated with salubrinal at the indicated concentrations for 18 h and then pulsed with rTNFα for 10 min. Cell lysates were prepared, and IκB protein levels determined by Western analysis. All results shown are representative of at least three independent experiments. *, p < 0.05 between the indicated groups; **, p > 0.05 between the indicated groups.
There was also a reduction in rTNFα-induced NF-κB activation in salubrinal-treated cells; this effect was specific for the NF-κB-regulated reporter gene and was not observed in the transfection control gene (Fig. 4B). Salubrinal treatment did not notably affect the basal expression levels of the NF-κB reporter gene. Additionally, there was a reduction in the rTNFα-induced degradation of endogenous IκB in salubrinal-treated cells (Fig. 4B, inset). These data are consistent with the reports cited above that similarly showed that salubrinal negatively affects β-amyloid- and free fatty acid-induced activation of NF-κB (29, 30). Although the mechanistic basis of how eIF2 signaling is linked to NF-κB is currently not known, the data shown here raise the possibility that the inhibitory effect of YopJ on the NF-κB activation and subsequent cytokine expression that occur during a Yersinia infection (22) is regulated by eIF2.
eIF2 Signaling Opposes Yersinia Invasion
We noted that, upon prolonged infection with Yersinia (∼4 h), the eIF2α(S51A) MEFs rounded up and detached from the substratum compared with the wild-type MEFs, which appeared relatively much less affected under similar infection conditions. Yersinia-directed cytotoxicity is primarily a result of the translocation of YopE into the host cell cytoplasm (31). A translocation assay (16) was employed to determine whether the enhanced cytotoxicity observed in the infected eIF2α(S51A) MEFs was due to higher levels of intracellular YopE. Using this assay, there was no detectable differences in the intracellular levels of YopE between wild-type and eIF2α(S51A) MEFs (Fig. 5A).
FIGURE 5.
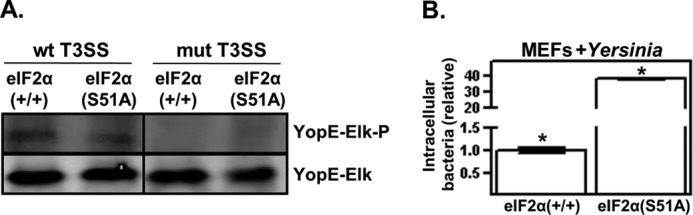
eIF2 signaling counteracts uptake of Y. pseudotuberculosis. A, wild-type and eIF2α(S51A) MEFs were infected with Y. pseudotuberculosis strains possessing either an intact or defective (mutant (mut)) type 3 secretion system (T3SS), each harboring a plasmid encoding a YopE-Elk translocation reporter. After 1 h of infection, the resulting whole cell lysates were examined by Western analysis using antibodies specific for the Elk epitope tag in either its phosphorylated (Elk-P) or non-phosphorylated form. Because YopE-Elk becomes phosphorylated exclusively within the eukaryotic cytosol, the phosphorylation status of the Elk epitope is readout for type 3 secretion system-mediated translocation). For presentation purposes, the lanes of a single blot were rearranged. B, three independent wells of wild-type and eIF2α(S51A) MEF cells were infected with Y. pseudotuberculosis at m.o.i. = 10, and after a 45-min infection period, excess bacteria were removed. Cells were then left untreated, or gentamycin was added to the wells to kill extracellular bacteria. The number of internalized bacteria was determined as described under “Experimental Procedures” and is shown relative to the internalization levels of the wild-type MEFs after 1 h. The results shown are representative of at least three independent experiments. *, p < 0.05 between the two groups.
