

Background: N-Acetylhexosamines are precursors of all major vertebrate glycan types.
Results: Mammalian cells can incorporate N-glycolylgalactosamine (GalNGc) and N-glycolylglucosamine (GlcNGc) into most major cellular glycans and utilize GalNGc for de novo biosynthesis of Neu5Gc.
Conclusion: N-Acetylhexosamine biosynthetic pathways and glycan assembly are mostly tolerant toward the N-glycolyl substituent.
Significance: Catabolism of Neu5Gc might create novel N-glycolylated glycan epitopes in vivo.
Keywords: Carbohydrate Metabolism, Glycobiology, Glycosylation, Mass Spectrometry (MS), Sialic Acid, N-Glycolylgalactosamine, N-Glycolylneuraminic Acid
Abstract
The outermost positions of mammalian cell-surface glycans are predominantly occupied by the sialic acids N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc). To date, hydroxylation of CMP-Neu5Ac resulting in the conversion into CMP-Neu5Gc is the only known enzymatic reaction in mammals to synthesize a monosaccharide carrying an N-glycolyl group. In our accompanying paper (Bergfeld, A. K., Pearce, O. M., Diaz, S. L., Pham, T., and Varki, A. (2012) J. Biol. Chem. 287, jbc.M112.363549), we report a metabolic pathway for degradation of Neu5Gc, demonstrating that N-acetylhexosamine pathways are tolerant toward the N-glycolyl substituent of Neu5Gc breakdown products. In this study, we show that exogenously added N-glycolylgalactosamine (GalNGc) serves as a precursor for Neu5Gc de novo biosynthesis, potentially involving seven distinct mammalian enzymes. Following the GalNAc salvage pathway, UDP-GalNGc is epimerized to UDP-GlcNGc, which might compete with the endogenous UDP-GlcNAc for the sialic acid biosynthetic pathway. Using UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase-deficient cells, we confirm that conversion of GalNGc into Neu5Gc depends on this key enzyme of sialic acid biosynthesis. Furthermore, we demonstrate by mass spectrometry that the metabolic intermediates UDP-GalNGc and UDP-GlcNGc serve as substrates for assembly of most major classes of cellular glycans. We show for the first time incorporation of GalNGc and GlcNGc into chondroitin/dermatan sulfates and heparan sulfates, respectively. As demonstrated by structural analysis, N-glycolylated hexosamines were found in cellular gangliosides and incorporated into Chinese hamster ovary cell O-glycans. Remarkably, GalNAc derivatives altered the overall O-glycosylation pattern as indicated by the occurrence of novel O-glycan structures. This study demonstrates that mammalian N-acetylhexosamine pathways and glycan assembly are surprisingly tolerant toward the N-glycolyl substituent.
Introduction
The cell surface of mammalian cells contains a variety of glycoconjugates, which are involved in modulating and mediating events such as cell-cell, cell-matrix, or cell-molecule interactions that are critical for the function and development of a complex organism (2). N-Glycans are covalently attached to asparagine residues within a specific consensus motif of proteins and share a common GlcNAc2Man3 core structure, which can be processed and/or elongated with additional monosaccharides such as galactose, GlcNAc, fucose, sialic acids, and less commonly by GalNAc (3). The most abundant O-glycans are characterized by an α-GalNAc residue covalently attached to serine or threonine residues of mammalian glycoproteins. GalNAc may be further extended with monosaccharides such as galactose, GlcNAc, fucose, and sialic acids, but not mannose or glucose (4). Many nucleocytoplasmic proteins contain single GlcNAc residues attached to serine or threonine residues (see our accompanying paper (5)). Glycosphingolipids are the most abundant class of glycolipids in mammals and commonly begin with a galactose (GalCer) or glucose (GlcCer) residue being linked to a ceramide backbone. In higher animals, most glycosphingolipids are built on GlcCer and may contain additional galactose, GalNAc, and sialic acid residues (6). Sialic acid-containing glycosphingolipids are referred to as gangliosides. Another class of glycans are glycosaminoglycans that are long unbranched polysaccharides consisting of repeating disaccharide building blocks. Examples are chondroitin sulfates (GalNAc/glucuronic acid), dermatan sulfates (GalNAc/iduronic acid and glucuronic acid), and heparan sulfates (GlcNAc and iduronic acid or glucuronic acid), which are covalently attached to serine residues in proteoglycans and represent a major component of the extracellular matrix (7).
Among the major mammalian classes of glycans, N-glycans, O-glycans, and gangliosides typically carry sialic acids at the outermost positions of a glycan chain (8). Sialic acids are a family of acidic nine-carbon backbone monosaccharides, with N-acetylneuraminic acid (Neu5Ac)2 and N-glycolylneuraminic acid (Neu5Gc) being the most prevalent in mammals. Because of their exposed position and quantity at the cell surface, they are involved in the stabilization of molecules and membranes as well as recognition and modulation of a multitude of cell-cell and cell-environment interactions. The only known cellular pathway for synthesis of Neu5Gc is the conversion of CMP-Neu5Ac into CMP-Neu5Gc catalyzed by the CMP-Neu5Ac hydroxylase (CMAH) enzyme (9–16). In contrast to other mammals studied to date, humans contain an inactivating frameshift mutation in the CMAH gene (17–20), and humans thus lost the ability to synthesize endogenous Neu5Gc. However, Neu5Gc has been found in various human cancers, in fetal meconium, and even in normal human tissues (21–27), apparently originating from Neu5Gc-rich animal-derived dietary sources such as red meats or dairy products (28, 29). Our accompanying paper (30) demonstrated that mice with a human-like defect in the Cmah gene incorporate Neu5Gc from a Neu5Gc-rich diet. All humans tested thus far display a polyclonal circulating anti-Neu5Gc antibody repertoire (31, 32), which suggests that Neu5Gc might be the first known example of a “xeno-autoantigen” (33).
As the CMAH enzyme is the only known mammalian biosynthetic pathway to synthesize a sugar carrying an N-glycolyl group, Neu5Gc is the only logical source for any other amino sugar carrying an N-glycolyl group. Thus far, uptake and recycling of Neu5Gc through the lysosomal transporter sialin have been well studied (29, 34), but turnover and putative degradative pathways are just beginning to be explored. In our accompanying paper (1), we report a pathway of Neu5Gc degradation. Therein, we show that mammalian cells can convert Neu5Gc into ManNGc, followed by epimerization to GlcNGc and phosphorylation into GlcNGc 6-phosphate, with final de-acylation of the latter, resulting in formation of the ubiquitous cellular metabolites glycolate and glucosamine 6-phosphate (1). This finding already suggests a certain level of tolerance of basic metabolic enzymes toward the N-glycolyl substituent and raises the question of whether the N-glycolylated breakdown products of Neu5Gc are able to be salvaged. The GalNAc salvage pathway has already been described to be very efficient in introducing artificial GalNAc derivatives, resulting in modified O-glycans on glycoproteins and nucleocytoplasmic proteins (35–40). In this study, we use exogenous GalNGc for metabolic labeling and demonstrate its incorporation in nearly all of the major glycans present in vertebrates.
EXPERIMENTAL PROCEDURES
Synthesis and Chemical Characterization
“Brine” refers to a saturated aqueous solution of sodium chloride. Proton nuclear magnetic resonance spectra (dH) were recorded on a Jeol ECA 500 (500 MHz). 500 MHz spectra were assigned using COSY. All chemical shifts were quoted on the δ-scale in ppm, using residual solvent as the internal standard. Low resolution mass spectra, obtained at the University of California San Diego, Chemistry and Biochemistry Molecular MS Facility, were recorded on a Micromass Platform 1 spectrometer using electron spray (ES) ionization with methanol as carrier solvent. Flash column chromatography was performed using Sorbsil C60 40/60 silica gel. Thin layer chromatography (tlc) was performed using Merck Kieselgel 60F254 pre-coated aluminum backed plates. Plates were visualized using 5% sulfuric acid in methanol.
2-Deoxy-2-[(hydroxyacetyl)amino]-d-galactopyranose (41)
The hydrochloride salt of 2-amino-2-deoxy-d-galactopyranose (1.0 g, 4.6 mmol) was dissolved in water (60 ml) with sodium bicarbonate (10.8 g, 130 mmol) and cooled in an ice bath. Acetoxyacetyl chloride (4.2 ml, 39 mmol) was added slowly to the reaction mixture. After 30 min, tlc (EtOAc/MeOH, 7:3, v/v) showed complete consumption of the starting material (Rf = 0.0) and the formation of the product (Rf = 0.3). The reaction mixture was neutralized with mixed bed resin and concentrated under vacuum. The product was purified through silica column and concentrated under vacuum to yield a white gum (1.0 g, 4.2 mm, 91%). For 1H NMR (D2O, 500 MHz) (assigned for the major anomer), δ = 3.53 (ddd, J = 1.1 Hz, J = 4.5 Hz, J = 8.0 Hz, 1H, H-6), 3.59–3.62 (m, 2H, H-5, H-6′), 3.64 (dd, J3,4 = 3.7 Hz, J4,5 = 14.0 Hz, 1H, H-4), 3.82 (dd, J3,4 = 2.8 Hz, J2,3 = 11.2 Hz, 1H, H-3), 3.97 (s, 2H, CH2OH), 4.04 (dd, J1,2 = 3.7 Hz, J2,3 = 10.9 Hz, 1H, H-2), 5.09 (d, J1,2 = 3.7 Hz, 1H, H-1). 13C-NMR (D2O, 500 MHz) δ = 53.5, 61.2, 67.4. 68.2, 70.9, 74.9, 95.1 (C-1), 175.8 (C = O). m/z (ESI+) was 260 (M + Na+, 100%), 238 (M + H+, 20%). The HRMS m/z (ES+) calculated for C8H15NO7Na (M + Na+) was 260.0741 and found was 260.0740.
1,3,4,6-Tetra-O-acetyl-2-deoxy-2-[(acetoxyacetyl)amino]-d-galactopyranose
2-Deoxy-2-[(hydroxyacetyl)amino]-d-galactopyranose (200 mg, 0.80 mmol) was mixed with acetic anhydride (5 ml) and pyridine (5 ml) to generate a slurry. After 24 h, tlc (petrol/EtOAc, 9:1) showed complete consumption of the starting material (Rf = 0.0) and formation of the product (Rf = 0.3). The clear/colorless solution was dried under reduced pressure to yield a sticky straw-colored gum. The residue was dissolved in chloroform and washed with sodium bicarbonate, water, and finally brine. The solution was then dried with sodium sulfite, filtered, and dried under vacuum. The crude product was purified through a silica column (petrol/EtOAc, 9:1) and concentrated under vacuum to yield a white gum (320 mg, 0.72 mm, 90%). For 1H NMR (CD3Cl, 500 MHz) (assigned for the major anomer), δ = 2.01 (s, 3H, CH3), 2.02 (s, 3H, CH3), 2.03 (s, 3H, CH3), 2.04 (s, 3H, CH3), 2.12 (s, 3H, CH3), 4.08–4.19 (m, 2H), 4.22 (t, J = 6.9 Hz, 1H), 4.52 (s, 2H), 4.65–4.70 (m, 1H), 5.26 (dd, J = 3.1 Hz, J = 11.4 Hz, 1H), and 6.24 (d, J = 3.8 Hz, 1H). For 13C NMR (CD3Cl, 500 MHz), δ = 20.6, 20.7, 49.7, 61.3, 62.8, 66.7, 67.5, 70.2, 74.0, 91.1, 169.0, 167.5, 169.7, 170.6, 171.0, and 171.8. m/z (ESI+) was 470 (M + Na+, 100%) and 465 (M + NH4+, 15%). The HRMS m/z (ES+) calculated for C18H25NO12Na (M + Na+) was 470.1269 and found was 470.1266.
