

Background: Elongation factor G of Escherichia coli is sensitive to oxidation.
Results: Elongation factor G is inactivated via the formation of an intramolecular disulfide bond.
Conclusion: Elongation factor G is a critical target during oxidative damage to the translation system.
Significance: Oxidation of elongation factor G suggests a novel mechanism for the redox regulation of translation.
Keywords: Protein Synthesis, Reactive Oxygen Species (ROS), Redox Regulation, Translation Elongation Factors, Translation Regulation, Elongation Factor G, In Vitro Translation, Oxidative Stress
Abstract
Elongation factor G (EF-G), a key protein in translational elongation, is known to be particularly susceptible to oxidation in Escherichia coli. However, neither the mechanism of the oxidation of EF-G nor the influence of its oxidation on translation is fully understood. In the present study, we investigated the effects of oxidants on the chemical properties and function of EF-G using a translation system in vitro derived from E. coli. Treatment of EF-G with 0.5 mm H2O2 resulted in the complete loss of translational activity. The inactivation of EF-G by H2O2 was attributable to the oxidation of two specific cysteine residues, namely, Cys114 and Cys266, and subsequent formation of an intramolecular disulfide bond. Replacement of Cys114 by serine rendered EF-G insensitive to oxidation and inactivation by H2O2. Furthermore, generation of the translation system in vitro with the mutated EF-G protected the entire translation system from oxidation, suggesting that EF-G might be a primary target of oxidation within the translation system. Oxidized EF-G was reactivated via reduction of the disulfide bond by thioredoxin, a ubiquitous protein that mediates dithiol-disulfide exchange. Our observations indicate that the translational machinery in E. coli is regulated, in part, by the redox state of EF-G, which might depend on the balance between the supply of reducing power and the degree of oxidative stress.
Introduction
Since the first demonstrations (1, 2) that protein synthesis in prokaryotic and eukaryotic organisms is suppressed by oxidizing reagents, the effects of oxidative stress on protein synthesis have been the focus of extensive analysis. In recent years, the effects of oxidative stress on elongation factors, which are components of the translational machinery, have received particular attention. Elongation factor G (EF-G),4 a key protein in translational elongation, was identified as one of proteins that were abundantly carbonylated in cells of Escherichia coli that had been exposed to H2O2 (3, 4) and in mutant E. coli cells that lacked a superoxide dismutase (5). Elongation factor 2 (EF-2), the eukaryotic counterpart of EF-G, was also identified as a protein that is susceptible to oxidation. Treatment of rabbit reticulocytes with the oxidant cumene hydroperoxide resulted in the inhibition of the phosphorylation of EF-2 (2). Treatment of rat liver with cumene hydroperoxide stimulated the carbonylation of amino acids and subsequent ADP-ribosylation of EF-2 (6). However, the effects of the oxidation of these elongation factors on translational activity remained to be clarified.
A clue to the significance of the oxidation of elongation factors was provided by studies of photosynthesis in the cyanobacterium Synechocystis sp. PCC 6803 (hereafter referred to as Synechocystis) (7). Recent studies of the effects of reactive oxygen species (ROS) on the photoinhibition of photosystem II (PSII) revealed that ROS act primarily by inhibiting the repair of photodamaged PSII (8, 9). Inhibition of the repair of PSII has been attributed to the suppression, at the level of translational elongation, of the synthesis of proteins that are required for the repair of PSII (8, 9). Biochemical studies using a translation system in vitro from Synechocystis revealed that EF-G is a primary target of inactivation by ROS, and that inactivation of EF-G by H2O2 is caused by the oxidation of two specific cysteine residues with the resultant formation of an intramolecular disulfide bond (10, 11).
Alignment of the deduced amino acid sequences of EF-G proteins from various organisms revealed that the two cysteine residues that correspond to the targets of oxidation by ROS in Synechocystis are strongly conserved in the EF-G proteins of several species of cyanobacteria, as well as in those of nonphotosynthetic prokaryotes, such as E. coli (11). The presence of these conserved cysteine residues led us to postulate that EF-G might also be a target during oxidative damage to the translation system in E. coli.
In the present study, we examined the effects of ROS on the redox state of cysteine residues and the activity of EF-G of E. coli, using a reconstituted translation system derived from E. coli. Treatment of EF-G with H2O2 resulted in the oxidation of two specific cysteine residues and subsequent formation of an intramolecular disulfide bond. This oxidation was responsible for the inactivation of EF-G in translation. The oxidized EF-G was reduced and reactivated by thioredoxin. Our observations suggest that regulation of translation via the redox state of EF-G might occur not only in photosynthetic prokaryotes but also in other prokaryotes, such as E. coli.
EXPERIMENTAL PROCEDURES
Preparation of Recombinant Proteins
The fusA, trxA, and grxA genes of E. coli, which encode EF-G, thioredoxin A, and glutaredoxin A, respectively, were cloned into the pET21b vector (Novagen, Darmstadt, Germany; sequences of primers are available on request). Proteins were expressed in E. coli BL21(DE3) as carboxyl-terminal histidine-tagged recombinant proteins and purified, in reduced form, as described previously (10). Each protein was stored in buffer that contained 20 mm HEPES-KOH (pH 7.5), 50 mm NaCl, and 20% (w/v) glycerol.
Site-directed Mutagenesis of Proteins
Site-directed mutagenesis of EF-G was performed as described previously (11), using a KOD-Plus Mutagenesis Kit (Toyobo, Osaka, Japan). The reaction mixture for PCR included a pair of complementary oligonucleotides of 25–30 bases that incorporated the desired mutation and pET21b, which harbored the gene for EF-G from E. coli, as the template (sequences of primers are available on request).
Modification of Thiol Groups of Cysteine Residues
The redox state of the cysteine residues in EF-G was monitored by modifying the thiol groups in EF-G with a maleimidyl reagent, methoxypoly(ethylene glycol)maleimide, which has an average molecular mass of 5 kDa (Nihon Yushi, Tokyo, Japan), and subsequent separation of modified proteins by nonreducing SDS-PAGE on a 7.5% polyacrylamide gel, as described previously (11).
