

Background: Microtubule plus-end-tracking proteins (+TIPs) use microtubule tip localization signals (MtLSs) to target growing microtubule ends in an end-binding protein (EB)-dependent manner.
Results: The data define the sequence determinants of a canonical MtLS.
Conclusion: EB binding affinity and microtubule-tip tracking activity correlate.
Significance: The data provide a basis to carry out genome-wide predictions of novel +TIPs.
Keywords: Cytoskeleton, Microtubules, Protein Chemistry, Protein Domains, Protein Motifs, End-binding Proteins, Microtubule Plus-end-tracking Proteins, Microtubule Tip Localization Signal, SXIP Motifs
Abstract
Microtubule plus-end-tracking proteins (+TIPs) specifically localize to the growing plus-ends of microtubules to regulate microtubule dynamics and functions. A large group of +TIPs contain a short linear motif, SXIP, which is essential for them to bind to end-binding proteins (EBs) and target microtubule ends. The SXIP sequence site thus acts as a widespread microtubule tip localization signal (MtLS). Here we have analyzed the sequence-function relationship of a canonical MtLS. Using synthetic peptide arrays on membrane supports, we identified the residue preferences at each amino acid position of the SXIP motif and its surrounding sequence with respect to EB binding. We further developed an assay based on fluorescence polarization to assess the mechanism of the EB-SXIP interaction and to correlate EB binding and microtubule tip tracking of MtLS sequences from different +TIPs. Finally, we investigated the role of phosphorylation in regulating the EB-SXIP interaction. Together, our results define the sequence determinants of a canonical MtLS and provide the experimental data for bioinformatics approaches to carry out genome-wide predictions of novel +TIPs in multiple organisms.
Introduction
Microtubules are key components of the cytoskeleton. They are highly dynamic protein filaments, which continuously switch between phases of growth and shrinkage (1). This dynamic behavior is fundamental to microtubule function and is essential to control diverse vital cellular processes, including mitosis, cell division, intracellular transport, cell motility, and cell organization. In living cells, microtubule dynamics is regulated both spatially and temporally by a large number of microtubule-associated proteins (2).
Microtubule plus-end-tracking proteins (+TIPs)3 are specialized microtubule-associated proteins that are conserved in all eukaryotes. They are characterized by their preferential accumulation at growing microtubule plus-ends (3). +TIPs regulate microtubule dynamics and mediate the anchorage of microtubules to different cellular structures, including kinetochores and membrane compartments. As such, +TIPs play an important role in almost all microtubule-based cellular processes. Consistent with their importance for normal cell physiology, aberrant +TIP functions are linked to human malignancies and neurodevelopmental disorders (4).
+TIPs comprise a structurally and functionally diverse group of multidomain and/or multisubunit proteins, ranging in size from a few hundred up to thousands of residues (4). An important feature of +TIPs is their remarkable ability to form dynamic interaction networks at growing microtubule ends. In this context, the members of the evolutionarily conserved end-binding protein (EB) family (5) act as master regulators of +TIP networks. EBs form homo- and heterodimers (6–9) and are able to autonomously track growing microtubule ends independently of any binding partner (10, 11). In contrast, most of the remaining +TIPs generally require EBs to efficiently localize to growing microtubule ends (reviewed in Refs. 4 and 12–14).
It has been recently demonstrated that EBs display a highly conserved docking site for multiple +TIPs containing conserved SXIP motifs found in intrinsically disordered sequence regions enriched in basic, serine, and proline residues. The SXIP motif specifically binds to the C-terminal EB homology (EBH) domain of EBs (Fig. 1) and is responsible for the localization of many diverse +TIPs to growing microtubule ends (15). Mutations within the SXIP motif abrogate EB binding and, as a consequence, microtubule plus-end tracking of +TIPs (15). The EB-SXIP complex is stabilized by long range electrostatic attractive interactions and can be regulated by phosphorylation (15, 16). Prominent examples of +TIPs containing the SXIP motif include the tumor suppressor adenomatous polyposis coli, APC (7); the spectraplakin microtubule-actin cross-linking factors, MACF1 and -2 (6); the mitotic centromere-associated kinesin, MCAK (17); CLIP-associating proteins, CLASP1 and -2 (18, 19); the stromal interaction molecule 1, STIM1 (20); SLAIN2 (21); Sentin (22); p140Cap (p130Cas-associated protein)/SNIP (SNAP25-interacting protein) (23); and the budding yeast kinase Aurora/Ipl1p (24, 25). Based on the significant number of proteins that are targeted to microtubule ends by an SXIP-dependent mechanism, its evolutionary conservation and the fact that a green fluorescent protein (GFP) artificially fused to a short SXIP-containing peptide displayed plus-end accumulation, it has been proposed that the SXIP motif acts as a general “microtubule tip localization signal” (MtLS) (15).