Preparation of [glycolyl-3H]GalNGc
To synthesize [glycolyl-3H]GalNGc, 18.5 MBq of [3H]glycolic acid (prepared as described in the companion paper (1) were incubated with 50 mm galactosamine in the presence of 50 mm ethyl-3-(3-dimethylaminopropyl)-carbodiimide and 2 mm N-hydroxysulfosuccinimide in 50 mm MOPS buffer (pH 7.5) in a final volume of 2 ml. The reaction mixture was incubated overnight at room temperature with gentle mixing, subsequently diluted to 10 ml with water, and applied to a 2 ml Dowex-50 column (AG-50W-X2, Bio-Rad) pre-equilibrated with water. The column was washed with 5 column volumes of water; the washes were pooled with the run-through, and the pool was applied onto a 2 ml AG-3X-4A (OH− form, Bio-Rad) column pre-equilibrated with water. The column was washed with 5 column volumes of water, and the washes were pooled with the run-through and dried down. The recovery was monitored by scintillation counting, and the final yield of [glycolyl-3H]GalNGc was 7.2 MBq (39.0%). The identity of synthesized [glycolyl-3H]GalNGc was confirmed by HPLC confirming co-elution with the nonlabeled chemically well characterized counterpart (data not shown).
Cell Culture
Human THP-I cells (42) as well as murine Emeg32−/− and Emeg32+/− embryonic fibroblasts (43) were cultivated in Dulbecco's modified Eagle's medium (DMEM high glucose, Invitrogen). Human M-21 cells (44) were grown in α-minimal essential medium (Invitrogen), and human B lymphoma cell line BJA-B (Burkitt's lymphoma-like (45), subclones K88 and K20 (46) was kept in RPMI 1640 medium (Invitrogen). Wild type CHO-K1 and mutant ldl-D cell lines (47) were cultured in DMEM/F-12 (Invitrogen). All media were supplemented with 5% Neu5Gc-free human serum (heat-inactivated and sterile-filtered, Valley Biomedical Inc.) instead of fetal bovine serum and 2 mm glutamine. Cells were cultivated in a humidified 5% CO2 atmosphere at 37 °C.
Flow Cytometry
Experiments were performed as described previously (1). For cell feedings, 1 × 106 suspension cells (THP-I, BJA-B subclones K20 or K88) were seeded into 6-well dishes (2 ml of feeding media/well), and adherent cells (CHO-K1, ldl-D, Emeg32−/−, and Emeg32+/− cells) were used in P-100 dishes 3 days prior to reaching confluence again. The feeding media were supplemented with compounds as indicated using the following pH neutral stock solutions either prepared in PBS (pH 7.4) (100 mm Neu5Gc and 1 m GalNGc) or in DMSO (100 mm GalNGc, 100 mm perGalNGc). Regarding cell feedings with GalNGc, the DMSO stock was used when aiming at a final concentration of 100 μm in the media. Cells were cultured for 3 days in the feeding media. Thereafter, adherent cells were detached with 20 mm EDTA in PBS, and all cells were washed three times with PBS.
1,2-Diamino-4,5-methylenedioxybenzene (DMB) Derivatization and HPLC
DMB-HPLC was performed as described in our accompanying paper (30). As starting material, ∼5% of cells from a confluent P-100 dish or 100 μl of a dense suspension cell culture were used in the protocol. Cell lysates thereof were prepared by repetitive freeze-thaw followed by sonication. An aliquot (10%) of each lysate was used to determine the protein concentration using the BCA protein assay kit (Pierce). The remaining lysates were hydrolyzed with 2 m acetic acid to release glycosidically bound Neu5Gc prior to derivatization with DMB and analysis by HPLC. The percentage of Neu5Gc was calculated from total sialic acid content in all samples.
Preparation and Analysis of UDP-sugars from Cells
Two T-75 flasks of BJA-B subclones K20 and K88 were set up each with 5 × 106 cells in 5 ml of culturing media. The media were supplemented with 1.036 MBq [3H]GalNAc (American Radiolabeled Chemicals, Inc.) or [3H]GalNGc, and cells were harvested after 3 days. Cell pellets were washed three times with PBS, resuspended in 30 μl of 10 mm Tris-HCl (pH 7.5), and lysed by repetitive freeze-thaw. After addition of 70 μl of ethanol (70% final) and incubation at −20 °C overnight, proteins and membrane debris were precipitated (20 min/20,000 × g/4 °C). The supernatants were transferred into fresh tubes, and samples were dried down. For analysis, samples were resuspended in a final volume of 110 μl of water, which was supplemented with internal standard [14C]GlcNGc (1). The sample was sterile-filtered prior to HPLC analysis using high performance anion exchange chromatography-pulsed amperometric detection and scintillation counting. Monosaccharides were separated by HPLC (Dionex DX-600 BioLC system equipped with an ED50 Electrochemical Detector, Dionex) using a CarboPac PA-1 column (Dionex) under alkaline conditions. Samples (100 μl) were injected, and an optimized gradient (supplemental Fig. S3) was applied at a flow rate of 1 ml/min, and 0.5-ml fractions were collected directly into scintillation vials. To each vial, 5 ml of scintillation mixture (Scintiverse BD, Fisher) was added prior to scintillation counting. Based on the elution of the internal standard, elution profiles were superimposed.
Isolation of Glycosaminoglycans (GAGs) from Cells
Purification of GAGs was performed based on established protocols (48). For radiolabeling studies, CHO-K1 and ldl-D cells were split into P-100 dishes and cultured in regular media until 3 days prior to reaching confluence. At this time point, the media were supplemented with 925 kBq [3H]GalNAc or [3H]GalNGc. Three days later, cells were detached using 20 mm EDTA in PBS. The culture media were saved, and cells were washed three times with PBS. Nonradioactive experiments were carried out similarly, but P-150 dishes were used, and cells were incubated at a final concentration of 10 mm GalNAc or 10 mm GalNGc. Cell pellets were lysed in 2.5 ml of 0.1 m NaOH for 15 min at 37 °C. After addition of 20 ml of buffer A (50 mm sodium acetate, 200 mm NaCl (pH 6.0)), the pH was adjusted to 8.0 with acetic acid. The pH of the culture media was adjusted to 8.0 as well. Pronase E (Sigma) was added at a final concentration of 100 μg/ml, and samples were incubated for 24 h in a end-over-end shaker. Additional 50 μg/ml of Pronase E was added, and samples were incubated overnight. Samples were boiled for 10 min to inactivate the protease, followed by centrifugation at 3000 × g for 1 h, and passed through a 5 μm low binding syringe filter. A polyprep column (1 ml) was prepared with DEAE-Sephacel (Sigma) and prewashed with 20 ml of buffer B (buffer A + 0.1% w/v Triton X-100) prior to loading the samples. Thereafter, columns were washed with 30 ml of buffer A, and GAGs were subsequently eluted with 5 ml of buffer C (50 mm sodium acetate, 1 m NaCl (pH 6.0)). The eluates were collected and desalted on PD-10 columns (GE Healthcare) following the manufacturer's guidelines. Thereafter, samples were frozen and lyophilized, and material was stored at −20 °C.
CL-6B Gel Filtration Chromatography
Isolated GAGs from radiolabeled cells (see above) were resuspended in 500 μl of water and split equally into two tubes. Samples were diluted 10-fold with buffer (0.5 m Tris-HCl, 0.5 m NaCl (pH 7.9)) and 20 milliunits of chondroitinase ABC (Proteus vulgaris, Sigma) was added to only one of the samples followed by incubation at 37 °C overnight. Thereafter, saturated phenol red solution (sterile filtered) was added to a final volume of 0.5 ml to each sample. A gel filtration column (1.7 cm outer diameter by 42 cm, CL-6B, Sigma) was poured and equilibrated in buffer A. Samples were loaded and column run with ∼40 μl/min using buffer A. Fractions were collected (15 min each, ∼120 fractions total), and radioactivity was determined by scintillation counting. The elution time of phenol red was set to 1, and relative elution times of all fractions were calculated accordingly.
LC/MS Analysis of GAGs
Enzymatically depolymerized GAG preparations were differentially mass labeled by reductive amination with [12C6]aniline as described previously (49). Each sample was mixed with commercially available standard unsaturated disaccharides that were tagged with [13C6]aniline. Samples were analyzed by liquid chromatography-mass spectrometry (LC/MS) using an LTQ Orbitrap Discovery electrospray ionization mass spectrometer (Thermo Scientific) equipped with an Ultimate 3000 quaternary high performance liquid chromatography micro pump (Dionex) and a C-18 reverse-phase micro-bore column (Higgins Analytical) as described previously (49). Disaccharides containing an N-glycolyl group were identified based on their unique mass. The Disaccharide Structure Code previously described (50) was amended to include N-glycolyl moieties denoted with an uppercase letter Q in heparan sulfate-derived disaccharides and a lowercase q in chondroitin sulfate-derived disaccharides. Thus, GlcA-GlcNGc is denoted as G0Q0 and GlcA-GalNGc as G0q0.
Isolation of O-Glycans from Cells
CHO-K1 and ldl-D cells were split into 2× P-150 dishes each and cultured in regular media until 3 days prior to reaching confluency. At this time point, the media were supplemented with 10 mm GalNAc or 10 mm GalNGc (from 1 m neutral stocks in PBS (pH 7.4)). Three days later, cells were detached using 20 mm EDTA in PBS and washed three times with PBS. For the following protocol, 60% of cells from a confluent P-150 dish were resuspended in 600 μl of water, ruptured by sonication, and incubated by stirring overnight at 45 °C in the presence of 50 mm NaOH and 1 m NaBH4 (51). Samples were neutralized by addition of 30% acetic acid on ice and desalted by passing over a 2 ml Dowex AG50–0W-8X column. The flow-through was lyophilized, and borates were removed by repeated co-evaporation under nitrogen flush four times with 2 ml of methanol, five times with 2 ml of 9:1 methanol/acetic acid, and five times with 2 ml of methanol. Finally, the sample was dissolved in 1 ml of water and purified using a C-18 column (100 mg, Waters), which was pre-washed as follows: four times with 1 ml of 10% acetic acid; four times with 1 ml of 50% methanol; four times with 1 ml of methanol; four times with 1 ml of 72% isopropyl alcohol + 28% methanol + 0.1% formic acid; four times with 1 ml of ethyl acetate; four times with 1 ml of chloroform; two times with 1 ml of methanol; four times with 1 ml of 50% methanol; eight times with 1 ml of water. After loading, C-18 column was washed three times with 1 ml of 0.1% TFA to elute released O-glycans, and the sample was lyophilized and stored at −80 °C.
Isolation of N-Glycans from Cells
CHO-K1 and ldl-D cells were split into 2 P-150 dishes each and cultured in regular media until 3 days prior to reaching confluency. At this time point, the media were supplemented with 10 mm GalNAc or 10 mm GalNGc. Three days later, cells were detached using 20 mm EDTA in PBS and washed three times with PBS. For this protocol, 30% of cells from a confluent P-150 dish were resuspended in 200 μl of buffer A (20 mm HEPES, 1% SDS (pH 8.2)) and lysed by sonication. The sample was heated for 20 min at 100 °C. Once the sample cooled down to room temperature, 800 μl of buffer B (20 mm HEPES, 1.25% Nonidet P-40 (pH 8.2)), 2 μl of β-mercaptoethanol, and 20 μl peptide:N-glycosidase F (5 milliunits; prepared as described previously (52, 53)) were added to the sample. The sample was incubated overnight at 37 °C in a head-over-head shaker followed by addition of another 5 μl of peptide:N-glycosidase F and further incubation for 4–5 h at 37 °C. A HyperSep Hypercarb charcoal cartridge (25 mg, Thermo Scientific) was pre-washed successively four times with 1 ml of acetonitrile, four times with 1 ml of 50% acetonitrile, and eight times with 1 ml of 0.1% TFA, and a C-18 column was pre-washed as described above under “Isolation of O-Glycans.” The sample was loaded onto C-18 column, allowed to elute directly onto the charcoal cartridge, and let pass through both columns completely. Columns were washed two times with 1 ml of water, and the upper C-18 was discarded thereafter. The charcoal cartridge was washed with another 1 ml of water, and the N-glycans were eluted with two times with 1 ml of 30% acetonitrile, two times with 1 ml of 60% acetonitrile, and 1 ml of 100% acetonitrile. The eluates were pooled, and acetonitrile was removed under nitrogen gas followed by lyophilizing the sample.