Quantitative Analysis of Thiol Groups
The number of thiol groups per EF-G molecule was determined as described previously (11). EF-G at 50 μm was incubated with 0.4 mm 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) (Sigma) in buffer that contained 100 mm Tris-HCl (pH 8.0), 10 mm EDTA, and 6 m guanidine HCl. Changes in absorbance at 412 nm were monitored, and the number of reactive thiol groups was determined.
Translation in Vitro
Translation in vitro was performed with the PURE system, a reconstituted translation system derived from E. coli (12). The translation system was generated, in the absence of EF-G and reducing reagents, by mixing 70 S ribosomes with the individual components that are required for transcription and translation in vitro. After EF-G had been treated with H2O2 in buffer that contained 50 mm HEPES-KOH (pH 7.5), residual H2O2 was removed by incubating the mixture of EF-G and H2O2 with catalase (Nacalai Tesque, Kyoto, Japan), as described previously (11). EF-G was then added to the translation system that had been prepared without EF-G. The resultant translation system was incubated at 37 °C in the presence of a plasmid DNA that harbored a gene for dihydrofolic acid reductase (DHFR) as template, 35S-labeled cysteine/methionine, and reagents required for translation (12).
Mass Spectrometry
EF-G that had been treated with 0.5 mm H2O2 or 5 mm dithiothreitol (DTT) was subjected to analysis by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry on an Autoflex system (Bruker Daltonics, Bremen, Germany). External calibration was performed using the peptide calibration standard (Bruker Daltonics), and the m/z value of 2466.74 of a product of autodigested trypsin was used for internal calibration.
Reduction of EF-G by Thioredoxin and by Glutaredoxin
EF-G at 2 μm was oxidized by incubation with 0.5 mm H2O2 for 5 min at 25 °C. Then 0.75 μm catalase was added to remove residual H2O2, and the oxidized EF-G was incubated for 15 min at 25 °C with DTT at various concentrations in the presence of 4 μm thioredoxin A or 4 μm glutaredoxin A. Proteins were then precipitated with 10% (w/v) trichloroacetic acid and subjected to the thiol modification assay for detection of thiol groups on cysteine residues.
Quantitative Analysis of GTPase Activity
The GTPase activity of EF-G was measured by monitoring the release of inorganic phosphate from GTP by the end-point assay with malachite green (13). The reaction mixture contained 30 nm 70 S ribosomes of E. coli, 20 μm GTP, and 60 nm wild-type EF-G or its mutant derivative, plus either 0.5 mm H2O2 or 5 mm DTT. Changes in absorbance at 650 nm were measured spectroscopically.
GTP-binding Assay
EF-G (6 μm) that had been treated with 0.5 mm H2O2 or 5 mm DTT was incubated at 37 °C for 5 min in the presence of 0.1 μm [α-32P]GTP (3 MBq ml−1, PerkinElmer Life Sciences) in blotting buffer that contained 50 mm Tris-HCl (pH 8.0), 5 mm MgCl2, 50 mm KCl, 200 μg ml−1 of bovine serum albumin, 10% (w/v) glycerol, and 10 μm GTP. An aliquot of 10 μl was blotted onto a nitrocellulose membrane (GE Healthcare) and the membrane was washed with 2 ml of blotting buffer that did not contain GTP. The radioactivity from the membrane was quantitated by liquid scintillation counting, as described previously (10).
RESULTS
Effects of Oxidants on the Translational Activity of EF-G and the Redox State of Its Cysteine Residues
We examined the effects of oxidants on the translational activity of EF-G using the PURE system, a translation system in vitro derived from E. coli that was generated by mixing the individual components required for translation (12). Addition of the reduced form of EF-G to a translation system that had been generated without EF-G resulted in the synthesis of DHFR (Fig. 1A). However, when EF-G had been treated with H2O2 for 5 min prior to its addition to the translation system, the synthesis of DHFR was suppressed (Fig. 1A). The extent of suppression depended on the concentration of H2O2 and, at 0.5 mm H2O2, the synthesis of DHFR was completely inhibited (Fig. 1B). These observations suggested that the translational activity of EF-G had been abolished by H2O2.
FIGURE 1.
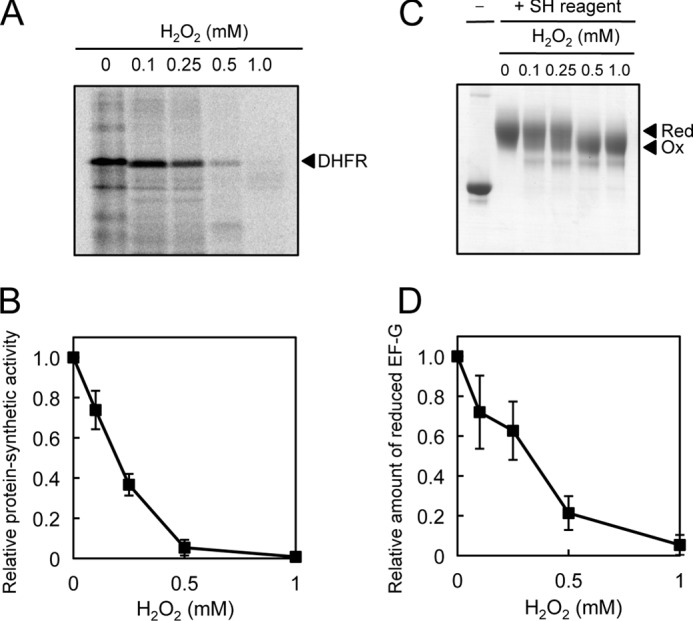
Effects of H2O2 on the translational activity and the redox state of cysteine residues of wild-type EF-G. A, wild-type EF-G (2 μm) was incubated for 5 min in the presence of H2O2 at the indicated concentrations. After residual H2O2 had been eliminated by addition of 0.75 μm catalase, EF-G was added to a translation system in vitro, derived from E. coli, that had been generated without EF-G. The resultant translation system was incubated at 30 °C for 60 min in the presence of template DNA that encoded DHFR, and proteins were then fractionated by SDS-PAGE. Translational activity was monitored in terms of the synthesis of 35S-labeled DHFR. B, quantitation of radioactivity from 35S-labeled DHFR as shown in A. C, after EF-G had been incubated for 10 min in the presence of H2O2 at the indicated concentrations, EF-G was treated with a maleimidyl thiol-modifying reagent (SH reagent). EF-G was then fractionated by nonreducing SDS-PAGE. Red, reduced EF-G; Ox, oxidized EF-G. D, quantitation of the ratio of reduced EF-G to oxidized EF-G as shown in C. Values are mean ± S.D. (bars) of results from three independent experiments.