FIGURE 1.

X-ray crystal structure of the EB1c-MACFp1 complex. A, representation of the complex between MACFp1 (cyan sticks) and EB1c (transparent surface and schematic representation). The transparent surface of EB1c is color-coded with its electrostatic potential (from −69.74 to 69.74 KbT; red and blue depict negative and positive electrostatic potentials, respectively). Hydrogen bonds are represented with dashed orange lines. The crystal structure of the EB1c-MACFp1 (Protein Data Bank entry 3GJO) complex has been relaxed by running a 1-ns molecular dynamics simulation. The figure was prepared using PyMOL (Schrödinger, LLC). B, schematic diagram of the EB1c-MACFp1 complex. Hydrogen bonds are indicated by dashed lines between atoms, including their distance in Å. Residue labels are shown in black for EB1 and blue for MACFp1. Hydrophobic contacts are represented by arcs with spokes radiating toward the ligand atoms, which are shown with spokes radiating back. The figure has been generated using LIGPLOT (48). The chain identifiers are shown in parentheses for EB1 residue labels.
The list of different +TIP families is still growing and currently comprises more than 20 representatives (12). How many different +TIPs are there in total? Detailed knowledge about MtLSs is expected to help in predicting the +TIP proteome in different species and can lead to the discovery of novel +TIPs. Toward this goal, we intend here to systematically assess the sequence determinants of a canonical MtLS. We started by using synthetic peptide arrays on membrane supports and binding free energy predictions to derive the residue preferences at every amino acid position of an MtLS-containing +TIP fragment. We further developed an in vitro fluorescence polarization assay to obtain detailed mechanistic insights into the interaction between the SXIP motif and EBs. By testing several MtLS-containing protein fragments derived from different +TIPs both in vitro and in vivo, we could establish a correlation between EB binding affinity and microtubule tip accumulation in cells. Finally, we investigated the role of phosphorylation in regulating the EB-SXIP interaction.
EXPERIMENTAL PROCEDURES
Cloning and Protein Purification
Human EB1 (accession number AAC09471) was cloned into a pET3d vector for bacterial expression (New England Biolabs). Expression in Escherichia coli strain BL21(DE3) was performed in autoinduction medium (26) for 40 h at 24 °C. All subsequent purification steps were performed at 4 °C. Cells were pelleted by centrifugation and resuspended in a buffer of 20 mm BisTris, pH 6.0, supplemented with one tablet of protease inhibitor mixture (Complete EDTA-free; Roche Applied Science). Cells were disrupted by mechanical pressure on an Emulsiflex homogenizer (Avestin), and the total protein extract was loaded onto an anion exchange chromatography column (Resource Q; GE Healthcare) equilibrated in the same buffer. The protein was eluted by applying a linear gradient from 0 to 500 mm NaCl over 20 column volumes. Fractions containing EB1 were pooled, concentrated, and applied onto a Superdex 200 column (GE Healthcare), equilibrated with 20 mm Tris-HCl, 300 mm NaCl, pH 7.5. The various human EB1 mutants and the Schizosaccharomyces pombe Mal3p and Arabidopsis thaliana AtEB1A EB1 orthologues were produced in an identical manner. Appropriate buffer pH and matrix used for the ion exchange chromatography step were chosen according to the pI predicted from the sequence of the different EBs.
The various SXIP-containing ∼40-amino acid residue-long polypeptides shown in Fig. 6A were cloned into modified pET15b or pET47b vectors (Invitrogen) using conventional cloning or a positive selection strategy (27), respectively. The SXIP constructs contained a His6 tag followed by thioredoxin (Trx) and either a thrombin or a precision cleavage site at their N termini (sequence of elements: His-Trx-precision cleavage site-SXIP). The dimeric version of TrxMACF (TrxMACF-GCN4 (general control nondepressible 4)) was obtained by introducing the leucine zipper domain of GCN4 (28) at the C terminus of HisTrx-MACF using a PCR- and homologous recombination-based cloning strategy (29). Expression in E. coli strain BL21(DE3) was performed in Luria-Bertani (LB) medium. Cells were grown at 37 °C until an A600 between 0.6 and 0.8 was reached. After induction with 1 mm isopropyl-d-thiogalactopyranoside, the cells were incubated for 1.5 h at 37 °C. The fusion proteins were purified at 4 °C by immobilized affinity chromatography using nickel-Sepharose columns (Invitrogen) followed by size exclusion chromatography (Superdex 75, GE Healthcare) in 20 mm Tris-HCl, pH 7.5, supplemented with 150 mm NaCl.