Permethylation of Glycans
Permethylation was performed based on previously described reports (54). Isolated O-glycans or N-glycans (see above) were dried in a desiccator and resuspended in 300 μl of DMSO; the sample was sealed and stirred for 45 min at RT. A slurry of 2–3 NaOH pellets in ∼500 μl of DMSO was freshly prepared using mortar and pestle and added to each sample. After stirring for 20–30 min at RT, the sample was put on ice prior to addition of 250 μl of CH3I to the frozen sample. The reaction mixture was put on a stirrer at RT for 20 min and then another 50 μl of CH3I was added following 10 min of stirring. The reaction was terminated by careful addition of 1 ml of water to the sample on ice and stirring for 5 min. Thereafter, permethylated N-/O-glycans were extracted by adding 1 ml of chloroform followed by stirring for 5 min and centrifuging at 2000 rpm for 5 min to separate the organic and the aqueous phases. The chloroform layer was washed five times with 1 ml of water to remove salts and DMSO and finally evaporated dry with nitrogen flush. Permethylated samples were stored at −20 °C until used.
Isolation of Gangliosides
M-21 cells were split into 2 P-150 dishes and cultivated in standard media until 3 days prior to reaching confluency. At this point, the media were supplemented with a final concentration of 10 mm GalNAc or 10 mm GalNGc (from 1 m neutral stocks in PBS (pH 7.4)). After 3 days of incubation, cells reached confluency and were detached using 20 mm EDTA in PBS, and the pellet was washed three times with PBS. After addition of 20 volumes of chloroform/methanol, 2:1, cells were mixed on ice for 30 s using the Polytron at high speed. The sample was incubated on ice for 10 min prior to mixing cells again on ice for 30 s using the Polytron at high speed. The sample was spun for 10 min at 2000 × g, and the organic supernatant was transferred into a clean tube. The remaining pellet was extracted again as described above with 20 volumes of chloroform/methanol, 1:1, followed by 20 volumes of chloroform/methanol, 1:2, and finally with 20 volumes of chloroform/methanol/water, 10:10:1. All extracts were pooled (pool 1). The remaining pellet was dissolved in 3 ml of 95% ethanol, stirred at 4 °C overnight, and spun 10 min at 2000 rpm. The supernatant was added to pool 1, and the original tube was washed with 2 ml of chloroform/methanol, 2:1, prior to adding it to the pellet and mixing. The tube was spun 10 min at 2000 rpm again, and the supernatant was added to pool 1 and dried under nitrogen gas. Total lipid extracts were then resuspended in 500 μl of potassium phosphate buffer (pH 7.5), sonicated for 10 min prior to addition of 6 units of phospholipase C (Sigma), and incubated for 3 h at 37 °C. Thereafter, the sample was dialyzed using 500 molecular weight cutoff tubing (Spectrapore, cellulose acetate) against 4 liters of water at 4 °C overnight. Water was changed once, and dialysis was continued for another 10 h. Sample was recovered from tubing, lyophilized, and dissolved in 1 ml of methanol. Sample was loaded onto a 1 ml DEAE-A25 column, which had been pre-equilibrated three times with 1 ml of methanol. Neutral glycolipids were eluted five times with 1 ml of methanol; monosialylgangliosides were eluted five times with 1 ml of 10 mm ammonium acetate in methanol, and disialylgangliosides were eluted with eluted five times with 1 ml of 200 mm ammonium acetate in methanol. An equal volume of water was added to each of the eluates; samples were purified using a C-18 column as described (48), and C-18 eluates were dried down with nitrogen gas.
MALDI Analysis
Permethylated or native N-/O-glycan samples were dried to completion before dissolving in methanol (55). The glycan sample was then mixed 1:1 (v/v) with super DHB matrix (Sigma; prepared by mixing a 1:5 molar mixture of 5-methylsalicylic acid and 2,5-dihydroxybenzoic acid in absolute methanol) and spotted on a stainless steel MALDI plate. The MALDI was run on an Applied Biosystem Q-StarXL instrument in positive mode. The collision-induced dissociation was done using argon as collision gas. The ganglioside samples were dissolved in 2:1 chloroform/methanol and mixed with 0.5 m trihydroxyacetophenone in a 1:1 (v/v) ratio. The mass spectral data of the ganglioside samples were acquired in both positive and negative ion mode. The MS/MS of selected ions was also performed in the negative ion mode using argon as collision gas. The complete set of data were analyzed using Data Explorer software from Applied Biosystem and GlycoWorkbench software (56).
RESULTS AND DISCUSSION
Mammalian Cells Are Capable of Converting Incorporated GalNGc into Neu5Gc
In our accompanying paper (1), we describe a metabolic pathway for degradation of Neu5Gc. We also demonstrated that the Neu5Gc-degrading pathway is partially reversible in that exogenously added GlcNGc or ManNGc resulted in de novo biosynthesis of Neu5Gc (1). As a presumed “negative control,” we had synthesized GalNGc, i.e. galactosamine carrying an N-glycolyl group. Surprisingly, we observed that supplemented GalNGc was not only incorporated by mammalian cells but also resulted in de novo Neu5Gc biosynthesis in various cell lines. This unanticipated finding prompted an additional study to further investigate the tolerance of mammalian N-acetylhexosamine pathways toward the N-glycolyl substituent, as presented in this paper.
Human THP-I cells were kept under Neu5Gc-free conditions, and the media were supplemented with either GalNGc or perGalNGc for 3 days. In parallel, non-fed cells were kept in culture to serve as a negative control, and Neu5Gc-loaded cells were used as a positive control. Loaded cells were analyzed by flow cytometry using anti-Neu5Gc IgY (1, 27). These human cells were able to use GalNGc as a precursor for de novo biosynthesis of Neu5Gc. Although 100 μm GalNGc in the feeding media was not sufficient to achieve detectable levels of cell-surface-exposed Neu5Gc, 100 μm perGalNGc resulted in a significant shift of the signal (Fig. 1A). Increasing the unmodified GalNGc concentration in the medium to 10 mm also resulted in significant synthesis of cell-surface Neu5Gc, confirming that peracetylation significantly enhances uptake and utilization of sugars (Fig. 1A) (57). This finding was further confirmed by analyzing acid-treated total cell lysates by DMB-HPLC, in which Neu5Gc was chemically detected (supplemental Fig. S1).
FIGURE 1.

Mammalian cells can incorporate GalNGc from the medium, convert it into Neu5Gc, and express it on the cell surface. A, human THP-I cells were cultivated under Neu5Gc-free conditions using 5% human serum. Thereafter, the growth medium was supplemented with either 5 mm Neu5Gc (positive control; shaded dark gray), 100 μm GalNGc (light blue line), 10 mm GalNGc (magenta line), or 100 μm per-O-acetylated GalNGc (perGalNGc, dark blue line). In parallel, cells were kept in culture without supplementation (negative control; black line). After 3 days, cells were harvested and analyzed by flow cytometry using polyclonal chicken anti-Neu5Gc IgY (27) for sensitive detection of cell-surface glycosidically bound Neu5Gc. As an additional negative control, cells fed 5 mm Neu5Gc were also stained with control chicken IgY antibody (control IgY; shaded light gray) to verify the absence of Neu5Gc on human THP-I cells. Also, EMeg32+/− (B) and EMeg32−/− cells (C) were analyzed for their ability to convert supplemented GalNGc into Neu5Gc and subsequently express Neu5Gc on the cell-surface by flow cytometry.
Possible Mammalian Cellular Pathway for the Conversion of GalNGc into Neu5Gc
Based on the well studied mammalian pathways of N-acetylhexosamines, we propose a putative metabolic pathway for enzymatic conversion of GalNGc into Neu5Gc (Fig. 2) including the following seven steps. 1) Phosphorylation of supplemented GalNGc by GalNAc kinase (GALK2, EC 2.7.1.157) resulted in the formation of GalNGc-1-P (58). 2) Subsequent conversion of GalNGc-1-P into UDP-GalNGc potentially catalyzed by UDP-N-acetylglucosamine pyrophosphorylase (AGX2, EC 2.7.7.23) (59); followed by 3) epimerization into UDP-GlcNGc by action of the UDP-GlcNAc 4-epimerase (EC 5.1.3.7) (60). 4/5) Next, bifunctional UDP-N-acetylglucosamine 2-epimerase/N-acylmannosamine kinase (GNE, EC 5.1.3.14 and EC 2.7.1.60) might catalyze conversion of UDP-GlcNGc to ManNGc-6-P via ManNGc (61). 6) Aldol condensation of ManNGc-6-P was with phosphoenolpyruvate, which may be catalyzed by Neu5Ac 9-phosphate synthase (EC 2.5.1.57) (62). 7) The final step would involve dephosphorylation of Neu5Gc 9-phosphate to result in Neu5Gc, potentially catalyzed by Neu5Ac 9-phosphate phosphatase (EC 3.1.3.29) (63). A recent publication revealed that metabolic cross-talk allowed exploitation of the GalNAc salvage pathway to tag and identify O-GlcNAcylated proteins (64). This study clearly demonstrated that synthetic GalNAz (N-azidoacetylgalactosamine) is converted into UDP-GalNAz, followed by epimerization to UDP-GlcNAz (64). Given the similar size of GalNAz and GalNGc, these data suggested that the N-glycolyl group might be tolerated as well by the first three steps of the predicted pathway from GalNGc toward Neu5Gc. To further study this putative pathway, we next analyzed Emeg32−/− mouse embryonic fibroblasts, which lack the glucosamine-6-phosphate acetyltransferase (emeg32) activity (5, 43). Because of a markedly reduced UDP-GlcNAc pool, the suggested pathway for conversion of GalNGc into Neu5Gc might be affected as well. EMeg32+/− cells, which were found to have normal UDP-GlcNAc levels, and mutant EMeg32−/− cells were cultured under Neu5Gc-free conditions using human serum, and the medium was supplemented with GalNGc or perGalNGc for 3 days. Loaded cells were analyzed by flow cytometry using anti-Neu5Gc IgY as described above (1, 27). Despite the presence of an intact Cmah gene in murine cells, Cmah expression and base-line endogenous Neu5Gc levels were found to be negligible, as demonstrated by the overlying flow cytometry signals of non-fed cells with the negative control cells (control IgY-stained Neu5Gc-fed cells, see Fig. 1, B and C). In contrast, supplementation with 100 μm perGalNGc (Fig. 1, B and C, dark blue line) or 10 mm GalNGc (magenta line) resulted in a significant shift of the signal in both EMeg+/− and EMeg−/− cells, into the range of the positive control (5 mm Neu5Gc). Interestingly, as little as 100 μm GalNGc (turquoise line) was sufficient to achieve Neu5Gc de novo biosynthesis in EMeg−/− cells (Fig. 1C), whereas EMeg32+/− cells did not reveal detectable amounts of newly synthesized Neu5Gc under these conditions (Fig. 1B). The differences between these two cell lines suggest that the pathway of conversion from GalNGc to Neu5Gc is affected by endogenous UDP-GlcNAc levels. This result further confirms that in addition to human THP-I cells, mouse cells are also capable of incorporating GalNGc and converting it into Neu5Gc. A potentially valuable cell line to further investigate the putative pathway would be CHO-K1-derived mutant cell line ldl-D (47, 65), which is deficient in UDP-GlcNAc 4-epimerase activity (step 3 of the predicted pathway, Fig. 2), and therefore should not be able to convert GalNGc into Neu5Gc. Unfortunately, these hamster cells were found to already have high endogenous Neu5Gc levels due to CMAH expression, as the non-fed cells and the positive control (5 mm Neu5Gc) have almost comparable levels of Neu5Gc (supplemental Fig. S2).
FIGURE 2.