We monitored the redox state of three cysteine residues of EF-G by modifying free thiol groups with a maleimidyl reagent (average molecular mass, 5 kDa) under the same oxidizing conditions. Modification of the fully reduced form of EF-G with this reagent resulted in a decrease in the electrophoretic mobility of EF-G on a nonreducing gel (Fig. 1C). Incubation of EF-G with H2O2 at various concentrations, prior to the modification, decreased the change in mobility, indicating that some fraction of the three cysteine residues had been oxidized by H2O2 and had failed to bind the maleimidyl reagent (Fig. 1C). The extent of oxidation of EF-G depended on the concentration of H2O2 and, at 0.5 mm H2O2, EF-G was fully oxidized (Fig. 1D). No oligomers of EF-G were detected after oxidation of EF-G with H2O2, suggesting that EF-G might be unable to form intermolecular disulfide bonds.
The similarity in terms of dependence on the concentration of H2O2 between the translational activity of EF-G and the redox state of its cysteine residues suggested that the inactivation of EF-G might have been due to the oxidation of specific cysteine residues. We also determined that not only H2O2 but also other oxidants, such as copper(II) chloride and AldrithiolTM-4, inhibited the translational activity of EF-G and oxidized specific cysteine residues (supplemental Fig. S1). Thus, it appeared that EF-G might be oxidized to yield an identical product under a variety of oxidizing conditions. The circular dichroism spectra revealed that the oxidation of EF-G with 0.5 mm H2O2 did not affect the secondary structure of EF-G (supplemental Fig. S2 and supplemental Experimental Procedures).
To determine the number of oxidized cysteine residues in EF-G, we compared the number of free thiol groups on cysteine residues in EF-G and mutant derivatives, in which specific cysteine residues had been replaced by serine, by monitoring changes in absorption after reaction of thiol groups with DTNB. The number of free thiol groups per molecule of reduced EF-G was 3.0, which corresponds to the number of cysteine residues in EF-G, whereas those of oxidized EF-G, single-mutated C114S, double-mutated C114S/C266S, and triple-mutated C114S/C266S/C398S EF-G were 1.2, 2.2, 1.3, and 0.51, respectively (Fig. 2A). These results suggested that two cysteine residues of EF-G had been oxidized by H2O2 to generate an intramolecular disulfide bond.
FIGURE 2.
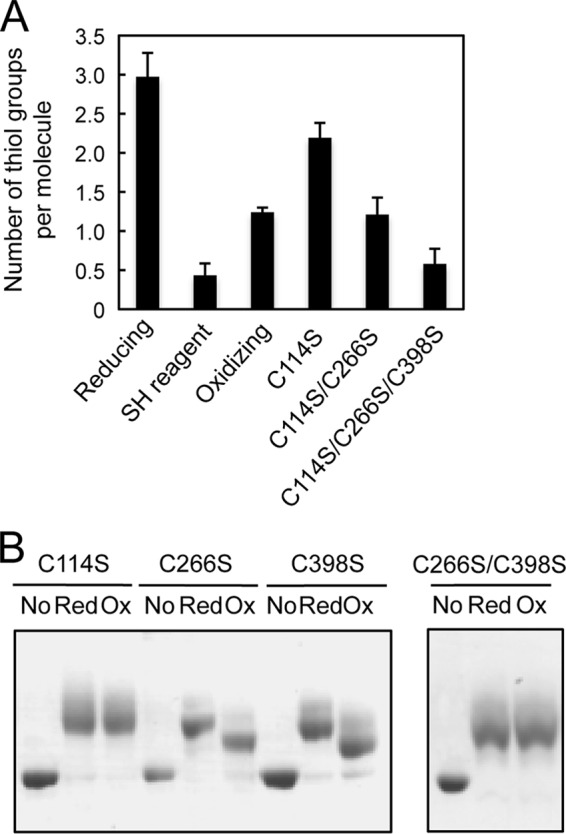
Quantitation of the H2O2-oxidized cysteine residues of EF-G. A, quantitative analysis of thiol groups. Wild-type EF-G (2 μm) was treated with 5 mm DTT (reducing), 0.5 mm H2O2 (oxidizing), or the thiol-modifying (SH) reagent. The number of thiol groups per protein molecule of wild-type EF-G and its mutant derivatives was determined with DTNB. Values are mean ± S.D. (bars) of results from three independent experiments. B, the effect of H2O2 on the redox state of mutant derivatives of EF-G. After the derivatives of EF-G (2 μm) had been incubated with 0.5 mm H2O2 for 5 min, they were treated with the thiol-modifying reagent. Proteins were then fractionated by nonreducing SDS-PAGE. No, no modification with SH reagent; Red, reducing conditions; Ox, oxidizing conditions.
Identification of the Cysteine Residues That Were Oxidized by H2O2
To identify the cysteine residues that were oxidized by H2O2, we examined the effects of oxidation on the redox state of cysteine residues of mutated EF-G by the thiol-modification assay. In the presence of 0.5 mm H2O2, the cysteine residues in the C266S and C398S mutant proteins were oxidized in a similar manner to those in wild-type EF-G, whereas those in the C114S mutant protein were no longer oxidized (Fig. 2B). We concluded, therefore, that Cys114 might be a target of oxidation by H2O2. However, we failed to identify the counterpart of Cys114 for the formation of a disulfide bond by this method. It is possible that Cys114 is extremely reactive and can readily react with another cysteine residue to form an abnormal disulfide bond in the absence of its true counterpart. As a consequence, both the single-mutated proteins, namely, C266S and C398S appeared to be susceptible to oxidation by H2O2. The double-mutated C266S/C398S protein, in which Cys114 is the sole remaining cysteine residue, was not oxidized by H2O2 (Fig. 2B). Apparently, Cys114 cannot be oxidized in the absence of other cysteine residues, and other types of oxidation of Cys114, such as the formation of sulfenic acid, might not occur under oxidizing conditions.