To determine whether the enhanced toxicity of the eIF2α(S51A) MEFs to Yersinia infection could be due to differences in bacterial invasion, the number of internalized bacteria was compared in wild-type and eIF2α(S51A) MEFs after a brief infection period. Using a standard bacterial invasion assay (32), the number of internalized bacteria recovered from eIF2α(S51A) MEFs was much greater (>25-fold) than the number of internalized bacteria recovered from wild-type MEFs (Fig. 5B). Because we could detect no differences in bacterial adhesion between wild-type and eIF2α(S51A) MEFs (data not shown), these data suggest that the internalization of Yersinia is regulated by eIF2 signaling.
eIF2 Signaling Opposes Invasion by Professional Intracellular Pathogens
The bacterial pathogens L. monocytogenes and C. trachomatis actively invade and proliferate within eukaryotic cells. With similar kinetics as in Yersinia-infected cells, infection of murine macrophage-like cells with L. monocytogenes induced phosphorylation of eIF2α (Fig. 6A) and expression of Atf3 (Fig. 6B). These findings are consistent with those recently reported by Pillich et al. (33), who showed that Listeria infection is associated with enhanced levels of phosphorylated eIF2α. L. monocytogenes also induced phosphorylation of eIF2α in wild-type MEFs with similar kinetics as in macrophages (data not shown). In cell invasion assays, the number of intracellular L. monocytogenes cells recovered from eIF2α(S51A) MEFs was severalfold greater than that recovered from wild-type MEFs (Fig. 6C). There was a similar level of enhanced invasion of eIF2α(S51A) MEFs by C. trachomatis (Fig. 6, D and E). Interestingly, although the number of C. trachomatis-containing inclusion-forming units was significantly greater in eIF2α(S51A) MEFs compared with wild-type MEFs (4.9-fold at 4 °C and 7.5-fold at 37 °C), the inclusions themselves were visibly comparable (i.e. contained similar numbers of bacterial cells) between the two cell types (Fig. 6D). Additionally, the progeny recovered for both MEF lines were equally infectious as determined by standard titering on HeLa cells (data not shown), indicating that a functioning eIF2 signaling pathway is not required for the intracellular development of C. trachomatis. Collectively, these data show that there is a host cell mechanism involving eIF2 signaling that plays a role in opposing invasion by bacterial pathogens.
FIGURE 6.

eIF2 signaling is activated and opposes invasion by intracellular bacterial pathogens. A, RAW 264.7 cells were left untreated (Mock), exposed to thapsigargin (Tg) or sodium arsenite (As), or infected with L. monocytogenes (Lm) for 30 min at m.o.i. = 50. The resulting whole cell lysates were examined by Western analysis using antibodies specific for eIF2α phosphorylated at Ser-51 (P-eIF2α) and actin. B, RAW 264.7 cells were infected with L. monocytogenes at m.o.i. = 50, and at the indicated times, cells were collected, and Atf3 and TNFα transcript levels were determined by qPCR. Shown are the average GAPDH-normalized signals of three separate wells relative to the uninfected controls. *, p < 0.05 between the indicated groups and transcript levels in uninfected cells. C, wild-type and eIF2α(S51A) MEFs were infected with L. monocytogenes, and after a 30-min attachment period, gentamycin was added to the wells to kill extracellular bacteria. The number of internalized bacteria was then determined 30 min later and is shown relative to the internalization levels of the wild-type MEFs. Each data point represents the average of three independent wells and is representative of three separate experiments (p < 0.05 between the two indicated groups). D, wild-type and eIF2α(S51A) MEFs were seeded on coverslips and infected with C. trachomatis L2 at m.o.i. = 1 for either 1.5 h (4 °C) or 1 h (37 °C). Excess bacteria were then removed, and after 24 h of infection at 37 °C, cells were fixed and stained for C. trachomatis, and the total number of inclusion-forming units (shown in D) per coverslip was determined by direct microscopic counting and is plotted in E. All results shown are representative of at least three independent experiments. * and **, p < 0.05 between co-labeled groups.
DISCUSSION
Recently, we discovered that, in yeast cells, eIF2 signaling is specifically activated and regulated by the bacterial virulence factors YpkA and YopJ (12), suggesting that this pathway may participate in antibacterial responses. Here, we have shown that eIF2 signaling in mammalian cells is activated following bacterial infection and that this activation is negatively regulated by YopJ (Fig. 7). An unexpected finding supported by five independent experiments (Fig. 3, A and B, and Fig. 4, A, B, and inset) is that eIF2 signaling is required for the infection-specific activation of NF-κB and proinflammatory gene expression. These finding suggest that cytokines should be classified as “stress recovery” genes, analogous to other genes that are expressed in an eIF2α-dependent manner in response to oxidative, osmotic, thermal, and endoplasmic reticulum stress.