Conceivable mechanism for conversion of GalNGc into Neu5Gc. Based on the well studied mammalian pathways of N-acetylhexosamines, the depicted metabolic pathway for successive enzymatic conversion of GalNGc into Neu5Gc, including the following 7 steps, is proposed: 1) phosphorylation of supplemented GalNGc by GalNAc kinase (GALK2, EC 2.7.1.157) resulting in the formation of GalNAc-1-P (58); 2) subsequent conversion of GalNAc-1-P into UDP-GalNAc potentially catalyzed by UDP-N-acetylglucosamine pyrophosphorylase (AGX2, EC 2.7.7.23, (59)) followed by 3) epimerization into UDP-GlcNAc by action of the UDP-GlcNAc 4-epimerase (EC 5.1.3.7, (60)). 4/5) Next, bifunctional UDP-N-acetylglucosamine 2-epimerase/N-acylmannosamine kinase (GNE, EC 5.1.3.14 and EC 2.7.1.60) would catalyze conversion of UDP-GlcNAc to ManNGc-6-P via ManNGc (61). 6) Aldol condensation of ManNGc-6-P with phosphoenolpyruvate, which might be catalyzed by Neu5Ac 9-phosphate synthase (EC 2.5.1.57 (62)). 7) Final step would involve dephosphorylation of Neu5Gc 9-phosphate to result Neu5Gc, potentially catalyzed by Neu5Ac 9-phosphate phosphatase (EC 3.1.3.29 (63)).
Conversion of GalNGc into Neu5Gc Is GNE-dependent
Another key enzyme of the predicted pathway would be the bifunctional GNE (UDP-GlcNAc 2-epimerase/ManNAc kinase; steps 4/5, Fig. 2) that acts in an irreversible manner. Therefore, we predicted that human B-cell lymphoma cell line BJAB-K20, which is devoid of UDP-GlcNAc 2′-epimerase activity (46) and lacks endogenous Neu5Gc, would be a valuable tool to study the predicted pathway. In parallel to the mutant BJAB-K20 cells, hypersialylated BJAB-K88 cells were also grown under Neu5Gc-free conditions to serve as a positive control. The media were supplemented with GalNGc, perGalNGc, or Neu5Gc for 3 days, and loaded cells were analyzed for cell-surface Neu5Gc by flow cytometry using anti-Neu5Gc IgY as described above. Control BJAB-K88 cells were found to synthesize cell-surface Neu5Gc after incubation with either 100 μm perGalNGc or 10 mm GalNGc (Fig. 3A), whereas no significant amount of Neu5Gc was detectable in the GNE-deficient BJAB-K20 cells (Fig. 3B). This result was confirmed by direct DMB-HPLC analysis of sialic acids (supplemental Fig. S1). Thus, conversion of exogenously supplied GalNGc to cell-surface Neu5Gc is GNE-dependent, which renders the predicted pathway very likely.
FIGURE 3.

Conversion from GalNGc into Neu5Gc is GNE-dependent. Human BJA-B K20 (A, GNE-deficient) and BJA-B K88 (B, hypersialylated) cells were cultivated under Neu5Gc-free conditions. Thereafter, the feeding media were supplemented with either 5 mm Neu5Gc (positive control; shaded dark gray), 100 μm GalNGc (light blue line), 10 mm GalNGc (magenta line), or 100 μm peracetylated GalNGc (perGalNGc, dark blue line). In parallel, cells were kept in culture without feeding (negative control; black line). After 3 days, cells were harvested and analyzed by flow cytometry using anti-Neu5Gc IgY for detection of cell-surface glycosidically bound Neu5Gc. As an additional negative control, cells fed 5 mm Neu5Gc were also stained with control IgY antibody (shaded light gray) to verify the absence of Neu5Gc on these human cells.
UDP-GalNGc and UDP-GlcNGc Are Formed in Cells
To further substantiate the putative pathway, we next analyzed the occurrence of cytosolic UDP-GalNGc and UDP-GlcNGc (steps 2 and 3 of the predicted pathway, Fig. 2). Therefore, a method was developed to separate cytosolic UDP-sugars by HPLC using a PA-1 column under alkaline conditions (supplemental Fig. S3). As standards, UDP-GalNAc, UDP-GlcNAc, UDP-GalNGc, and UDP-GlcNGc separated well under these conditions (Fig. 4A). To track the formation of UDP-sugars, the culture media of BJAB cells were supplemented with [3H]GalNGc or [3H]GalNAc for 3 days. Harvested cells were lysed followed by protein precipitation, and the supernatants were analyzed by HPLC. Although UDP-sugars in the crude cell lysate did not separate as distinctly as the pure standards (Fig. 4), there were strikingly different elution patterns in cells incubated in the presence of [3H]GalNGc or [3H]GalNAc for the two subclones of BJA-B cells. The majority of recovered counts from [3H]GalNGc-fed cells co-eluted with UDP-GalNGc/UDP-GlcNGc standards, whereas the majority of counts aligned with UDP-GalNAc/UDP-GlcNAc in samples from cells incubated in the presence of [3H]GalNAc (Fig. 4B). This finding showed that human cells are capable of incorporating GalNGc and converting it into UDP-GalNGc as well as UDP-GlcNGc. This observation is consistent with separate observations made in the preceding accompanying papers, wherein we showed that GlcNGc is a metabolite of the Neu5Gc-degrading pathway (1) and that exogenously added GlcNGc can be activated to UDP-GlcNGc and UDP-GalNGc to serve as a substrate to O-GlcNAc transferase (5).
FIGURE 4.
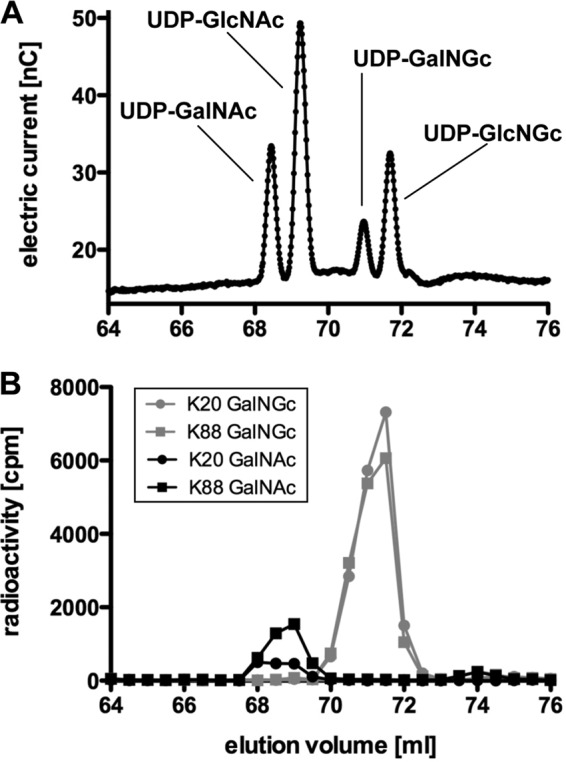
Mammalian cells incorporate exogenous GalNGc into UDP-GalNGc and convert it to UDP-GlcNGc. A, UDP-GalNAc, UDP-GlcNAc, UDP-GalNGc, and UDP-GlcNGc were analyzed by HPLC under alkaline conditions using a PA-1 column with an optimized gradient (supplemental Fig. S3). B, human BJA-B K20 (GNE-deficient) and BJA-B K88 (hypersialylated) cells were cultured under Neu5Gc-free conditions. The media were supplemented with [3H]GalNGc or [3H]GalNAc for 3 days. Cells were harvested, washed well, lysed, and subjected to 70% ethanol followed by precipitation at −20 °C overnight. The supernatants were analyzed by HPLC as described in A. Fractions (0.5 ml) were collected, and radioactivity was determined by scintillation counting.
Cells Incorporate UDP-GalNGc into GAGs
We next asked if GalNGc could be incorporated into various cellular glycoconjugates by supplementing the media of wild type CHO-K1 and ldl-D cells with [3H]GalNGc and control [3H]GalNAc. ldl-D cells are deficient in UDP-GlcNAc 4-epimerase activity, and therefore, they should only synthesize [3H]UDP-GalNGc but not [3H]UDP-GlcNGc from [3H]GalNGc, whereas wild type CHO-K1 cells can potentially synthesize both activated sugars. As only chondroitin sulfate (CS) and dermatan sulfate (DS) polymers contain GalNAc residues among GAGs, only ldl-D cells should incorporate radioactivity into CS/DS, whereas wild type CHO-K1 cells might incorporate counts into both CS/DS and HS. Wild type CHO-K1 and mutant ldl-D cells were therefore cultivated under Neu5Gc-free conditions, and the culture medium was supplemented with [3H]GalNGc or [3H]GalNAc for 3 days. Thereafter, cells were harvested and lysed, and GAGs were purified using DEAE-Sepharose chromatography after proteolysis (48). Desalted GAGs were then treated with chondroitinase ABC to specifically depolymerize CS/DS into disaccharides, which were analyzed by gel filtration and scintillation counting. Both wild type CHO-K1 and ldl-D cells appeared to incorporate the radiolabeled substrates into GAGs as seen by the radioactivity eluting at a relative elution time of ∼0.5–0.8 (black symbols and line) for samples that were not treated with chondroitinase ABC (Fig. 5). Treatment of samples from ldl-D cells with chondroitinase ABC caused a significant shift of counts eluted at a relative elution time of ∼0.9, where disaccharides elute (gray symbols and line; Fig. 5, A and B). This finding indicates that all radioactivity of this sample was incorporated into CS/DS, likely in the form of GalNAc or GalNGc. About 90% of the radioactivity from wild type CHO-K1 cells labeled with [3H]GalNGc was sensitive to chondroitinase ABC treatment (relative elution time = 0.9), whereas a portion eluted as intact chains (relative elution time = 0.6–0.7) (gray symbols and line; Fig. 5, C and D). When cells were labeled with [3H]GalNAc, a large portion of material was resistant to chondroitinase ABC, consistent with its identification as heparan sulfate. These findings suggest that some [3H]GalGc was incorporated into heparan sulfate, which we confirmed by mass spectrometry (see below).
FIGURE 5.

Mammalian cells incorporate exogenous GalNGc into GAGs. Wild type CHO-K1 and ldl-D cells were cultured in Neu5Gc-free medium. The media were then supplemented with [3H]GalNGc or [3H]GalNAc for the last 3 days before cells reached confluence. GAGs were isolated by DEAE chromatography and treated with chondroitinase ABC (gray symbols and line) or buffer (black symbols and line). The disaccharides were separated from the intact chains by CL-6B gel filtration chromatography. Relative elution times of all samples were calculated by setting the elution time of phenol red to 1.