To identify the counterpart of Cys114 for formation of a disulfide bond, we performed peptide-mapping analysis of EF-G. We digested the reduced and oxidized forms of EF-G, which had been obtained by incubation of EF-G with 5 mm DTT and 0.5 mm H2O2, respectively, with trypsin and measured the mass of the resulting peptide fragments by mass spectrometry. In the analysis of the reduced form of EF-G, peptide fragments that included Cys114, Cys266, and Cys398 were detected (supplemental Fig. S3 and Table 1). By contrast, in the analysis of the oxidized form of EF-G, levels of these peptide fragments were considerably lower and a peptide fragment with a mass of 4282.49 was newly detected. The mass of the new peptide corresponded to that of a peptide fragment that resulted from the combination of two peptides, one including Cys114 and one including Cys266, via formation of a disulfide bond (supplemental Fig. S3 and Table 1). These observations support the hypothesis that Cys114 and Cys266 might form an intramolecular disulfide bond under oxidizing conditions. In the oxidized form of EF-G, we did not detect the peptide including Cys114 that was oxidized to sulfenic acid under the conditions that we tested.
TABLE 1.
Theoretical and observed monoisotopic mass values for peptide fragments that included cysteine residues
Peptide fragments that included Cys266, Cys398, and Cys114 and a disulfide-linked peptide were obtained from EF-G that had been treated with 5 mm DTT (reducing) or 0.5 mm H2O2 (oxidizing). Peaks 1 through 3 correspond to those shown in supplemental Fig. S3.
| Peak 1 (Cys266) | Peak 2 (Cys398) | Peak 3 (Cys114) | Peak 4 (Cys114-Cys266) | |||||
|---|---|---|---|---|---|---|---|---|
| Expected | 1721.04 | Δa | 2044.26 | Δ | 2564.96 | Δ | 4283.01 | Δ |
| Reducing | 1721.58 | +0.54 | 2045.34 | +0.08 | 2565.20 | +0.06 | NDb | |
| Oxidizing | 1721.45 | +0.41 | 2045.31 | +0.05 | 2565.12 | +0.16 | 4282.49 | −0.52 |
a Δ, difference between given value and expected value.
b ND, not detected.
Effects of Mutation of EF-G on Translational Activity in Vitro
To clarify the relationship between the redox state of individual cysteine residues and translational activity, we investigated the activities of our mutant derivatives of EF-G in the translation system prepared without EF-G. Treatment of neither the C266S nor the C398S mutant protein with 0.5 mm H2O2 resulted in the synthesis of detectable DHFR, whereas the C114S mutant protein that had been treated with 0.5 mm H2O2 was able to support translation at the same rate as its reduced form (Fig. 3), indicating that Cys114 is both a target of oxidation by H2O2 and a site of regulation of the activity of EF-G. The double-mutated C266S/C398 protein, with an intact Cys114 residue, was also insensitive to oxidation by H2O2 (Fig. 3), suggesting that EF-G cannot be inactivated unless Cys114 forms a disulfide bond with another cysteine residue. Given the results of our analysis of the redox state of cysteine residues, we concluded from these observations that EF-G might be active in its reduced form and inactive in its oxidized form.
FIGURE 3.
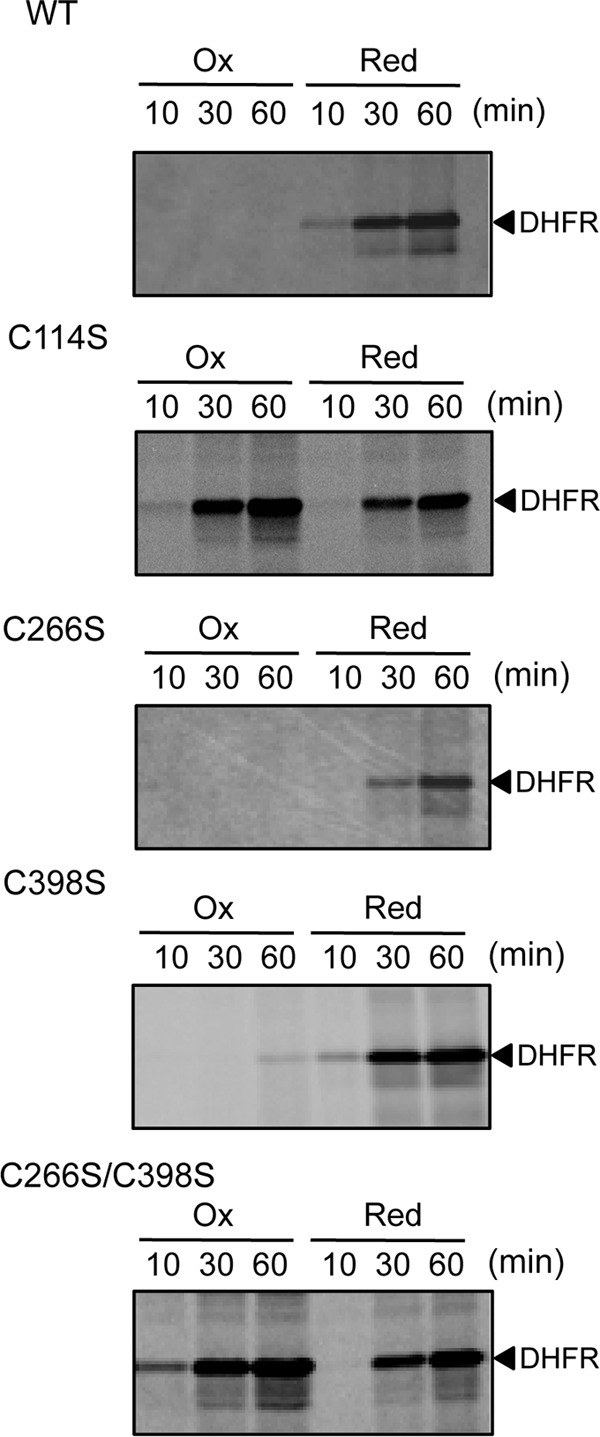
Effects of H2O2 on the translational activity of mutant derivatives of EF-G. Derivatives of EF-G (2 μm) were treated, separately, with 0.5 mm H2O2 (Ox) and 5 mm DTT (Red) for 5 min. After removal of residual H2O2 by catalase, the derivatives were subjected to the translational assay in the translation system from E. coli that had been prepared without EF-G. Translation was allowed to proceed at 30 °C for the indicated times and translational activity was monitored in terms of the synthesis of 35S-labeled DHFR, as indicated in the legend to Fig. 1.