FIGURE 6.

EB1 binding affinities of different MtLS-containing +TIPs peptides. A, sequence alignment of +TIP fragments encompassing an MtLS: MACF2, accession number NP_899236 (6); APC, accession number P25054 (49); melanophilin, accession number AAH01653 (50); MCAK, accession number NP_006836 (51); Ipl1, accession number P38991 (24, 25); CLASP2, accession number NP_055912 (19); STIM1, accession number Q13586 (20); DDA3, accession number AAN73431 (52); FILIP, accession number NP_056502 (Jiang et al.)4; SLAIN2, accession number NP_065897 (21); Navigator, accession number NP_065176 (53); TIP150 (Tip-interacting protein of 150 kDa), accession number Q5JR59 (54); p140Cap, accession number P30622 (23); and CLIP170, accession number P30622 (55). The sequences are grouped into EB binders (Kd in the range between 1 and 140 μm) and non-binders (Kd in the millimolar range or higher). The residues proposed to compensate for the unfavorable residue at the X position of the SXIP motif in melanophilin, and DDA3 are underlined. APC residues encompassing the SXIP motif that were shown to interact with EB1c by NMR (15) are underlined with a discontinuous line (see “Results” for more details). B, displacement isotherms obtained by FP at 30 °C. Shown is the displacement of EB1-bound FC-MACFp1 by increasing amounts of MACFp1 (circles), APCp1 (squares), TrxMACF (diamonds), TrxAPC (triangles), and Trx alone (inverted triangles). C, binding isotherms obtained by ITC at 25 °C. MACFp1 (circles) or TrxMACF (diamonds) were titrated into a solution containing EB1. Data points (symbols) were fitted (solid line) by using the “one set of sites” binding model, following Equation 7, as described under “Experimental Procedures.” D, displacement isotherms obtained by FP at 25 °C. Shown is the displacement of EB1-bound FC-MACFp1 by increasing amounts of TrxMACF (diamonds), TrxCLASP (triangles), and TrxSLAIN (squares). E, displacement isotherms obtained by FP at 25 °C. Shown is the displacement of EB1-bound FC-MACFp1 by increasing amounts of unlabeled TrxMACF (diamonds), TrxIpl1-p1 (inverted triangles), TrxIpl1-p2 (circles), TrxMACF-GCN4 (triangles), and tandem TrxIpl1-p12 (squares). When shown, error bars represent S.E. values. FP-derived fractional saturation values (symbols) were calculated from the FP data, according to Equation 1, and fitted (solid line) to Equation 4, as described under “Experimental Procedures.”
Protein samples were concentrated with Centricon concentrators (Millipore) to 10–20 mg/ml, aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C until use. Protein concentrations were determined by UV absorption at 280 nm on a Nanodrop spectrophotometer (Thermo Scientific) using the theoretical molar extinction coefficient calculated from the corresponding amino acid sequence.
Peptide Synthesis
Soluble peptides used for biophysical measurements were prepared (50-μmol scale) by automatic solid-phase peptide synthesis on Tentagel-SRam resin (Rapp Polymere, Tübingen, Germany) using the Fmoc chemistry. The chemical reactions were performed in plastic syringes at room temperature on a multipeptide synthesis robot (Syro2000, MultiSynTech, Witten, Germany). Couplings were achieved by reacting 4 eq of Fmoc-AA-OH with 4 eq of benzotriazol-1-yl-oxytripyrroiidinophosphonium hexafluorophosphate and 8 eq of N-methylmorpholine in dimethylformamide. A solution of piperidine (20%) in dimethylformamide was used to remove the Fmoc-protecting group. The final peptides were deprotected and cleaved from the resin using a mixture of 10 ml of TFA, 0.75 g of phenol, 0.5 ml of water, 0.5 ml of methylphenylsulfide, and 0.25 ml of 1,2-ethandithiol. After 3 h at room temperature, the cleavage solution was collected, and the crude peptides were precipitated from the solution with dry ether at 0 °C. HPLC purification and analyses were achieved using a linear solvent gradient: A, 0.05% TFA in water; B, 0.05% TFA in acetonitrile; gradient 5–60% B over 30 min. HPLC conditions were as follows: UV detector 214 nm, RP-18 column. Identity of the peptides was validated by mass spectrometry using MALDI-TOF (VoyagerLT, Applied Biosystems, Weiterstadt, Germany) or electrospray ionization-mass spectrometry (Q-TOFmicroTM, Micromass, Manchester, UK).