Exogenous GalNAc Derivatives Incorporate into Cellular Chondroitin Sulfate and Dermatan Sulfate Polymers
To further substantiate our finding that exogenously added GalNGc is incorporated into cellular CS/DS, samples were analyzed by mass spectrometry. Wild type CHO-K1 and ldl-D cells were cultured for 3 days in the presence of either 10 mm GalNAc or 10 mm GalNGc. No significant growth difference of the cell lines was observed in the presence of these compounds compared with non-fed cells (data not shown). GAGs of cultures supplemented with GalNGc and control cultures incubated with GalNAc were isolated and depolymerized by chondroitinase ABC, and released disaccharides were analyzed by glycan reductive isotope labeling-liquid chromatography mass spectrometry (GRIL-LC/MS) (49). To identify the resulting disaccharide residues, differentially stable isotope-labeled internal standards of N-acetylated species were added to each sample, and peaks were assigned based on co-elution (49). In samples from wild type CHO-K1 cells cultured in the presence of GalNAc, three primary disaccharides were observed, which all harbor an N-acetyl group and differ only in the presence and/or location of sulfate groups (Fig. 6A). In contrast, CS/DS disaccharides derived from GalNGc-fed cells give rise to two additional disaccharides with m/z values consistent with ΔUA-GalNGc (D0q0) and ΔUA-GalNGc4S (D0q4). Further structural analysis of the most predominant species was carried out by tandem mass spectrometry. The product ions derived from fragmentation of D0q4 (Fig. 6D) were most similar to the product ions of ΔGalNAc4S (D0a4, Fig. 6C), suggesting that it is structurally similar, consistent with its assignment as D0q4 and not another isobaric species. Both spectra show a product ion with identical m/z value consistent with the B1 ion from the nonreducing end (Fig. 6, C and D, see insets). All of the remaining product ions found in D0a4 have corresponding product ions in D0q4 that differ by 16 mass units as expected for species that differ only in the substitution of an N-glycolyl group for an N-acetyl group. Compared with wild type CHO-K1 samples, CS/DS disaccharides from control GalNAc-fed ldl-D cells revealed a similar extracted ion current chromatograph (Fig. 6, A and E). In contrast, ldl-D cells fed GalNGc should not compose any GalNAc-containing CS/DS disaccharides due to the lack of UDP-GlcNAc 4-epimerase activity. As expected, the two CS/DS disaccharides harboring an N-glycolyl group, which were detected in small amounts in samples from wild type cells fed GalNGc (Fig. 6B), became the predominant species found in CS/DS disaccharides from the corresponding ldl-D cell sample (Fig. 6F). As the cell lines were cultured in the presence of 5% human serum at all times, low levels of GalNAc-containing CS/DS disaccharides may result from incorporation of GalNAc-containing serum components. Together, these findings show for the first time that exogenously added GalNAc derivatives can be successfully used to incorporate into cellular GAGs via the GalNAc-salvage pathway. Significant incorporation of exogenous GalNGc into cellular CS/DS was even observed in wild-type cells despite competition with UDP-GalNAc derived from the metabolic flux of the cellular UDP-GlcNAc pool (Fig. 6A).
FIGURE 6.

Disaccharide analysis of chondroitin sulfate extracted from wild type and mutant CHO cells after feeding GalNGc or GalNAc. Wild type CHO-K1 and ldl-D cells were kept under Neu5Gc-free conditions and supplemented with 10 mm GalNGc or 10 mm GalNAc for the last 3 days before cells reach confluence. GAGs were purified from the culture media using DEAE chromatography, and chondroitin sulfate was enzymatically depolymerized and subjected to compositional disaccharide analysis by LC/MS. The extracted ion current for internal disaccharide containing both GalNGc and GalNAc is shown for each cell line and feeding condition. The disaccharide structure code was adapted to include lowercase q to designate N-glycolyl with the rest of the code left as described (50). Peak assignments for N-acetylated species were made based on co-elution and mass analysis of differentially stable isotope-labeled standards added to each sample. A, extracted ion current chromatograph for CHO-K1 cells fed GalNAc. B, extracted ion current chromatograph for wild type CHO-K1 cells fed GalNGc. The peak marked with an asterisk is isobaric with D0q4. C and D, tandem mass spectra of D0a4 and D0q4 in chondroitin sulfate from GalNGc-fed CHO cells. The daughter ion profiles of both D0a4 in GalNAc fed ldl-D cells (C) and the putative D0q4 detected in GalNGc fed ldl-D cells (D) were compared. The overall CID spectrum of the putative D0q4 ion is most similar to that of D0a4 (49). Both spectra show a product ion with identical m/z value consistent with the B1 ion from the nonreducing end (see insets). All of the remaining product ions found in D0a4 have corresponding product ions in D0q4 that differ by 16 mass units expected for species that differ only in the substitution of an N-glycolyl group for an N-acetyl group. E, extracted ion current chromatograph for ldl-D cells fed GalNAc. F, extracted ion current chromatograph for ldl-D cells fed GalNGc. The peak marked with an asterisk is isobaric with D0q4.
Exogenous GalNGc Incorporates as GlcNGc into Cellular Heparan Sulfate Polymers
As described above, only a subfraction of radiolabeled GAGs isolated from CHO-K1 cells fed [3H]GalNGc or control [3H]GalNAc was susceptible to chondroitinase ABC treatment, suggesting that the chondroitinase-resistant radioactivity had been incorporated into GAGs other then CS/DS (i.e. as [3H]GlcNGc or [3H]GlcNAc into heparan sulfate). To verify this conclusion, GAGs were isolated from wild type CHO and ldl-D cells fed either 10 mm GalNAc or GalNGc as described above. Purified GAGs were incubated with heparin lyases to specifically depolymerize heparan sulfate (HS) polymers followed by mass spectrometry analysis to determine the structure of the resulting HS disaccharides. Compositional analysis by GRIL-LC/MS was performed using differentially stable isotope-labeled standards of the N-acetylated species as internal standards. Three HS disaccharides from wild type cells supplemented with 10 mm GalNAc were detected which differed only in the presence and/or positioning of sulfate groups (Fig. 7A). Interestingly, HS disaccharides from wild type cells supplemented with GalNGc contained two additional species, suggesting that UDP-GlcNGc had been synthesized from exogenous GalNGc and was incorporated into cellular HS. The identity of all peaks was confirmed by tandem mass spectrometry using CID as described above. The fragmentation pattern is shown for the two major species harboring either an N-acetyl group (D0A0, Fig. 7C) or an N-glycolyl group (D0Q0, Fig. 7D). The CID profiles suggest that the peaks labeled D0A0 and D0Q0 differ only in the presence of an N-acetyl group or N-glycolyl group, respectively. Both tandem mass spectra reveal three product ions with identical m/z values, which are consistent with the B1, C1, and 2A2 ions from the nonreducing end (Fig. 7, C and D, see insets). For all other product ions found in D0A0, the corresponding product ions are found in D0Q0 that differ by 16 mass units as expected for disaccharides that differ only in the substitution of an N-glycolyl group for an N-acetyl group.
FIGURE 7.

Disaccharide analysis of heparan sulfate extracted from wild type and mutant CHO cells after feeding GalNGc or GalNAc. Wild type CHO-K1 and ldl-D cells were kept under Neu5Gc-free conditions and supplemented with 10 mm GalNGc or 10 mm GalNAc for the last 3 days before cells reach confluence. Heparan sulfate from the medium of both wild type CHO-K1 and ldl-D cells was enzymatically depolymerized and subjected to compositional disaccharide analysis by GRIL-LC/MS. The extracted ion current for internal disaccharide containing both GalNGc and GalNAc is shown for each cell line and feeding condition. Peak assignments for N-acetylated species were made based on co-elution with differentially stable isotope-labeled standards added to each sample. A, extracted ion current chromatograph for wild type CHO-K1 cells fed GalNAc. B, extracted ion current chromatograph for wild type CHO-K1 cells fed GalNGc. C and D, tandem mass spectra of D0A0 and D0Q0 in heparan sulfate from GalNGc fed CHO cells. The daughter ion profiles of both the D0A0 in GalNAc fed CHO-K1 cells (C) and the putative D0Q0 detected in GalNGc fed CHO-K1 cells (D) were compared. The overall CID spectrum of the putative D0Q0 ion is most similar to that of D0A0, suggesting that it is structurally similar and consistent with its assignment as D0Q0 and not another isobaric species. Both spectra show three product ions with identical m/z values consistent with the B1, C1, and 2A2 ions from the nonreducing end (see insets). All of the remaining product ions found in D0A0 have corresponding product ions in D0Q0 that differ by 16 mass units expected for species that differ only in the substitution of an N-glycolyl group for an N-acetyl group. E, extracted ion current chromatograph for ldl-D cells fed GalNAc. F, extracted ion current chromatograph for ldl-D cells fed GalNGc. Species marked in CID spectra with single and double asterisks are corresponding undefined daughter ions that differ by 16 amu.
HS samples from ldl-D cells were expected to contain GlcNAc-containing structures exclusively, because of their inability to epimerize UDP-GalNAc to UDP-GlcNAc. As expected, similar extracted ion current chromatographs were observed for ldl-D cells supplemented with GalNAc or GalNGc, and only GlcNAc-containing HS-disaccharides were detected (Fig. 7, E and F).
Taken together, we show for the first time that exogenously added GalNGc can be successfully incorporated into cellular GAGs via the GalNAc-salvage pathway, not only as GalNGc but also as GlcNGc. Remarkably, a quite significant amount of GlcNGc was detected in HS of wild type CHO-K1 cells after feeding GalNGc (Fig. 7A). This finding means that UDP-GlcNGc resulting from the GalNAc salvage pathway constitutes a significant portion of the central cellular UDP-GlcNAc pool and can be utilized efficiently by the downstream enzymes involved in GAG polymerization. This finding is in line with our earlier observation that exogenously added GalNGc even results in de novo Neu5Gc biosynthesis (Fig. 1), which critically depends on the cellular UDP-GlcNAc pool (Fig. 2). Thus, the UDP-GlcNGc levels must be significant as well.
Exogenously Added GalNGc Incorporates into Cellular Gangliosides
Gangliosides represent another major class of cellular glycan structures, which may include GalNAc residues. Gangliosides of human myeloma cell line M-21 have been well characterized in the past (44) to include GalNAc-containing gangliosides. Thus, this cell line was chosen to investigate if cells incorporate exogenously provided GalNGc into gangliosides. M-21 cells were cultured for 3 days in the presence of 10 mm GalNAc or 10 mm GalNGc followed by purification of cellular gangliosides as described under “Experimental Procedures.” Isolated gangliosides from loaded M-21 cells were than analyzed by MALDI. The resulting MALDI spectra from gangliosides varied significantly between GalNAc-fed cells and GalNGc-fed cells. The predominant ion detected for GalNAc-fed cells (m/z 1354.98, Fig. 8A) most likely corresponds to GM2 as shown by MS/MS fragmentation analysis (Fig. 8B). By contrast, the major ion found among purified gangliosides from GalNGc-fed M-21 cells (m/z 1370.96, Fig. 8C) differs by +16 mass units, which would be expected for species that differ only in the substitution of an N-glycolyl group for an N-acetyl group. MS/MS fragmentation analysis of these two ions clearly shows that major fragment ions harboring a GalNAc residue for the m/z 1354.98 species (m/z 202.15 or m/z 1083.89; Fig. 8B) are shifted by +16 mass units for the m/z 1370.96 ion (m/z 218.15 or m/z 1079.84; Fig. 8D). MS/MS fragmentation patterns of the two structures are similar, suggesting a putative 536 atomic mass units ceramide backbone (supplemental Fig. S4). Together, these findings show for the first time that exogenously supplemented GalNGc may incorporate into cellular gangliosides as shown here for GM2 of M-21 cells.
FIGURE 8.

Exogenous GalNGc is incorporated into cellular gangliosides. Human melanoma cell line M-21 was cultured under Neu5Gc-free conditions. The media were supplemented with 10 mm GalNGc or 10 mm GalNAc for 3 days. Gangliosides were isolated from cell lysates and analyzed by MALDI. The common monosaccharide code (69) was extended by a red square representing N-glycolylhexosamine (HexNGc). A, MALDI spectrum of gangliosides isolated from GalNAc-fed M-21 cells (m/z 1000–2000). B, MS/MS fragmentation pattern of m/z 1354.98 ion from GalNAc-fed ldl-D cells. C, MALDI spectrum of gangliosides isolated from GalNGc-fed M-21 cells (m/z 1000–2000). D, MS/MS fragmentation pattern of m/z 1370.96 ion of gangliosides isolated from GalNGc-fed M-21 cells. Critical MS/MS fragment ions indicating the presence of HexNGc residues are highlighted with arrows.