Reduction and Reactivation of Oxidized EF-G by Thioredoxin
The redox behavior of EF-G that depended on its cysteine residues suggested that EF-G might interact with thioredoxin or glutaredoxin, two ubiquitous proteins that regulate the activity of target proteins by reducing disulfide bonds (14, 15). We examined whether these redox proteins might be able to reduce the disulfide bond in oxidized EF-G. The results of a thiol modification assay showed that the oxidized cysteine residues in EF-G (2 μm) that had been incubated with 0.5 mm H2O2 were partially reduced by subsequent exposure to a relatively high concentration of DTT, such as 5 mm (Fig. 4A). By contrast, complete reduction of EF-G was achieved by the addition of 4 μm thioredoxin A, even in the presence of only 50 μm DTT. Thus, thioredoxin was able to interact with EF-G and to reduce an intramolecular disulfide bond. However, when we tested glutaredoxin A in a similar manner, we failed to reduce the oxidized EF-G (Fig. 4A).
FIGURE 4.

Effects of thioredoxin and glutaredoxin on the redox state of cysteine residues in oxidized EF-G and on its translational activity. A, changes in the redox state of cysteine residues of wild-type EF-G. EF-G (2 μm) was incubated with 0.5 mm H2O2 or in its absence for 5 min. After removal of residual H2O2 by catalase, EF-G was incubated with DTT at the indicated concentrations plus 4 μm thioredoxin (Trx) or 4 μm glutaredoxin (Grx), as indicated. After proteins had been treated with the thiol-modifying reagent, they were fractionated by nonreducing SDS-PAGE. Red, reducing conditions; Ox, oxidizing conditions. B, activation of oxidized EF-G by thioredoxin. After oxidized EF-G had been incubated with 50 μm DTT plus 4 μm thioredoxin or plus 4 μm glutaredoxin for 5 min, each mixture was added to the translation system in vitro that had been incubated with H2O2. Translational activity was monitored as indicated in the legend to Fig. 1. C, the effects of DTT, thioredoxin, and glutaredoxin on the activity of the translation system, as controls. The complete translation system, with EF-G, was supplemented with the same amounts of DTT, thioredoxin, and glutaredoxin as those used in B to serve as controls in assays of the translational activity of oxidized and then reduced EF-G.
We next examined whether the reduction of EF-G by thioredoxin might affect the activity of EF-G in translation. We incubated 2 μm oxidized EF-G with 4 μm thioredoxin A and 50 μm DTT, namely, under the conditions used for the thioredoxin-assisted reduction of EF-G. When we added the reduced EF-G to the translation system in vitro, translational activity was restored (Fig. 4B). By contrast, after incubation of 2 μm oxidized EF-G with 50 μm DTT alone or with 4 μm glutaredoxin A alone, no translation was observed. In these experiments, low concentrations of DTT, thioredoxin, or glutaredoxin were added to the translation system concomitantly with EF-G. Control assays indicated that the addition of these components at such low concentrations to the translation system with EF-G did not affect translational activity (Fig. 4C). Thus, it was evident that oxidized EF-G could be reactivated by thioredoxin.
Effects of the Oxidation of Cysteine Residues in EF-G on its GTPase Activity
Vacant 70 S ribosomes are known to stimulate the GTPase activity of EF-G via formation of a 70S·EF-G·GTP complex (16). We investigated whether the redox state of EF-G might affect this GTPase activity. We incubated EF-G with 5 mm DTT or 0.5 mm H2O2 and then measured the GTPase activity by monitoring the formation of a malachite green complex. When we added reduced EF-G to 70 S ribosomes in the presence of GTP, we observed the hydrolysis of GTP (Fig. 5A). By contrast, the addition of oxidized EF-G resulted in 60% less hydrolysis of GTP. Furthermore, the C114S mutant protein that had been treated with 0.5 mm H2O2 was able to support the hydrolysis of GTP at the same rate as the reduced EF-G (Fig. 5A). Thus, the inactivation of EF-G by oxidation might be due, in part, to the suppression of its GTPase activity as a consequence of the formation of an intramolecular disulfide bond. However, the GTP-binding ability of EF-G was hardly affected by oxidation with 0.5 mm H2O2 (Fig. 5B).
FIGURE 5.

Effects of H2O2 on the GTPase activity and GTP-binding ability of EF-G. A, wild-type EF-G and its mutant C114S derivative (2 μm) were treated with 0.5 mm H2O2 (■) and 5 mm DTT (♦). Then the treated EF-G, at 60 nm, was incubated with 70 S ribosomes (30 nm) and 20 mm GTP. The GTPase activity was determined by the malachite green colorimetric assay. B, wild-type EF-G (6 μm) that had been treated with 0.5 mm H2O2 (oxidizing) and 5 mm DTT (reducing) was incubated in the presence of 0.1 μm 32P-labeled GTP. The mixture was blotted onto a nitrocellulose membrane, and the radioactivity from proteins bound to the membrane was measured by liquid scintillation counting. Values are mean ± S.D. (bars) of results from three independent experiments.