Multiangle Light Scattering
Standard multiangle light scattering was performed in 20 mm Tris, pH 7.5, supplemented with 150 mm NaCl using a Superdex 200 analytical size exclusion chromatography column connected in-line to miniDAWN TREOS light scattering and Optilab T-rEX refractive index detectors (Wyatt Technology). Samples of 2–4 mg/ml concentration were used for multiangle light scattering.
Fluorescence Polarization Assay
For assessing the direct binding of FC-MACFp1, a 1.5 μm concentration of the peptide was titrated with increasing concentrations of EB1 (up to 300 μm) in fluorescence polarization (FP) buffer (20 mm Tris-HCl, pH 7.5, supplemented with 150 mm NaCl, 0.05% Tween 20, 0.1% BSA). A total sample volume of 50 μl was dispensed on 384-well black polystyrene microtiter plates (Greiner Bio-one). FP was monitored using 485- and 535-nm polarization filters (20- and 25-nm cut-off, respectively) on a Tecan UltraEvolution system at temperatures calibrated between 26 and 42 °C. Usually, 20 measurements/well were recorded (integration time of 10 μs), and a G value of 0.942 was used. Gain values were adjusted for each plate. Blanks, consisting of buffer and EB1 protein, were subtracted from each data point. Fractional saturation values were calculated from the FP data as follows,
where FS represents the fractional saturation (i.e. [FC-MACFp1]bound/[EB1]total), r is the experimental fluorescence anisotropy value, and rmin and rmax are the minimum and maximum fluorescence anisotropy values, respectively (corresponding to the free and bound states of FC-MACFp1). Assuming a 1:1 stoichiometry (i.e. one MACFp1 per EB1 monomer) and in the absence of cooperativity, the experimental FP data can be fitted to the simple ligand binding equation,
where [EB1]free is the concentration of free monomeric EB1, and Kb is the binding constant of the FC-MACFp1-EB1 interaction. SigmaPlot (Jandel Scientific) was used to simultaneously fit Kb (Kd = 1/Kb), rmax, and rmin to Equation 2.
To perform a competitive displacement experiment, the concentration of EB1 (binding sites) was adjusted to a concentration that was about twice the Kd value. This ensures a high fractional saturation value in the absence of competitor and, thus, an optimal difference for the fluorescent signal between the starting (FC-MACFp1 bound without competitor) and the final (FC-MACFp1 fully displaced by the competitor) states. We optimized these concentrations based on the Z′ factor (see below) and found 1.5 μm FC-MACFp1 and 5 μm EB1 as optimal concentrations. At these concentrations, the initial fractional saturation was estimated to be 0.635, assuming a Kd = 2.3 μm as determined by isothermal titration calorimetry (ITC); see “Results.” Fractional saturation values were calculated from the FP data as follows,
where FS0 is the initial fractional saturation (i.e. where no competitor has been added).
The Ki and EC50 values of unlabeled competitors were calculated using SigmaPlot (Systat Software Inc.) by simultaneously fitting EC50, rmax, and rmin to the equation,
Ki can be easily derived from the next equation (30).
Competitive displacement FP experiments with all HisTrx-tagged SXIP-containing polypeptides were performed at 25 °C in FP buffer on a Cary Eclipse fluorescence spectrophotometer (Varian Inc.) connected to a Microlab500 titrator (Hamilton). The excitation and emission wavelengths were 485 and 530 nm, respectively, and the slit width for both of the wavelengths was 20 nm. The photomultiplier voltage was 600 V. An integration time of 5 s was used for each data point. The G factor of the instrument was determined to be 1.51, which was used for all measurements. The initial concentration of the ligand in the syringe was 500 μm; the cuvette contained 10 μm EB1 (monomer concentration) and 100 nm FC-MACFp1. Data analysis was performed using the software Datafitter.4
Validation of the FP Assay for High Throughput Screening of Chemical Libraries
To evaluate the suitability of the FP displacement assay described above for high throughput screening, we determined its Z′ factor, which is frequently used as a quality indicator (31) and is defined as follows,
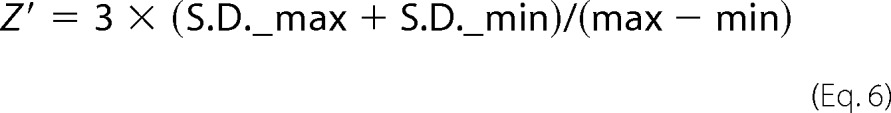 |
where S.D._max and S.D._min are the S.D. values for the positive and negative controls, and max and min are the mean values of the positive and negative controls, respectively. A Z′ value between 0.5 and 1 is indicative of high quality and suitable for high throughput screening (31). Positive (1.5 μm FC-MACFp1 and 5 μm EB1 in the absence of competitor (i.e. corresponding to the bound fluorophor state)) and negative controls (1.5 μm FC-MACFp1, corresponding to the free fluorophor state)) were measured in a 384-well microtiter plate with 50 μl/well (supplemental Fig. 4A). Evaluation of the data yielded a Z′ factor of 0.89, demonstrating the high quality and suitability of the assay for high throughput screening (31).