Exogenously Added GalNGc Incorporates into Cellular O-Glycans but Not N-Glycans
After demonstrating that exogenously added GalNGc serves as a precursor for UDP-GalNGc and UDP-GlcNGc and incorporates in significant amounts into cellular GAG polymers, we next asked whether the unnatural UDP-sugars are also incorporated into other glycan structures. Mammalian N-glycans are characterized by two GlcNAc residues in the core and may be modified further with branched GlcNAc and, to a lesser extent, also outer GalNAc residues. To investigate whether cells would synthesize GlcNGc-containing N-glycans after supplementing the culture media with GalNGc, N-glycans were released by peptide:N-glycosidase F from CHO-K1 and ldl-D cells fed with 10 mm GalNAc or 10 mm GalNGc as described above. Released N-glycans were purified from cell lysates, permethylated, and analyzed by MALDI-qTOF mass spectrometer. Detectable N-glycan structures revealed known CHO cell N-glycans (66), and no significant differences were observed between GalNGc-fed cells and control GalNAc-fed cells (data not shown). Thus, in contrast to GAG and ganglioside biosynthesis, glycosyltransferases involved in N-glycan assembly have a more narrow substrate specificity and cannot easily accommodate the N-glycolyl moiety on HexNGc.
We next investigated if exogenous GalNGc serves as a precursor for cellular O-glycan biosynthesis. The major class of mammalian O-glycans contains a core GalNAc residue, which is attached to the underlying protein structure and may include additional GalNAc as well as GlcNAc residues. O-Glycans were released by alkaline borohydride treatment from CHO-K1 and ldl-D cells fed with 10 mm GalNGc or 10 mm GalNAc as described above. Released O-glycans were purified via a Dowex-50 column, followed by removal of borates and a C-18 column. Isolated O-glycans were permethylated and analyzed by MALDI-qTOF mass spectrometer as described under “Experimental Procedures.” Wild-type CHO cells are known to synthesize a simple set of core 1 based structures, which are mono- or disialylated with Neu5Ac (66). However, culturing CHO cells in the presence of 10 mm GalNAc or GalNGc significantly altered O-glycan biosynthesis, and the resulting mass spectra of CHO-cell O-glycans were more complex. This will require future analyses, which is beyond the scope of this study. Here, we demonstrate incorporation of GalNGc into O-glycans, showing the example of the known tetrasaccharide structure GalNAc1Gal1Neu5Ac2 (m/z 1256.73) (66), which was detected in wild type CHO-K1 cells (data not shown) as well as in ldl-D cells (Fig. 9A) cultured in the presence of 10 mm GalNAc. Tandem mass fragmentation analysis of this ion confirmed the predicted disialylated species GalNAc1Gal1Neu5Ac2 (Fig. 9B). Interestingly, ldl-D cells cultured in the presence of GalNGc revealed an additional peak appearing at m/z 1286.75, which is +30 mass units more than the previously described m/z 1256.73 structure (Fig. 9C). All structures assigned represent sodium adducts of the glycans. For permethylated O-glycans, a 30-mass unit difference would be expected for species that differ only in the substitution of an N-glycolyl group for an N-acetyl group. Besides the exchange of a core GalNAc by GalNGc, this may also reflect substitution of Neu5Gc for Neu5Ac in this disialylated tetrasaccharide. However, MS/MS fragmentation analysis clearly shows that major fragment ions harboring a GalNAc residue (but no sialic acid) for the parent ion with m/z 1256.73 species (m/z 284.18 or m/z 506.31; Fig. 9B) are shifted by +30 mass units for the parent ion with m/z 1286.75 ion (m/z 314.18 or m/z 536.32; Fig. 9D). These data clearly indicate that GalNGc was incorporated as the core residue into CHO cell O-glycans. Furthermore, MS/MS analysis m/z 1256.73 ion from GalNAc-fed cells revealed a significant B1-type ion for terminal Neu5Ac (m/z 398.23; Fig. 9B), but no corresponding structure with terminal Neu5Gc was detectable at a +30 mass, which is in line with previous reports (66). However, MS/MS fragmentation analysis of the m/z 1286.75 ion from GalNGc-fed cells reveals a significant signal for terminal Neu5Gc (m/z 428.24) besides Neu5Ac (m/z 398.23), which indicates that only in GalNGc-fed cells is Neu5Gc incorporated into O-glycans at detectable levels (Fig. 9). This is in line with our previous finding showing that cells can also convert GalNGc into Neu5Gc (Fig. 1), which might result in an increased intracellular pool of Neu5Gc and subsequent incorporation of this sialic acid into O-glycans. The second known O-glycan structure of CHO cells is GalNAc1Gal1Neu5Ac1 (m/z 895.0) (66). In GalNAc-fed ldl-D cells, this ion is observed (m/z 895.53), and the MS/MS fragmentation analysis suggests a linear structure and no unambiguous fragment ions point to a mix with a branched structure as reported for CHO Pro5 cells (Fig. 10, A and B). In contrast to published reports, we also identified another ion at +30 mass difference (m/z 925.55, Fig. 10A) at similar intensity, which corresponds to a GalNAc1Gal1Neu5Gc1 ion as confirmed by MS/MS fragmentation analysis (supplemental Fig. S5). Interestingly, both ions are also present in the GalNGc-fed ldl-D cell, but the ratio between the two structures is significantly altered with m/z 925.55 being the predominant ion. In contrast to GalNAc-fed ldl-D cells, MS/MS fragmentation analysis clearly indicates that m/z 925.55 ion for GalNGc-fed ldl-D cells also included GalNGc1Gal1Neu5Ac1 (critical fragment ions, m/z 550.33 and m/z 398.20; Fig. 10D). Interestingly, GalNGc-fed ldl-D cells seem to have branched rather than linear GalNGc1Gal1Neu5Ac1 O-glycan structures, whereas the Neu5Gc-containing GalNAc1Gal1Neu5Gc1 ion appears to be predominantly branched for both GalNAc- and GalNGc-fed cells (Fig. 10, B and D, and supplemental Fig. S5). Additional novel O-glycan structures were detected in cells cultured in the presence of GalNAc or GalNGc, which were absent in all non-fed CHO cells. For example, significant amounts of m/z 779.47 ion were detected in GalNAc-fed ldl-D cells (Fig. 11A) and at significantly lower intensity also in GalNAc-fed CHO-K1 cells (data not shown). This m/z 779.47 ion was found to correspond to GalNAc1Gal1HexNAc1 by MS/MS fragmentation analysis (Fig. 11B). In contrast, GalNGc-fed ldl-D cells included a +30 mass units ion (m/z 809.49) instead, which corresponds to GalNAc1Gal1HexNGc1 as shown by MS/MS fragmentation analysis (Fig. 11C). The fragment ion pattern indicates that HexNGc is exclusively found as the nonreducing terminal residue but not as the core GalNGc residue (Fig. 11D). Non-fed ldl-D cells should per se be devoid of O-glycans due to the lack of endogenously biosynthesized GalNAc. However, cells were cultured in the presence of 5% human serum at all times, which represents a source of GalNAc and may likely explain the presence of GalNAc-containing O-glycan structures (Figs. 9–11) and GAGs (Figs. 6 and 7) found in GalNGc-fed ldl-D cells. Together, our analysis of CHO cell O-glycans by mass spectrometry strongly suggests that GalNGc can serve as the core GalNGc residue for the two known O-glycan structures and furthermore maybe incorporated as the terminal HexNGc residue onto novel O-glycan structures such as GalNAc1Gal1HexNGc1 (Fig. 11). Future studies will reveal the impact of media supplemented GalNAc derivatives onto the overall complexity of O-glycan structural diversity in cells. After this study was finalized, another research group showed predominantly by lectin binding analyses that various artificial GalNAc derivatives, including GalNGc, seem to incorporate into O-glycans via the GalNAc salvage pathway (36, 37).
FIGURE 9.

Mammalian cells incorporate exogenous GalNGc into O-glycans 1. CHO ldl-D cells were kept under Neu5Gc-free culture conditions. The media were supplemented with 10 mm GalNGc or 10 mm GalNAc for the last 3 days before cells reached confluence. Thereafter, cells were harvested with 20 mm EDTA in PBS, pelleted, and washed well. O-Glycans were released from cell lysates by alkaline borohydrate treatment, and purified O-glycans were permethylated and analyzed by MALDI. N-Glycolylhexosamine (HexNGc) residues are depicted as red squares in addition to the common monosaccharide code (69). A, zoom into MALDI spectrum of O-glycans isolated from GalNAc-fed ldl-D cells (m/z 1230 −1300). Peak assignment was based on earlier reports on CHO cell O-glycans (66) and reassured by MS/MS fragmentation analysis. B, fragmentation pattern of m/z 1256.73 ion from GalNAc-fed ldl-D cells. C, zoom into MALDI spectrum of O-glycans isolated from GalNGc-fed ldl-D cells (m/z 1230–1300). D, MS/MS fragmentation pattern of m/z 1286.75 ion from GalNGc-fed ldl-D cells. The asterisk represents the assumed prevalent structure based on the nature of the fragment ions. Most relevant differences in the MS/MS fragment ions are marked with an arrows.
FIGURE 10.

Mammalian cells incorporate exogenous GalNGc into O-glycans 2. O-Glycans from CHO ldl-D cells were isolated and analyzed by MALDI as described in the legend of Fig. 9. The common monosaccharide code (69) was extended by a red square representing N-glycolylhexosamine (HexNGc). A, zoom into MALDI spectrum of O-glycans isolated from GalNAc-fed ldl-D cells (m/z 850–935). Peak assignment was based on earlier reports on CHO cell O-glycans (66) and reconfirmed by MS/MS fragmentation analysis. B, fragmentation pattern of m/z 895.53 ion from GalNAc-fed ldl-D cells. C, zoom into MALDI spectrum of O-glycans isolated from GalNGc-fed ldl-D cells (m/z 850–935). D, MS/MS fragmentation pattern of m/z 925.55 ion from GalNGc-fed ldl-D cells. MS/MS fragment ions clearly associated with the presence of HexNGc are marked with an arrow.
FIGURE 11.

Exogenously supplemented GalNAc or GalNGc gives rise to novel O-glycan structures in CHO cells. O-Glycans from CHO ldl-D cells were isolated and analyzed by MALDI as described in the legend of Fig. 9. N-Glycolylhexosamine (HexNGc) is depicted as red squares to extend the common monosaccharide code (69). Peak assignment was based on MS/MS fragmentation analysis for these novel O-glycan structures. A, zoom into MALDI spectrum of O-glycans isolated from GalNAc-fed ldl-D cells (m/z 760–850). B, fragmentation pattern of m/z 779.47 ion from GalNAc-fed ldl-D cells. C, zoom into MALDI spectrum of O-glycans isolated from GalNGc-fed ldl-D cells (m/z 760–850). D, MS/MS fragmentation pattern of m/z 809.49 ion from GalNGc-fed ldl-D cells. Most relevant differences in the MS/MS fragment ions pointing toward the presence of HexNGc are highlighted with arrows.
Conclusion and Future Perspectives
In this study, we demonstrate that mammalian cells are able to take up exogenous GalNGc to enter the GalNAc salvage pathway. Here, we show for the first time that exogenously added GalNGc can be incorporated into cellular GAGs. After feeding CHO cells with [3H]GalNGc, we isolated radiolabeled GAGs, which proved to be partially sensitive toward chondroitinase ABC (Fig. 5). Additional structural analysis by mass spectrometry confirmed that significant amounts of GalNGc were incorporated into cellular CS/DS (Fig. 6). Besides glycosaminoglycans, we could also confirm that exogenous GalNGc was incorporated into cellular gangliosides (Fig. 8), which represent the second major glycan species harboring GalNAc residues. Furthermore, we show that cells synthesize UDP-GalNGc and UDP-GlcNGc from exogenous GalNGc (Fig. 4) and confirm by mass spectrometry the presence of significant amounts of GlcNGc in cellular heparan sulfates (Fig. 7) after feeding GalNGc. In addition, our accompanying paper (5) demonstrates that UDP-GlcNGc can also successfully enter the O-GlcNAcylation pathway resulting in O-GlcNGc modifications. We found GalNGc to be incorporated into O-glycans as also reported very recently by another group (36, 37). Going beyond these published reports, we also isolated cellular O-glycans from cells grown in the presence of either GalNAc or GalNGc and performed structural analyses using mass spectrometry. As expected, we found that CHO cells are able to incorporate GalNGc as the core residue at the reducing end of known O-glycan structures (Figs. 9 and 10) (66). However, we also observed additional O-glycan structures that were not found in non-fed CHO cells and thus conclude that exogenously added GalNAc and GalNGc in high concentration could significantly alter the O-glycosylation pattern of mammalian cells. This unexpected finding requires further work and is beyond the scope of this study.