FIGURE 7.
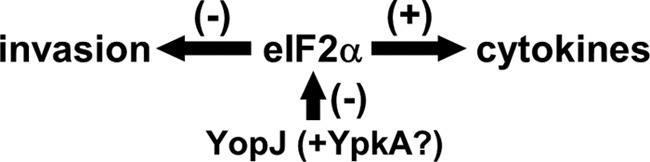
eIF2α functions during bacterial infection. Bacterial infection activates eIF2 signaling required for cytokine expression as well as opposing bacterial invasion. In cells infected with Yersinia, the virulence factor YopJ is translocated into the cytosol, where it inhibits eIF2-mediated functions. YopJ may collaborate with the coexpressed YpkA virulence factor (12).
The linking of cytokine expression to eIF2 signaling provides a clear rationale for why this signaling pathway would be targeted by a bacterially encoded virulence factor. It is currently thought that YopJ blocks NF-κB by directly modifying signaling proteins (e.g. IκB) that prevent its activation (34, 35). These latter studies were based on transfection expression systems that so far have not been extended to infection models. Because we have shown that YopJ inhibits eIF2 signaling and that infection-induced eIF2 signaling is required for NF-κB activation and proinflammatory gene expression, we propose that YopJ acts through eIF2 to repress proinflammatory signaling pathways.
Activation of eIF2 signaling leads to the expression of stress recovery genes, which, as far as we are aware, have not been linked to morphology-based activities. By comparing cells possessing intact versus defective eIF2 signaling, we made the unanticipated discovery that eIF2 signaling is linked to bacterial internalization. This property was observed in what is considered a facultative intracellular pathogen, Yersinia (Fig. 5), as well as two pathogens, Listeria and Chlamydia (Fig. 6), which actively invade cells as part of their infection cycle. To our knowledge, this is the first demonstration of a direct linkage between eIF2 signaling and cellular events at the morphological level. In the case of the Gram-negative Yersinia, the YpkA-mediated activation of eIF2 signaling (which is tempered by YopJ) may contribute to the well documented antiphagocytic activity of Yersinia (36, 37). With regard to the professional intracellular pathogens Listeria (Gram-positive) and Chlamydia (phylogenetically distinct, Gram-negative), invasion is clearly to the detriment of the host cell. The fact that there was a consistent pattern between these three very diverse pathogens indicates that eIF2 signaling is part of a general antibacterial defense system.
Acknowledgments
We thank Drs. Greg Plano, Sara Schesser Bartra, and Jane-Jane Chen for generous assistance.
This work was supported, in whole or in part, by United States Public Health Service Grant AI53459 (to K. S.) from NIAID.
- PKR
- protein kinase R
- MEF
- mouse embryonic fibroblast
- qPCR
- real-time quantitative PCR
- Luc
- luciferase
- rTNFα
- recombinant TNFα
- m.o.i.
- multiplicity of infection.
REFERENCES
- 1. Sonenberg N., Hinnebusch A. G. (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. García M. A., Meurs E. F., Esteban M. (2007) The dsRNA protein kinase PKR: virus and cell control. Biochimie 89, 799–811 [DOI] [PubMed] [Google Scholar]
- 3. Hsu L. C., Park J. M., Zhang K., Luo J. L., Maeda S., Kaufman R. J., Eckmann L., Guiney D. G., Karin M. (2004) The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature 428, 341–345 [DOI] [PubMed] [Google Scholar]
- 4. Subrahmanyam Y. V., Yamaga S., Prashar Y., Lee H. H., Hoe N. P., Kluger Y., Gerstein M., Goguen J. D., Newburger P. E., Weissman S. M. (2001) RNA expression patterns change dramatically in human neutrophils exposed to bacteria. Blood 97, 2457–2468 [DOI] [PubMed] [Google Scholar]
- 5. Bohn E., Müller S., Lauber J., Geffers R., Speer N., Spieth C., Krejci J., Manncke B., Buer J., Zell A., Autenrieth I. B. (2004) Gene expression patterns of epithelial cells modulated by pathogenicity factors of Yersinia enterocolitica. Cell. Microbiol. 6, 129–141 [DOI] [PubMed] [Google Scholar]