A significant fraction of compounds present in the most commonly used chemical screening libraries are poorly soluble in water and are thus supplied in DMSO. To test whether DMSO affects the results of the FP assay, we titrated 1.5 μm FC-MACFp1 with increasing concentrations of EB1 and in the presence of different concentrations of DMSO. As shown in supplemental Fig. 4B, only small effects on the binding isotherms were observed for DMSO concentrations up to 10%. We also tested whether DMSO could have an effect on the assay over time. We found that the fluorescence signal is stable in 10% DMSO for at least 3 h (data not shown). Together, these results show that the FP displacement assay is scalable to high throughput and suitable for screening chemical libraries of druglike molecules.
Isothermal Titration Calorimetry
Standard ITC experiments were performed using an ITC200 system (Microcal). Briefly, a 10 μm EB1 solution in PBS buffer (10 mm NaHPO4, 2 mm KHHPO4, 137 mm NaCl, 2.7 mm KCl, pH 7.4) was titrated at 25 °C with increasing concentrations of the various proteins or peptides (0.5–1 mm start concentration). The resulting heats were integrated using Origin (OriginLab) and fitted with the “one set of sites” binding model provided by the software package, following the equation,
SPOT Synthesis
Cellulose membrane-bound peptide arrays were prepared according to standard SPOT synthesis protocols using a SPOT synthesizer as described in detail (32). The syntheses were performed on Whatman-50 cellulose membranes (Whatman, Maidstone, UK) using an automatic SPOT synthesizer (INTAVIS AG, Köln, Germany). The peptides were synthesized on amino-functionalized cellulose membranes of the ester type prepared by modifying a cellulose paper with Fmoc-β-alanine as the first spacer residue. In the second coupling step, the anchor position Fmoc-β-alanine-O-pentafluorophenyl in DMSO was used. Residual amino functions between the spots were capped by acetylation. The Fmoc group was cleaved using 20% piperidine in dimethylformamide. The cellulose-bound peptide arrays were assembled on these membranes by using 0.3 m solutions of Fmoc-amino acid-O-pentafluorophenyl in N-methylpyrolidone. Side-chain protection of the used Fmoc-amino acids was as follows: Glu, Asp (O-tert-butyl); Ser, Thr, Tyr (tert-butyl); His, Lys, Trp (butyloxycarbonyl); Asn, Gln, Cys (trityl); Arg (pentamethyl-5-benzofuranyl). After the last coupling step, the acid-labile protection groups of the amino acid side chains were cleaved using 90% TFA for 30 min and 60% TFA for 3 h.
Binding Assay on Cellulose Membranes
Membrane-bound MACFp1 variants were washed with dimethylformamide and ethanol and three times with TBS buffer (13.7 mm NaCl, 0.27 mm KCl, 5 mm Tris (pH 8.0)) for 10 min each. Membranes were incubated for 3 h with 50 ml of blocking buffer (5 ml of blocking buffer (Sigma-Aldrich) contained 2.5 g of sucrose, 5 ml of 10× TBS buffer (137 mm NaCl, 2.7 mm KCl, 50 mm Tris, pH 8.0), filled up to 50 ml with water). Arrays were incubated with a solution of His-tagged EB1 at a concentration of 10 μg/ml in blocking buffer at room temperature overnight. After three washes with TBS buffer (10 min each), the arrays were incubated with a solution of a mouse anti-polyhistidine IgG antibody (H-1029, Sigma) in blocking buffer (dilution 1:10,000) for 3 h at room temperature and then washed three times with TBS. Afterward, the arrays were treated with a solution of a horseradish peroxidase-conjugated anti-mouse IgG antibody (A-5906, Sigma) in blocking buffer (dilution 1:1000) at room temperature for 1.5 h followed by three times washing with TBS (10 min each). Binding was visualized by using a chemiluminescent substrate (Uptilight HRP, Uptima) and a Lumi-Imager (Roche Applied Science). The signal intensities were recorded as Boehringer light units using the LumiAnalystTM software. Further analysis of spot signal intensities were executed with the Genespotter software (MicroDiscovery GmbH).