Effects of Mutation of EF-G on the Entire Translation System under Oxidizing Conditions
We examined the effect of replacement of Cys114 by serine on the sensitivity of the entire translation system to oxidation by H2O2. The translation system that had been generated with the C114S mutant protein, instead of wild-type EF-G, was able to synthesize DHFR in the presence of 100 μm H2O2, whereas the translation system with wild-type EF-G was completely inactive in the presence of 50 μm H2O2 (Fig. 6). Note that EF-G was diluted 20-fold when it was added to the translation system. The apparent sensitivity of the translation system to H2O2, as compared with the sensitivity of EF-G shown in Fig. 1, was due to the low molecular ratio of EF-G to H2O2. Thus, replacement of a single specific cysteine residue, Cys114, of EF-G by serine rendered the entire translation system of E. coli more resistant to oxidation. In other words, EF-G is likely to be a critical target of oxidation within the translation system of E. coli.
FIGURE 6.
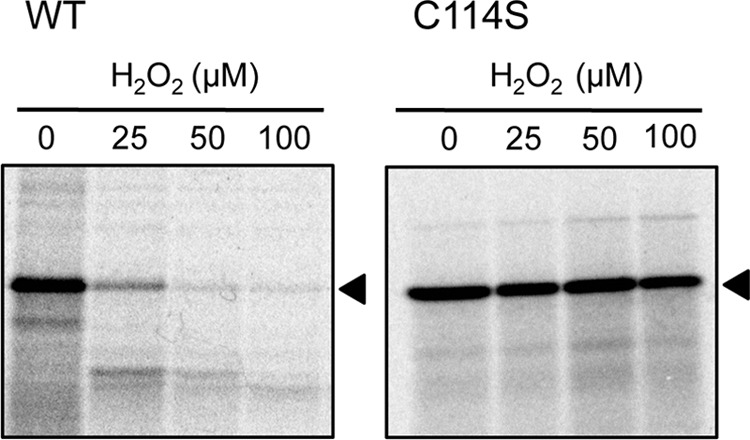
Effects of H2O2 on the activity of the translation system in vitro that had been generated with EF-G in which Cys114 had been replaced by serine. The translation system that had been generated with wild-type EF-G or with its mutant C114S derivative was incubated for 5 min in the presence of H2O2 at various concentrations. Then translational activity was monitored in terms of the synthesis of 35S-labeled DHFR, as shown in the legend to Fig. 1. Note that the final concentration of EF-G in the translation system was 0.1 μm. The arrowheads indicate DHFR.
DISCUSSION
Oxidation of Two Specific Cysteine Residues Inactivates EF-G
In the present study, we demonstrated that the inactivation of EF-G of E. coli by H2O2 was due to oxidation of two specific cysteine residues, namely, Cys114 and Cys266, and the subsequent formation of an intramolecular disulfide bond. The carbonylation of amino acid residues, which was previously proposed as the mechanism of oxidative damage to EF-G (3), occurs under much more severely oxidizing conditions.
Of the two redox-active cysteine residues, Cys114 was much more sensitive than Cys266 to H2O2. When Cys114 remained active in the absence of Cys266, Cys114 was able to react with Cys398 to form an abnormal disulfide bond under oxidizing conditions, yielding an inactive EF-G. Therefore, both the C266S and C398S mutant proteins were susceptible to oxidation by H2O2 (Figs. 2 and 3). By contrast, Cys266 was able only to react with Cys114. The different redox properties of the cysteine residues might explain why only the absence of Cys114 rendered EF-G insensitive to oxidation by H2O2. A similar hierarchy of reactivity of redox-active cysteine residues was observed in EF-G of Synechocystis (11) and also in PhoA, an alkaline phosphatase of E. coli (17). The insensitivity to oxidation of the double-mutated EF-G, C226S/C398S, in which Cys114 was present and the other cysteine residues had been replaced by serine suggests that Cys114 might not undergo some other types of oxidation, such as the formation of sulfenic acid, under the oxidizing conditions that we tested. In addition, the reduction of oxidized EF-G by thioredoxin suggests that Cys114 might not be oxidized to form sulfinic acid, which requires a specific enzyme, such as sulfiredoxin, and ATP for reduction (18).
However, in the crystal structure of EF-G of E. coli, which is bound to the ribosome-recycling factor (19), it appears that Cys266 is not located in the close vicinity of Cys114, whereas Cys398 is apart from Cys114 (supplemental Fig. S4). Thus, the distances between cysteine residues in the crystal structure cannot fully explain the formation of disulfide bonds, although these distances might have been changed by some conformational change in EF-G after the binding of the ribosome-recycling factor. From our biochemical evidence, we propose that the formation of disulfide bond is most likely, but we cannot thoroughly exclude the possibility that cysteine residues undergo some other types of oxidation, including the formation of sulfenic acid. Crystallographic studies of the reduced and oxidized forms of EF-G will be necessary for a full understanding of the mechanism of the oxidation of cysteine residues.
Possible Mechanism of the Inactivation of EF-G
EF-G translocates peptidyl-tRNA from the A site to the P site in the ribosome, upon a conformational change that is driven by the hydrolysis of GTP. Treatment of EF-G with H2O2 resulted in a decrease in the GTPase activity of EF-G, whereas replacement of Cys114 by serine rendered the GTPase activity insensitive to H2O2 (Fig. 5). The GTP-binding ability of EF-G and its global structure were hardly affected by oxidation by H2O2 (Figs. 5 and supplemental Fig. S2). These observations suggest that the formation of the intramolecular disulfide bond might inhibit the GTPase activity of EF-G via interference with an appropriate conformational change in EF-G that is induced after EF-G binds to the ribosome. However, 40% of the GTPase activity of EF-G remained when the translational activity of EF-G had been completely abolished in the presence of 0.5 mm H2O2. This apparent discrepancy suggests that the formation of an intramolecular disulfide bond might also inhibit a conformational change in EF-G after the hydrolysis of GTP. Fusidic acid, an antibiotic specific to EF-G, inhibits the conformational change in EF-G that facilitates the dissociation of EF-G from the ribosome, without affecting the hydrolysis of GTP (20, 21).