- 6. Li X., Fusco W. G., Seo K. S., Bayles K. W., Mosley E. E., McGuire M. A., Bohach G. A. (2009) Epithelial cell gene expression induced by intracellular Staphylococcus aureus. Int. J. Microbiol. 2009, 753278–753289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seimon T. A., Kim M. J., Blumenthal A., Koo J., Ehrt S., Wainwright H., Bekker L. G., Kaplan G., Nathan C., Tabas I., Russell D. G. (2010) Induction of ER stress in macrophages of tuberculosis granulomas. PLoS ONE 5, e12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu S., Suragani R. N., Wang F., Han A., Zhao W., Andrews N. C., Chen J. J. (2007) The function of heme-regulated eIF2α kinase in murine iron homeostasis and macrophage maturation. J. Clin. Invest. 117, 3296–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Woo C. W., Cui D., Arellano J., Dorweiler B., Harding H., Fitzgerald K. A., Ron D., Tabas I. (2009) Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by Toll-like receptor signaling. Nat. Cell Biol. 11, 1473–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nakayama Y., Endo M., Tsukano H., Mori M., Oike Y., Gotoh T. (2010) Molecular mechanisms of the LPS-induced non-apoptotic ER stress-CHOP pathway. J. Biochem. 147, 471–483 [DOI] [PubMed] [Google Scholar]
- 11. Wiley D. J., Nordfeldth R., Rosenzweig J., DaFonseca C. J., Gustin R., Wolf-Watz H., Schesser K. (2006) The Ser/Thr kinase activity of the Yersinia protein kinase A (YpkA) is necessary for full virulence in the mouse, mollifying phagocytes, and disrupting the eukaryotic cytoskeleton. Microb. Pathog. 40, 234–243 [DOI] [PubMed] [Google Scholar]
- 12. Wiley D. J., Shrestha N., Yang J., Atis N., Dayton K., Schesser K. (2009) The activities of the Yersinia protein kinase A (YpkA) and outer protein J (YopJ) virulence factors converge on an eIF2α kinase. J. Biol. Chem. 284, 24744–24753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scheuner D., Song B., McEwen E., Liu C., Laybutt R., Gillespie P., Saunders T., Bonner-Weir S., Kaufman R. J. (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165–1176 [DOI] [PubMed] [Google Scholar]
- 14. Bölin I., Norlander L., Wolf-Watz H. (1982) Temperature-inducible outer membrane protein of Yersinia pseudotuberculosis and Yersinia enterocolitica is associated with the virulence plasmid. Infect. Immun. 37, 506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Galyov E. E., Håkansson S., Wolf-Watz H. (1994) Characterization of the operon encoding the YpkA Ser/Thr protein kinase and the YopJ protein of Yersinia pseudotuberculosis. J. Bacteriol. 176, 4543–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Day J. B., Ferracci F., Plano G. V. (2003) Translocation of YopE and YopN into eukaryotic cells by Yersinia pestis yopN, tyeA, sycN, yscB, and lcrG deletion mutants measured using a phosphorylatable peptide tag and phospho-specific antibodies. Mol. Microbiol. 47, 807–823 [DOI] [PubMed] [Google Scholar]
- 17. Håkansson S., Galyov E. E., Rosqvist R., Wolf-Watz H. (1996) The Yersinia YpkA Ser/Thr kinase is translocated and subsequently targeted to the inner surface of the HeLa cell plasma membrane. Mol. Microbiol. 20, 593–603 [DOI] [PubMed] [Google Scholar]
- 18. Caldwell H. D., Kromhout J., Schachter J. (1981) Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect. Immun. 31, 1161–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin R., Génin P., Mamane Y., Hiscott J. (2000) Selective DNA binding and association with the CREB-binding protein coactivator contribute to differential activation of α/β-interferon genes by interferon regulatory factors 3 and 7. Mol. Cell. Biol. 20, 6342–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang H. Y., Wek S. A., McGrath B. C., Lu D., Hai T., Harding H. P., Wang X., Ron D., Cavener D. R., Wek R. C. (2004) Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 24, 1365–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palmer L. E., Hobbie S., Galán J. E., Bliska J. B. (1998) YopJ of Yersinia pseudotuberculosis is required for the inhibition of macrophage TNFα production and down-regulation of the MAP kinases p38 and JNK. Mol. Microbiol. 27, 953–965 [DOI] [PubMed] [Google Scholar]