Molecular Dynamics (MD) Simulation and Binding Free Energy Calculation
Molecular dynamics simulations were performed with GROMACS (33), using the all atom CHARMM force field (34). All C termini were amidated, and all N termini were acetylated to prevent biologically irrelevant charges. The EB1 C terminus (chain A in Protein Data Bank entry 3GJO) was extended with the two wild-type residues (glutamate and glycine, not defined in the x-ray structure) following Asp-257. The structures were first minimized (500 steps of steepest descent) and then equilibrated (400 ps) at 300 K before starting 1-ns MD simulation for the parent sequence and each mutant (the protein was solvated in a cubic box of TIP3 water molecules, with at least 14 Å distance between the box and the protein, Nose-Hover thermostat and Parrinello-Rahman barostat were used). Binding free energy differences were calculated with the MM-GBSA method (35) by taking 100 equally distributed snapshots along the MD trajectories and averaging the binding free energy (ΔG) between the EB1c-MACFp1 complex and the two separated entities. The final binding free energy differences (ΔΔG) were computed by comparing the results for the parent sequence and each mutant. The contribution of residues 12–19 in MACFp1 mutants was included in the energy calculations because the other flanking amino acids display very large fluctuations or poor electron densities in the crystal structure. Similarly, residues 218, 222, 225, 229, 247–249, and 252–256 in chain A and 212, 213, 216, and 217 in chain B from the EB1 dimer were considered in the energy calculations because they are in direct contact with the ligand. In Fig. 2C, different MACFp1 mutants were selected: first, all mutants with an increased SI values compared with the wild type in the SPOT array; second, all mutants to alanine; and third, a set of 10 mutants manually selected to better sample the range of SPOT signals.
FIGURE 2.

Sequence profiling of a canonical MtLS. A, complete single-point substitution analysis of the peptide MACFp1. Black spots indicate interactions between EB1 and membrane-bound MACFp1 variants. Each spot corresponds to a variant in which one residue of the MACFp1 sequence (given on the left) was replaced by one of the 20 gene-encoded amino acids (shown at the top). Spot SI values were determined by densitometry. The spots in the first column and the ones marked by white crosses represent replicas of the wild type MACFp1 sequence and were used to define the threshold between binding and non-binding (binding spot: SI ≥ ½(mean SI of wild type spots)). The box highlights the sequence 12PSKIPTPQRKSP23, which is the region of MACFp1 most sensitive to substitutions. The SKIP motif (i.e. which corresponds to the SXIP motif of MACFp1) is indicated by a vertical bar on the left. B, percentage of replacement variability (V) at each position of the MACFp1 sequence. The V value was calculated as V = (number of binding spots/20) × 100 and plotted against the MACFp1 sequence. The dashed lines divide sequence regions into low (V ≤ 25%), medium (75% ≥ V ≥ 25%), and high residue variability (V ≥ 75%). C, correlation between the SPOT signal (ln(SI/SIWT) and the predicted binding free energy differences. The dashed line represents the lineal regression of the different data points. The two crossed squares (P16A and K14A) indicate outliers for which the predictions failed.
Cell Culture, Transfections, and Live Cell Imaging
GFP fusions of +TIP fragments were generated using a PCR- and recombination-based cloning strategy, where PCR fragments with homologous flanking sites were integrated into a pEGFP-C2 (Clontech). The dimeric versions of +TIP fragments were obtained by introducing the leucine zipper domain of GCN4 (28) at the N terminus of the respective constructs using a PCR- and homologous recombination-based cloning strategy (29). Expression constructs were transfected into COS-7 cells as described previously (15). Live cell imaging using total internal reflection fluorescence (TIRF) microscopy was performed on an inverted research microscope Nikon Eclipse Ti-E (Nikon) with perfect focus system (Nikon). The microscope was equipped with a Nikon CFI Apo TIRF ×100, 1.49 numerical aperture oil objective (Nikon) and a QuantEM 512SC EMCCD camera (Roper Scientific) and was controlled with the MetaMorph 7.5 software package (Molecular Devices). The 16-bit images were projected onto the charge-coupled device chip with intermediate lens ×2.5 at a magnification of 0.065 μm/pixel. The microscope was further equipped with a TIRF-E motorized TIRF illuminator. Excitation of GFP was performed using a 491-nm 50-milliwatt Calypso (Cobolt) laser; imaging was carried out using an ET-GFP filter set (Chroma). To keep cells at 37 °C, we used a stage top incubator (model INUG2E-ZILCS, Tokai Hit). Images were analyzed with the MetaMorph software package. Live cell images were prepared for publication using Adobe Photoshop. Details of image adjustment are indicated in the figure legends.