We also found that all analyzed mammalian cells use artificial GalNGc as a source for Neu5Gc de novo biosynthesis (Fig. 1) in a GNE-dependent manner (Fig. 3); therefore, we propose a conceivable pathway requiring seven distinct, basic, and conserved metabolic enzymes to be most tolerant toward the N-glycolyl group (Fig. 2). Although we use exogenously added artificial GalNGc in this study, the amazing tolerance of cellular metabolism toward the N-glycolyl substituent demands an explanation.
The only known mammalian biosynthetic pathway to synthesize a sugar carrying an N-glycolyl group is the conversion of CMP-Neu5Ac into CMP-Neu5Gc (9–16), with Neu5Gc being the only logical source for any other amino sugar carrying an N-glycolyl group. Although the predicted pathway (Fig. 2) is not considered reversible due to involvement of GNE, there are likely other pathways for the turnover of Neu5Gc that are just starting to be exploited. The breakdown of Neu5Gc into ManNGc is well described (67, 68), and in our preceding paper (1), we propose a pathway for degradation of Neu5Gc, also demonstrating subsequent turnover of ManNGc into GlcNGc. In addition, our accompanying paper (5) shows that mammalian cells are able to incorporate exogenous GlcNGc to form UDP-GlcNGc via the GlcNAc salvage pathway and subsequent incorporation as O-GlcNGc modification. Besides degradation of Neu5Gc involving release of the N-glycolyl group (1), the combination of our above findings suggests yet another possible biochemical route for excess cellular Neu5Gc, i.e. conversion into UDP-GlcNGc to be part of the central cellular UDP-GlcNAc pool. From this pool, either incorporation of GlcNGc (and possibly GalNGc) into cellular glycan structures would be plausible and is worthy of further study. In addition, de novo biosynthesis of Neu5Gc in times of need would be conceivable. If this was to happen in vivo, mammalian cells might naturally harbor a so far unnoticed small subset of glycans containing GlcNGc and GalNGc as a result of cellular Neu5Gc turnover. This is beyond the scope of this study, but we are currently investigating the natural occurrence of such glycan structures in mammalian tissues and cells known to be rich in Neu5Gc in the first place. If present in vivo, such GlcNGc- or GalNGc-containing glycan structures are likely of low abundance as they have not yet been described. However, just as Neu5Gc itself represents a diet-acquired xeno-autoantigen in humans (33) with all humans having circulating antibodies against this epitope (31, 32), even low amounts of its breakdown products (GlcNGc and GalNGc) would theoretically represent additional putative diet-acquired xeno-autoantigens in humans. We intend to also address this possibility in our future study.
Interestingly, cellular N-glycans represent the single major class of glycans that did not incorporate detectable amounts of GalNGc or GlcNGc after cells were grown in the presence of GalNGc (data not shown). In the light of all other glycosylation pathways being tolerant toward the N-glycolyl substituent, one can only speculate why glycosyltransferases involved in N-glycan assembly might have a more narrow substrate specificity to exclude N-glycolylated species. One hypothesis would be that even small amounts of N-glycans harboring modified GalNAc or GlcNAc derivatives have the potential to impact critical cellular functions. The more pronounced tolerance of certain metabolic routes toward modified GalNAc or GlcNAc derivatives may also reflect a possible route for cells to handle rare modified or partially damaged amino sugars rather then accumulating them. As glycans such as GAGs are shed from cells in significant amounts, this might be an elegant way for mammalian cells to release modified or damaged amino sugars.
Supplementary Material
Acknowledgments
We thank Monty Krieger of the Massachusetts Institute of Technology for kindly providing ldl-D cells and Tak Mak of the Ontario Cancer Institute for Emeg32 cells. We also thank William C. Lamanna for helpful discussions regarding the purification of glycosaminoglycans. Mass spectrometry analysis of N-glycans, O-glycans, and gangliosides was performed by the Glycotechnology Core Resource at the University of California, San Diego.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM32373 and R01CA38701 (to A. V.). This work was also supported by Deutsche Forschungsgemeinschaft Research Fellowship BE 4643/1-1 (to A. K. B.) and a Cancer Research Institute/Samuel and Ruth Engelberg Fellowship (to O. M. T. P.).

This article contains supplemental Figs. S1–S5.
- Neu5Ac
- N-acetylneuraminic acid
- CID
- collision-induced dissociation
- CMAH
- CMP-Neu5Ac hydroxylase
- CS
- chondroitin sulfate
- DMB
- 1,2-diamino-4,5-methylenedioxybenzene
- DS
- dermatan sulfate
- GAG
- glycosaminoglycan
- GalNAz
- N-azidoacetylgalactosamine
- GalNGc
- N-glycolylgalactosamine
- GlcNGc
- N-glycolylglucosamine
- GNE
- UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase
- Hex
- hexose
- HS
- heparan sulfate
- ManNGc
- N-glycolylmannosamine
- Neu5Gc
- N-glycolylneuraminic acid
- UDP
- uridine diphosphate
- perGalNGc
- per-O-acetylated GalNGc
- GRIL
- glycan reductive isotope labeling.
REFERENCES
- 1. Bergfeld A. K., Pearce O. M., Diaz S. L., Pham T., Varki A. (2012) Metabolism of vertebrate amino sugars with N-glycolyl groups. Elucidating the intracellular fate of the non-human sialic acid N-glycolylneuraminic acid. J. Biol. Chem. 287, jbc.M112.363549(1801) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E. (2009) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 3. Stanley P., Schachter H., Taniguchi N. (2009) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds) pp. 101–114, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 4. Brockhausen I., Schachter H., Stanley P. (2009) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds) pp. 115–128, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 5. Macauley M. S., Chan J., Zandberg W., He Y., Whitworth G. A., Stubbs K. A., Yuzwa S., Varki A., Davies G. J., Vocadlo D. J. (2012) Metabolism of vertebrate amino sugars with N-glycolyl groups. Intracellular O-GlcNGc, UDP-GlcNGc, and the biochemical and structural rationale for the substrate tolerance of O-GlcNAcase. J. Biol. Chem. 287, jbc.M112.363721 (1802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schnaar R. L., Suzuki A., Stanley P. (2009) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds) pp. 129–142, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 7. Esko J. D., Kimata K., Lindahl U. (2009) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds) pp. 229–248, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 8. Cohen M., Varki A. (2010) The sialome. Far more than the sum of its parts. OMICS 14, 455–464 [DOI] [PubMed] [Google Scholar]
- 9. Shaw L., Schauer R. (1988) The biosynthesis of N-glycoloylneuraminic acid occurs by hydroxylation of the CMP-glycoside of N-acetylneuraminic acid. Biol. Chem. Hoppe-Seyler 369, 477–486 [DOI] [PubMed] [Google Scholar]
- 10. Kozutsumi Y., Kawano T., Yamakawa T., Suzuki A. (1990) Participation of cytochrome b5 in CMP-N-acetylneuraminic acid hydroxylation in mouse liver cytosol. J. Biochem. 108, 704–706 [DOI] [PubMed] [Google Scholar]
- 11. Shaw L., Schneckenburger P., Carlsen J., Christiansen K., Schauer R. (1992) Mouse liver cytidine-′-monophosphate-N-acetylneuraminic acid hydroxylase. Catalytic function and regulation. Eur. J. Biochem. 206, 269–277 [DOI] [PubMed] [Google Scholar]
- 12. Kawano T., Kozutsumi Y., Takematsu H., Kawasaki T., Suzuki A. (1993) Regulation of biosynthesis of N-glycolylneuraminic acid-containing glycoconjugates. Characterization of factors required for NADH-dependent cytidine 5′-monophosphate-N-acetylneuraminic acid hydroxylation. Glycoconj. J. 10, 109–115 [DOI] [PubMed] [Google Scholar]
- 13. Shaw L., Schneckenburger P., Schlenzka W., Carlsen J., Christiansen K., Jürgensen D., Schauer R. (1994) CMP-N-acetylneuraminic acid hydroxylase from mouse liver and pig submandibular glands. Interaction with membrane-bound and soluble cytochrome b5-dependent electron transport chains. Eur. J. Biochem. 219, 1001–1011 [DOI] [PubMed] [Google Scholar]
- 14. Takematsu H., Kawano T., Koyama S., Kozutsumi Y., Suzuki A., Kawasaki T. (1994) Reaction mechanism underlying CMP-N-acetylneuraminic acid hydroxylation in mouse liver. Formation of a ternary complex of cytochrome b5, CMP-N-acetylneuraminic acid, and a hydroxylation enzyme. J. Biochem. 115, 381–386 [DOI] [PubMed] [Google Scholar]
- 15. Kawano T., Koyama S., Takematsu H., Kozutsumi Y., Kawasaki H., Kawashima S., Kawasaki T., Suzuki A. (1995) Molecular cloning of cytidine monophospho-N-acetylneuraminic acid hydroxylase. Regulation of species- and tissue-specific expression of N-glycolylneuraminic acid. J. Biol. Chem. 270, 16458–16463 [DOI] [PubMed] [Google Scholar]
- 16. Schlenzka W., Shaw L., Kelm S., Schmidt C. L., Bill E., Trautwein A. X., Lottspeich F., Schauer R. (1996) CMP-N-acetylneuraminic acid hydroxylase. The first cytosolic Rieske iron-sulfur protein to be described in Eukarya. FEBS Lett. 385, 197–200 [DOI] [PubMed] [Google Scholar]
- 17. Chou H. H., Takematsu H., Diaz S., Iber J., Nickerson E., Wright K. L., Muchmore E. A., Nelson D. L., Warren S. T., Varki A. (1998) A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc. Natl. Acad. Sci. U.S.A. 95, 11751–11756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Irie A., Koyama S., Kozutsumi Y., Kawasaki T., Suzuki A. (1998) The molecular basis for the absence of N-glycolylneuraminic acid in humans. J. Biol. Chem. 273, 15866–15871 [DOI] [PubMed] [Google Scholar]
- 19. Hayakawa T., Aki I., Varki A., Satta Y., Takahata N. (2006) Fixation of the human-specific CMP-N-acetylneuraminic acid hydroxylase pseudogene and implications of haplotype diversity for human evolution. Genetics 172, 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Varki A. (2001) N-Glycolylneuraminic acid deficiency in humans. Biochimie 83, 615–622 [DOI] [PubMed] [Google Scholar]
- 21. Higashi H., Hirabayashi Y., Fukui Y., Naiki M., Matsumoto M., Ueda S., Kato S. (1985) Characterization of N-glycolylneuraminic acid-containing gangliosides as tumor-associated Hanganutziu-Deicher antigen in human colon cancer. Cancer Res. 45, 3796–3802 [PubMed] [Google Scholar]
- 22. Miyoshi I., Higashi H., Hirabayashi Y., Kato S., Naiki M. (1986) Detection of 4-O-acetyl-N-glycolylneuraminyl lactosylceramide as one of tumor-associated antigens in human colon cancer tissues by specific antibody. Mol. Immunol. 23, 631–638 [DOI] [PubMed] [Google Scholar]
- 23. Hirabayashi Y., Higashi H., Kato S., Taniguchi M., Matsumoto M. (1987) Occurrence of tumor-associated ganglioside antigens with Hanganutziu-Deicher antigenic activity on human melanomas. Jpn. J. Cancer Res. 78, 614–620 [PubMed] [Google Scholar]