Physiological Implications of the Redox Regulation of EF-G
Some metabolic processes in a variety of organisms are known to be controlled by regulatory systems that involve redox reactions. A fundamental example of such a redox reaction is the reversible reduction of disulfide bonds between cysteine residues by thioredoxin or glutaredoxin (14, 15, 22). Assays of thiol modification and translation in vitro revealed that the disulfide bond in peroxide-oxidized EF-G was reduced by thioredoxin but not glutaredoxin and that the newly reduced form of EF-G was able to function in translation (Fig. 4). It has also been reported that EF-G was one of a number of proteins from E. coli that bound to a thioredoxin-affinity column (23). The activation of EF-G by thioredoxin suggests that EF-G might be involved in the redox regulation of cellular metabolism in E. coli.
In E. coli, thiol-dependent redox systems in the cytoplasm include thioredoxin- and glutaredoxin-dependent pathways that correspond to the flows of electrons that reflect gradients in redox potentials. The main source of electrons for these reducing pathways is the oxidation of hexose phosphates in the pentose phosphate pathway, which regenerates NADPH from NADP+ (22, 24). The reducing power of NADPH is used for the reduction of thioredoxin by thioredoxin reductase. If these reducing pathways are suppressed, cellular homeostasis is greatly perturbed. For example, deletion of the gene for thioredoxin reductase significantly suppresses the growth of E. coli under oxidative stress (25, 26).
The activation of EF-G by the reduced form of thioredoxin suggests that the translational machinery might be regulated, via EF-G, by the reducing power that is generated by the pentose phosphate pathway and mediated by thioredoxin in E. coli. Under oxidative conditions, for example, when the supply of reducing power is limited and the production of ROS from respiration is stimulated, the specific pair of cysteine residues in EF-G might be oxidized. As a consequence, both protein synthesis and those aspects of cell metabolism that are supported by protein synthesis would be suppressed.
In the photosynthetic organism Synechocystis, the reducing power that results from the photosynthetic transport of electrons is transmitted to EF-G via thioredoxin and activates the synthesis of proteins de novo, which enhances the repair of PSII. However, under strong light, which induces oxidative stress, the oxidation of EF-G by ROS competes with its reduction, suppressing protein synthesis (7, 11, 27). Thus, the ROS-sensitive cysteine residues in EF-G might play a key role in the regulation of protein synthesis, via either activation or suppression, in both Synechocystis and E. coli.
The locations of cysteine residues are strongly conserved in the EF-G proteins of various prokaryotes, in the chloroplast EF-G proteins of some plants, and in the mitochondrial EF-G proteins of some mammals, such as human and Drosophila (supplemental Fig. S5). The conserved cysteine residues in EF-G might play an evolutionarily conserved and important role in the regulation of the translational machinery.
Improvement of Translation Systems under Oxidizing Conditions
Most translation systems in vitro are susceptible to oxidation, and the appropriate synthesis of proteins under oxidizing conditions is technically difficult. For example, translation systems that are derived from E. coli (12), Drosophila melanogaster (28), wheat germ (29), and the choloroplasts of tobacco (30) require reducing reagents, such as DTT, to drive translational reaction. The human Cu,Zn-superoxide dismutase SOD1 was not synthesized successfully in the translation system of E. coli, mainly because of the absence of the appropriate formation of disulfide bonds (31). The synthesis of active SOD1 has been achieved with an insect translation system from Spodoptera frugiperda 21, which does not require reducing reagents (31).
Replacement of Cys114 by serine in EF-G altered the sensitivity of the entire translation system of E. coli to H2O2 in vitro, and the modified translation system was capable of operating under moderately oxidizing conditions. Appropriate alteration of EF-G in the translational system of E. coli should allow the synthesis of active proteins with the formation of the necessary disulfide bonds.
Supplementary Material
Acknowledgments
We thank Shinichi Takeuchi (Ehime University) and Tomohisa Niimi (Saitama University) for their skilled technical assistance.
This work was supported, in part, by a Grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (21570033 and 24570039) (to Y. N.), the Asahi Glass Foundation (to Y. N.), and the Cooperative Research Program of the Network Joint Research Center for Materials and Devices (to Y. N. and T. H.).

This article contains supplemental “Experimental Procedures” and Figs. S1–S5.
- EF-G
- elongation factor G
- DHFR
- dihydrofolic acid reductase
- DTNB
- 5,5-dithiobis(2-nitrobenzoic acid)
- DTT
- dithiothreitol
- EF-2
- elongation factor 2
- PSII
- photosystem II
- ROS
- reactive oxygen species.
REFERENCES
- 1. Poot M., Verkerk A., Koster J. F., Esterbauer H., Jongkind J. F. (1988) Reversible inhibition of DNA and protein synthesis by cumene hydroperoxide and 4-hydroxynonenal. Mech. Ageing Dev. 43, 1–9 [DOI] [PubMed] [Google Scholar]
- 2. Nilsson A., Nygård O. (1995) Effect of oxidizing agents and haemin on the phosphorylation of eukaryotic elongation factor 2 in rabbit reticulocyte lysates. Biochim. Biophys. Acta 1260, 200–206 [DOI] [PubMed] [Google Scholar]
- 3. Tamarit J., Cabiscol E., Ros J. (1998) Identification of the major oxidatively damaged proteins in Escherichia coli cells exposed to oxidative stress. J. Biol. Chem. 273, 3027–3032 [DOI] [PubMed] [Google Scholar]
- 4. Cabiscol E., Piulats E., Echave P., Herrero E., Ros J. (2000) Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J. Biol. Chem. 275, 27393–27398 [DOI] [PubMed] [Google Scholar]
- 5. Dukan S., Nyström T. (1999) Oxidative stress defense and deterioration of growth-arrested Escherichia coli cells. J. Biol. Chem. 274, 26027–26032 [DOI] [PubMed] [Google Scholar]