- 22. Schesser K., Spiik A. K., Dukuzumuremyi J. M., Neurath M. F., Pettersson S., Wolf-Watz H. (1998) The yopJ locus is required for Yersinia-mediated inhibition of NF-κB activation and cytokine expression: YopJ contains a eukaryotic SH2-like domain that is essential for its repressive activity. Mol. Microbiol. 28, 1067–1079 [DOI] [PubMed] [Google Scholar]
- 23. Lu P. D., Harding H. P., Ron D. (2004) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 167, 27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo S., Baumeister P., Yang S., Abcouwer S. F., Lee A. S. (2003) Induction of Grp78/BiP by translational block. Activation of the Grp78 promoter by ATF4 through an upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 278, 37375–37385 [DOI] [PubMed] [Google Scholar]
- 25. McEwen E., Kedersha N., Song B., Scheuner D., Gilks N., Han A., Chen J. J., Anderson P., Kaufman R. J. (2005) Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 280, 16925–16933 [DOI] [PubMed] [Google Scholar]
- 26. Lu J., O'Hara E. B., Trieselmann B. A., Romano P. R., Dever T. E. (1999) The interferon-induced double-stranded RNA-activated protein kinase PKR will phosphorylate serine, threonine, or tyrosine at residue 51 in eukaryotic initiation factor 2α. J. Biol. Chem. 274, 32198–32203 [DOI] [PubMed] [Google Scholar]
- 27. Sweet C. R., Conlon J., Golenbock D. T., Goguen J., Silverman N. (2007) YopJ targets TRAF proteins to inhibit TLR-mediated NF-κB, MAPK, and IRF3 signal transduction. Cell. Microbiol. 9, 2700–2715 [DOI] [PubMed] [Google Scholar]
- 28. Boyce M., Bryant K. F., Jousse C., Long K., Harding H. P., Scheuner D., Kaufman R. J., Ma D., Coen D. M., Ron D., Yuan J. (2005) A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 307, 935–939 [DOI] [PubMed] [Google Scholar]
- 29. Huang X., Chen Y., Zhang H., Ma Q., Zhang Y. W., Xu H. (2012) Salubrinal attenuates β-amyloid-induced neuronal death and microglial activation by inhibition of the NF-κB pathway. Neurobiol. Aging 33, 1007.e9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuo T. F., Tatsukawa H., Matsuura T., Nagatsuma K., Hirose S., Kojima S. (2012) Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J. Cell Physiol. 227, 1130–1137 [DOI] [PubMed] [Google Scholar]
- 31. Rosqvist R., Forsberg A., Rimpiläinen M., Bergman T., Wolf-Watz H. (1990) The cytotoxic protein YopE of Yersinia obstructs the primary host defense. Mol. Microbiol. 4, 657–667 [DOI] [PubMed] [Google Scholar]
- 32. Disson O., Grayo S., Huillet E., Nikitas G., Langa-Vives F., Dussurget O., Ragon M., Le Monnier A., Babinet C., Cossart P., Lecuit M. (2008) Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455, 1114–1118 [DOI] [PubMed] [Google Scholar]
- 33. Pillich H., Loose M., Zimmer K. P., Chakraborty T. (2012) Activation of the unfolded protein response by Listeria monocytogenes. Cell. Microbiol. 14, 949–964 [DOI] [PubMed] [Google Scholar]
- 34. Mukherjee S., Keitany G., Li Y., Wang Y., Ball H. L., Goldsmith E. J., Orth K. (2006) Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214 [DOI] [PubMed] [Google Scholar]
- 35. Mittal R., Peak-Chew S. Y., McMahon H. T. (2006) Acetylation of MEK2 and IκB kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 18574–18579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trülzsch K., Sporleder T., Igwe E. I., Rüssmann H., Heesemann J. (2004) Contribution of the major secreted Yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect. Immun. 72, 5227–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Viboud G. I., Bliska J. B. (2005) Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu. Rev. Microbiol. 59, 69–89 [DOI] [PubMed] [Google Scholar]