- 24. Kawachi S., Saida T., Uhara H., Uemura K., Taketomi T., Kano K. (1988) Heterophile Hanganutziu-Deicher antigen in ganglioside fractions of human melanoma tissues. Int. Arch. Allergy Appl. Immunol. 85, 381–383 [DOI] [PubMed] [Google Scholar]
- 25. Devine P. L., Clark B. A., Birrell G. W., Layton G. T., Ward B. G., Alewood P. F., McKenzie I. F. (1991) The breast tumor-associated epitope defined by monoclonal antibody 3E1.2 is an O-linked mucin carbohydrate containing N-glycolylneuraminic acid. Cancer Res. 51, 5826–5836 [PubMed] [Google Scholar]
- 26. Malykh Y. N., Schauer R., Shaw L. (2001) N-Glycolylneuraminic acid in human tumors. Biochimie 83, 623–634 [DOI] [PubMed] [Google Scholar]
- 27. Diaz S. L., Padler-Karavani V., Ghaderi D., Hurtado-Ziola N., Yu H., Chen X., Brinkman-Van der Linden E. C., Varki A., Varki N. M. (2009) Sensitive and specific detection of the non-human sialic acid N-glycolylneuraminic acid in human tissues and biotherapeutic products. PLoS ONE 4, e4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tangvoranuntakul P., Gagneux P., Diaz S., Bardor M., Varki N., Varki A., Muchmore E. (2003) Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc. Natl. Acad. Sci. U.S.A. 100, 12045–12050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bardor M., Nguyen D. H., Diaz S., Varki A. (2005) Mechanism of uptake and incorporation of the non-human sialic acid N-glycolylneuraminic acid into human cells. J. Biol. Chem. 280, 4228–4237 [DOI] [PubMed] [Google Scholar]
- 30. Banda K., Gregg C. J., Chow R., Varki N., Varki A. (2012) Metabolism of vertebrate amino sugars with N-glycolyl groups. Mechanisms underlying gastrointestinal incorporation of the non-human sialic acid xeno-autoantigen, N-glycolylneuraminic acid. J. Biol. Chem. 287, J. Biol. Chem.287, jbc.M112.364182 (1803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Padler-Karavani V., Yu H., Cao H., Chokhawala H., Karp F., Varki N., Chen X., Varki A. (2008) Diversity in specificity, abundance, and composition of anti-Neu5Gc antibodies in normal humans. Potential implications for disease. Glycobiology 18, 818–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nguyen D. H., Tangvoranuntakul P., Varki A. (2005) Effects of natural human antibodies against a nonhuman sialic acid that metabolically incorporates into activated and malignant immune cells. J. Immunol. 175, 228–236 [DOI] [PubMed] [Google Scholar]
- 33. Varki N. M., Strobert E., Dick E. J., Jr., Benirschke K., Varki A. (2011) Biomedical differences between human and nonhuman hominids: potential roles for uniquely human aspects of sialic acid biology. Annu. Rev. Pathol. 6, 365–393 [DOI] [PubMed] [Google Scholar]
- 34. Verheijen F. W., Verbeek E., Aula N., Beerens C. E., Havelaar A. C., Joosse M., Peltonen L., Aula P., Galjaard H., van der Spek P. J., Mancini G. M. (1999) A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat. Genet. 23, 462–465 [DOI] [PubMed] [Google Scholar]
- 35. Dube D. H., Prescher J. A., Quang C. N., Bertozzi C. R. (2006) Probing mucin-type O-linked glycosylation in living animals. Proc. Natl. Acad. Sci. U.S.A. 103, 4819–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pouilly S., Piller V., Piller F. (2012) Metabolic glycoengineering through the mammalian GalNAc salvage pathway. FEBS J. 279, 586–598 [DOI] [PubMed] [Google Scholar]
- 37. Pouilly S., Bourgeaux V., Piller F., Piller V. (2012) Evaluation of analogues of GalNAc as substrates for enzymes of the mammalian GalNAc pathway. ACS Chem. Biol. 7, 753–760 [DOI] [PubMed] [Google Scholar]
- 38. Hang H. C., Yu C., Kato D. L., Bertozzi C. R. (2003) A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl. Acad. Sci. U.S.A. 100, 14846–14851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Laughlin S. T., Bertozzi C. R. (2007) Metabolic labeling of glycans with azido sugars and subsequent glycan-profiling and visualization via Staudinger ligation. Nat. Protoc. 2, 2930–2944 [DOI] [PubMed] [Google Scholar]
- 40. Laughlin S. T., Baskin J. M., Amacher S. L., Bertozzi C. R. (2008) In vivo imaging of membrane-associated glycans in developing zebrafish. Science 320, 664–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jourdian G. W., Roseman S. (1962) The sialic acids. II. Preparation of N-glycolylhexosamines, N-glycolylhexosamine 6-phosphates, glycolyl coenzyme A, and glycolyl glutathione. J. Biol. Chem. 237, 2442–2446 [PubMed] [Google Scholar]
- 42. Auwerx J. (1991) The human leukemia cell line, THP-1. A multifaceted model for the study of monocyte-macrophage differentiation. Experientia 47, 22–31 [DOI] [PubMed] [Google Scholar]
- 43. Boehmelt G., Wakeham A., Elia A., Sasaki T., Plyte S., Potter J., Yang Y., Tsang E., Ruland J., Iscove N. N., Dennis J. W., Mak T. W. (2000) Decreased UDP-GlcNAc levels abrogate proliferation control in EMeg32-deficient cells. EMBO J. 19, 5092–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sjoberg E. R., Manzi A. E., Khoo K. H., Dell A., Varki A. (1992) Structural and immunological characterization of O-acetylated GD2. Evidence that GD2 is an acceptor for ganglioside O-acetyltransferase in human melanoma cells. J. Biol. Chem. 267, 16200–16211 [PubMed] [Google Scholar]
- 45. Menezes J., Leibold W., Klein G., Clements G. (1975) Establishment and characterization of an Epstein-Barr virus (EBC)-negative lymphoblastoid B cell line (BJA-B) from an exceptional EBV-genome-negative African Burkitt's lymphoma. Biomedicine 22, 276–284 [PubMed] [Google Scholar]
- 46. Keppler O. T., Hinderlich S., Langner J., Schwartz-Albiez R., Reutter W., Pawlita M. (1999) UDP-GlcNAc 2-epimerase. A regulator of cell-surface sialylation. Science 284, 1372–1376 [DOI] [PubMed] [Google Scholar]
- 47. Kingsley D. M., Krieger M. (1984) Receptor-mediated endocytosis of low density lipoprotein. Somatic cell mutants define multiple genes required for expression of surface-receptor activity. Proc. Natl. Acad. Sci. U.S.A. 81, 5454–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Esko J. D. (2001) Special considerations for proteoglycans and glycosaminoglycans and their purification. Curr. Protoc. Mol. Biol. Chapter 17, Unit 17.2 [DOI] [PubMed] [Google Scholar]
- 49. Lawrence R., Olson S. K., Steele R. E., Wang L., Warrior R., Cummings R. D., Esko J. D. (2008) Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem. 283, 33674–33684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lawrence R., Lu H., Rosenberg R. D., Esko J. D., Zhang L. (2008) Disaccharide structure code for the easy representation of constituent oligosaccharides from glycosaminoglycans. Nat. Methods 5, 291–292 [DOI] [PubMed] [Google Scholar]
- 51. Carlson D. M. (1968) Structures and immunochemical properties of oligosaccharides isolated from pig submaxillary mucins. J. Biol. Chem. 243, 616–626 [PubMed] [Google Scholar]
- 52. Plummer T. H., Jr., Tarentino A. L. (1991) Purification of the oligosaccharide-cleaving enzymes of Flavobacterium meningosepticum. Glycobiology 1, 257–263 [DOI] [PubMed] [Google Scholar]
- 53. Hayes B. K., Freeze H. H., Varki A. (1993) Biosynthesis of oligosaccharides in intact Golgi preparations from rat liver. Analysis of N-linked glycans labeled by UDP-[6-3H]N-acetylglucosamine. J. Biol. Chem. 268, 16139–16154 [PubMed] [Google Scholar]
- 54. Dell A., Khoo K. H., Panico M., McDowell R. A., Etienne A. T., Reason A. J., Morris H. R. (1993) in Glycobiology: A Practical Approach (Fukuda M., Kobata A., eds) pp. 187–222, Oxford University Press, Oxford, UK [Google Scholar]
- 55. Jang-Lee J., North S. J., Sutton-Smith M., Goldberg D., Panico M., Morris H., Haslam S., Dell A. (2006) Glycomic profiling of cells and tissues by mass spectrometry. Fingerprinting and sequencing methodologies. Methods Enzymol. 415, 59–86 [DOI] [PubMed] [Google Scholar]
- 56. Ceroni A., Maass K., Geyer H., Geyer R., Dell A., Haslam S. M. (2008) GlycoWorkbench. A tool for the computer-assisted annotation of mass spectra of glycans. J. Proteome Res. 7, 1650–1659 [DOI] [PubMed] [Google Scholar]
- 57. Sarkar A. K., Fritz T. A., Taylor W. H., Esko J. D. (1995) Disaccharide uptake and priming in animal cells. Inhibition of sialyl Lewis X by acetylated Galβ1→4GlcNAc β-O-naphthalenemethanol. Proc. Natl. Acad. Sci. U.S.A. 92, 3323–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pastuszak I., Drake R., Elbein A. D. (1996) Kidney N-acetylgalactosamine (GalNAc)-1-phosphate kinase, a new pathway of GalNAc activation. J. Biol. Chem. 271, 20776–20782 [DOI] [PubMed] [Google Scholar]
- 59. Wang-Gillam A., Pastuszak I., Stewart M., Drake R. R., Elbein A. D. (2000) Identification and modification of the uridine-binding site of the UDP-GalNAc (GlcNAc) pyrophosphorylase. J. Biol. Chem. 275, 1433–1438 [DOI] [PubMed] [Google Scholar]
- 60. Głowacka D., Zwierz K., Gindzieński A., Gałasiśki W. (1978) The metabolism of UDP-N-acetyl-d-glucosamine in the human gastric mucous membrane. II. The activity of UDP-N-acetylglucosamine 4-epimerase (EC 5.1.3.7.). Biochem. Med. 19, 202–210 [DOI] [PubMed] [Google Scholar]
- 61. Hinderlich S., Stäsche R., Zeitler R., Reutter W. (1997) A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J. Biol. Chem. 272, 24313–24318 [DOI] [PubMed] [Google Scholar]
- 62. Roseman S., Jourdian G. W., Watson D., Rood R. (1961) Enzymatic synthesis of sialic acid 9-phosphates. Proc. Natl. Acad. Sci. U.S.A. 47, 958–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maliekal P., Vertommen D., Delpierre G., Van Schaftingen E. (2006) Identification of the sequence encoding N-acetylneuraminate-9-phosphate phosphatase. Glycobiology 16, 165–172 [DOI] [PubMed] [Google Scholar]
- 64. Boyce M., Carrico I. S., Ganguli A. S., Yu S. H., Hangauer M. J., Hubbard S. C., Kohler J. J., Bertozzi C. R. (2011) Metabolic cross-talk allows labeling of O-linked β-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 3141–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kingsley D. M., Kozarsky K. F., Hobbie L., Krieger M. (1986) Reversible defects in O-linked glycosylation and LDL receptor expression in a UDP-Gal/UDP-GalNAc 4-epimerase-deficient mutant. Cell 44, 749–759 [DOI] [PubMed] [Google Scholar]
- 66. North S. J., Huang H. H., Sundaram S., Jang-Lee J., Etienne A. T., Trollope A., Chalabi S., Dell A., Stanley P., Haslam S. M. (2010) Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J. Biol. Chem. 285, 5759–5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bulai T., Bratosin D., Artenie V., Montreuil J. (2002) Characterization of a sialate pyruvate-lyase in the cytosol of human erythrocytes. Biochimie 84, 655–660 [DOI] [PubMed] [Google Scholar]
- 68. Schauer R., Sommer U., Krüger D., van Unen H., Traving C. (1999) The terminal enzymes of sialic acid metabolism: acylneuraminate pyruvate-lyases. Biosci. Rep. 19, 373–383 [DOI] [PubMed] [Google Scholar]
- 69. Varki A., Sharon N. (2009) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds) pp. 1–22, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.