RESULTS
Sequence Profiling of the MtLS Sequence from MACF2
To identify the sequence determinants of MtLSs, we employed the technology of synthetic peptide arrays on cellulose membrane supports (SPOT (32, 36, 37)). We used a 30-residue SXIP-containing peptide (MACFp1) derived from the C terminus of MACF2 as the parent sequence for the substitution analysis (residues 5468–5497 of human MACF2). MACFp1 was chosen because high resolution structural information on the peptide in complex with the C-terminal domain of EB1 (EB1c) is available (15). Moreover, fusing the polypeptide sequence corresponding to MACFp1 to the C terminus of GFP is sufficient to target the fluorescent protein to growing microtubule ends (15). MACFp1 thus represents a favorable platform to systematically probe the sequence determinants of a canonical MtLS. For simplicity, the amino acid residues of MACFp1 are numbered 1–30 from here on, whereas the residues of EB1 are numbered according to the human EB1 sequence.
The peptide array shown in Fig. 2A represents a complete amino acid substitution analysis in which all 30 MACFp1 residues were mutated, one at a time, to each of the 20 gene-encoded amino acids. The array was subsequently probed for EB1 binding, which is reflected by black spots of variable intensities. Visual inspection of the SPOT array results reveals that both the N-terminal (residues 1–10) and the C-terminal sequence regions (residues 24–30) of MACFp1 tolerate any substitution. The signal intensities of individual spots (referred to as SI values) can be used as an objective criteria to assess the binding efficiency of peptide variants (38). We defined the SI threshold that discriminates between binding and non-binding as half of the mean value of the wild type SI. Subsequent calculations of the percentage of residue replacement variability (referred to as V value; see Fig. 2B) at each residue position of MACFp1 enables the classification of sequence positions with low (V ≤ 25%), medium (75% ≥ V ≥ 25%) and high residue variability (V ≥ 75%), respectively.
The medium to low V values for spots corresponding to sequence positions 12–23 (with the exception of position 22) indicate that these regions are the most sensitive to substitutions (Fig. 2B). Notably, these positions encompass the SXIP motif of MACFp1 (SKIP; residues 13–16). All four residues of the SKIP sequence are members of the low variability group. Remarkably, the proline of SKIP was found to be invariant. This can be understood by observing that the proline ring fills a hydrophobic pocket in the crystal structure of the EB1c-MACFp1 complex, whereas any other side chain would have the polar amide group of the backbone pointing into this pocket. Moreover, this proline helps in stabilizing the peptide in a polyproline conformation at this position (φ = −41° and ϕ = 126°). The aliphatic hydroxyl-bearing residue serine of the SKIP sequence can only be replaced by its physicochemical similar counterpart threonine. Likewise, the hydrophobic residue isoleucine can be replaced by its physicochemically similar counterpart leucine although at the cost of a significant loss in EB1 binding capacity. This lower affinity can be explained by observing that leucine is a β-branched aliphatic amino acid and thus exhibits a different conformation than isoleucine, which is an aliphatic β-branched residue. Moreover, valine, the other hydrophobic β-branched residue, cannot fill the hydrophobic binding pocket observed in the crystal structure, which results in an unfavorable packing, whereas bulkier hydrophobic side chains (e.g. phenylalanine) generate clashes that destabilize the complex and decrease its binding free energy. Notably, the lysine residue occupying the X position of the SXIP motif in MACFp1 also belongs to the low variability group (Fig. 2B). Because the lysine side chain is not directly pointing toward the EB1 surface, its importance can be explained by long range electrostatic attractive interactions with the negatively charged surface of the C-terminal domain of EB1 (Fig. 1; also see below).
As illustrated by Fig. 2B, the N- and C-terminal SKIP flanking regions of MACFp1 displayed different tolerance to substitutions. With the exception of position 12, the N-terminal proximal region of SKIP tolerates virtually any residue replacement. In contrast, the C-terminal proximal region of SKIP, comprising residue positions 17, 18, and 20, displays medium-to-low variability. This analysis suggests that the 13 residues spanning the sequence region 12–23 of MACFp1 code for the main determinants of the MACFp1-EB1 interaction. It is noteworthy that the replacement of residues in this sequence region by negatively charged glutamates or aspartates severely impaired EB1 binding (Fig. 2A). Basic lysine and arginine residues are preferred instead at many of these positions, including position 14 (the X position of SXIP), and hydrophobic ones are favored at positions 17–19 (Fig. 2A). Based on our substitution analysis, a residue preference list was generated that assigns the probability of finding a residue in a certain position within the core sequence of the MtLS of MACFp1 (Table 1).
TABLE 1.
Residue preference at each amino acid position of a canonical MtLS
The table, which is derived from the SPOT substitution analysis (Fig. 2A), ranks all naturally occurring amino acids for every position of the MtLS of MACFp1. Original MACFp1 residues are highlighted in red.