- 6. Ayala A., Parrado J., Bougria M., Machado A. (1996) Effect of oxidative stress, produced by cumene hydroperoxide, on the various steps of protein synthesis. Modifications of elongation factor-2. J. Biol. Chem. 271, 23105–23110 [DOI] [PubMed] [Google Scholar]
- 7. Nishiyama Y., Allakhverdiev S. I., Murata N. (2011) Protein synthesis is the primary target of reactive oxygen species in the photoinhibition of photosystem II. Physiol Plant 142, 35–46 [DOI] [PubMed] [Google Scholar]
- 8. Nishiyama Y., Yamamoto H., Allakhverdiev S. I., Inaba M., Yokota A., Murata N. (2001) Oxidative stress inhibits the repair of photodamage to the photosynthetic machinery. EMBO J. 20, 5587–5594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nishiyama Y., Allakhverdiev S. I., Yamamoto H., Hayashi H., Murata N. (2004) Singlet oxygen inhibits the repair of photosystem II by suppressing the translation elongation of the D1 protein in Synechocystis sp. PCC 6803. Biochemistry 43, 11321–11330 [DOI] [PubMed] [Google Scholar]
- 10. Kojima K., Oshita M., Nanjo Y., Kasai K., Tozawa Y., Hayashi H., Nishiyama Y. (2007) Oxidation of elongation factor G inhibits the synthesis of the D1 protein of photosystem II. Mol. Microbiol. 65, 936–947 [DOI] [PubMed] [Google Scholar]
- 11. Kojima K., Motohashi K., Morota T., Oshita M., Hisabori T., Hayashi H., Nishiyama Y. (2009) Regulation of translation by the redox state of elongation factor G in the cyanobacterium Synechocystis sp. PCC 6803. J. Biol. Chem. 284, 18685–18691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T. (2001) Cell-free translation reconstituted with purified components. Nat. Biotechnol. 19, 751–755 [DOI] [PubMed] [Google Scholar]
- 13. Starosta A. L., Qin H., Mikolajka A., Leung G. Y., Schwinghammer K., Nicolaou K. C., Chen D. Y., Cooperman B. S., Wilson D. N. (2009) Identification of distinct thiopeptide-antibiotic precursor lead compounds using translation machinery assays. Chem. Biol. 16, 1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hisabori T., Nishiyama Y. (2009) in Advances in Botanical Research (Jacquot J.-P., ed) Vol. 52, pp. 187–205, Academic Press, Burlington, VT [Google Scholar]
- 15. Hisabori T., Motohashi K., Hosoya-Matsuda N., Ueoka-Nakanishi H., Romano P. G. (2007) Towards a functional dissection of thioredoxin networks in plant cells. Photochem. Photobiol. 83, 145–151 [DOI] [PubMed] [Google Scholar]
- 16. Ramakrishnan V. (2002) Ribosome structure and the mechanism of translation. Cell 108, 557–572 [DOI] [PubMed] [Google Scholar]
- 17. Kadokura H., Beckwith J. (2009) Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell 138, 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roussel X., Béchade G., Kriznik A., Van Dorsselaer A., Sanglier-Cianferani S., Branlant G., Rahuel-Clermont S. (2008) Evidence for the formation of a covalent thiosulfinate intermediate with peroxiredoxin in the catalytic mechanism of sulfiredoxin. J. Biol. Chem. 283, 22371–22382 [DOI] [PubMed] [Google Scholar]
- 19. Yokoyama T., Shaikh T. R., Iwakura N., Kaji H., Kaji A., Agrawal R. K. (2012) Structural insights into initial and intermediate steps of the ribosome-recycling process. EMBO J. 31, 1836–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Inoue-Yokosawa N., Ishikawa C., Kaziro Y. (1974) The role of guanosine triphosphate in translocation reaction catalyzed by elongation factor G. J. Biol. Chem. 249, 4321–4323 [PubMed] [Google Scholar]
- 21. Martemyanov K. A., Liljas A., Yarunin A. S., Gudkov A. T. (2001) Mutations in the G-domain of elongation factor G from Thermus thermophilus affect both its interaction with GTP and fusidic acid. J. Biol. Chem. 276, 28774–28778 [DOI] [PubMed] [Google Scholar]
- 22. Meyer Y., Buchanan B. B., Vignols F., Reichheld J. P. (2009) Thioredoxins and glutaredoxins. Unifying elements in redox biology. Annu. Rev. Genet. 43, 335–367 [DOI] [PubMed] [Google Scholar]
- 23. Kumar J. K., Tabor S., Richardson C. C. (2004) Proteomic analysis of thioredoxin-targeted proteins in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 101, 3759–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Toledano M. B., Kumar C., Le Moan N., Spector D., Tacnet F. (2007) The system biology of thiol redox system in Escherichia coli and yeast. Differential functions in oxidative stress, iron metabolism, and DNA synthesis. FEBS Lett. 581, 3598–3607 [DOI] [PubMed] [Google Scholar]
- 25. Vlamis-Gardikas A., Potamitou A., Zarivach R., Hochman A., Holmgren A. (2002) Characterization of Escherichia coli null mutants for glutaredoxin 2. J. Biol. Chem. 277, 10861–10868 [DOI] [PubMed] [Google Scholar]
- 26. Derman A. I., Prinz W. A., Belin D., Beckwith J. (1993) Mutations that allow disulfide bond formation in the cytoplasm of Escherichia coli. Science 262, 1744–1747 [DOI] [PubMed] [Google Scholar]
- 27. Ejima K., Kawaharada T., Inoue S., Kojima K., Nishiyama Y. (2012) A change in the sensitivity of elongation factor G to oxidation protects photosystem II from photoinhibition in Synechocystis sp. PCC 6803. FEBS Lett. 586, 778–783 [DOI] [PubMed] [Google Scholar]
- 28. Tuschl T., Zamore P. D., Lehmann R., Bartel D. P., Sharp P. A. (1999) Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev. 13, 3191–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Madin K., Sawasaki T., Ogasawara T., Endo Y. (2000) A highly efficient and robust cell-free protein synthesis system prepared from wheat embryos. Plants apparently contain a suicide system directed at ribosomes. Proc. Natl. Acad. Sci. U.S.A. 97, 559–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hirose T., Sugiura M. (1996) Cis-acting elements and trans-acting factors for accurate translation of chloroplast psbA mRNAs. Development of an in vitro translation system from tobacco chloroplasts. EMBO J. 15, 1687–1695 [PMC free article] [PubMed] [Google Scholar]
- 31. Ezure T., Suzuki T., Ando E., Nishimura O., Tsunasawa S. (2009) Expression of human Cu,Zn-superoxide dismutase in an insect cell-free system and its structural analysis by MALDI-TOF MS. J Biotechnol 144, 287–292 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.