To complement the SPOT analysis, we performed computational simulations to predict the affinities of selected MACFp1 mutants for EB1. The MACFp1-EB1c complex structure (Protein Data Bank entry 3GJO) was used as a template to generate in silico MACFp1 mutant variants showing either higher or lower EB1 binding compared with the parent peptide. SI values were expressed as the logarithm of the normalized signal intensity ratio of the mutant versus the parent sequence (ln(SI/SIwt)). Free energy differences (ΔΔG) between mutants and parent sequence were evaluated by running 1-ns MD simulations to sample the structural environment and computing the binding free energy differences along these trajectories. As shown in Fig. 2C, the agreement between the experimental SI values and the computational predictions is good, except for two mutants (K14A and P16A). The overall correlation coefficient between SPOT signals and binding free energy differences is 0.58 and rises to 0.76 when these two outliers are excluded. Mutations of Pro-16 probably affect the overall conformation and flexibility of the peptide, an effect that is difficult to detect with short MD simulations. Similarly, Lys-14 makes long range electrostatic interactions with the flexible parts of EB1, such as the C-terminal tail (15), and these interactions are more difficult to accurately estimate in silico.
Our computational analysis enables rationalizing some of the SPOT observations. For instance, only serine and threonine are tolerated at position 13 of MACFp1. We observe that the hydroxyl group of Ser-13 makes very stable contacts with the backbone amide of Ile-15 as well as the carboxylate of Glu-225 on the EB1 protein (Fig. 1 and supplemental Fig. 1, A and B). Position 17 (threonine in the parent sequence) accommodates different residues ranging from polar side chains (threonine or glutamine) to hydrophobic side chains (valine, isoleucine, leucine, or methionine) or to long basic side chains (arginine or lysine). For the latter, our simulations suggest that the positively charged side chain can make transient interactions with negatively charged residues, such as Glu-258, present on the surface of the EB1 dimer (supplemental Fig. 1C). This provides an explanation for the larger diversity of amino acids observed at this position. Finally, position 18 (proline in the parent sequence) displays medium variability in the SPOT data, suggesting that protecting the backbone nitrogen is not a critical feature at this position. In our simulations, we observe that this atom can interact with the polar side chain of either Glu-213 or Tyr-217 on the EB1 dimer, confirming the SPOT observations.
Collectively, these results define the sequence 12PSKIPTPQRKSP23, in particular the SKIP motif, as the most substitution-sensitive region of MACFp1 with respect to EB binding. They further reveal the residue preference at every amino acid position of a canonical MtLS.
Biophysical Analysis of the MACFp1-EB Interaction
To quantitatively assess the interaction between EB and SXIP-containing proteins or peptides, we developed an in vitro assay based on FP. We first assessed the binding properties of 5(6)-carboxyfluorescein (FC) N-terminally labeled MACFp1. Fig. 3A shows the binding isotherm at 30 °C obtained by titrating 1.5 μm FC-MACFp1 with increasing concentrations of EB1 (up to 150 μm). From these data, a dissociation constant, Kd, of 3.2 ± 0.2 μm was calculated, assuming two independent binding sites on the EB1 dimer (7, 8, 15). This value is in good agreement with the one determined by ITC at 25 °C, which yielded a Kd of 5.5 ± 0.1 μm (Fig. 3B). Furthermore, it is consistent with the one determined by ITC using an unlabeled MACFp1 peptide version (Kd = 2.3 ± 0.2 μm; Fig. 3B), demonstrating that the FC label at the N terminus of MACFp1 does not significantly perturb the EB1-MACFp1 binding reaction.
FIGURE 3.

Binding of FC-MACFp1 to EB1. A, binding isotherms obtained by FP at 30 °C. FC-MACFp1 was titrated into a solution containing either wild type EB1 (circles) or EB1-Y217A/E225A (triangles). Fractional saturation values were calculated from the FP data using equation 1 and fitted to the simple ligand binding model described by Equation 2 (see “Experimental Procedures”). B, binding isotherms obtained by ITC at 32 °C. MACFp1 (circles) or FC-MACFp1 (squares) were titrated into a solution containing EB1. Data points were fitted by using the “one set of sites” binding model described by Equation 7 (see “Experimental Procedures”). C, displacement isotherms obtained by FP at 30 °C. Shown is the displacement of EB1-bound FC-MACFp1 by increasing amounts of MACFp1 (circles) or MACFp1-NN (triangles). FP displacement data points were fitted using Equation 4 (see “Experimental Procedures”). D, binding isotherms obtained by FP at 32 °C. FC-MACFp1 was titrated into a solution containing either human EB1 (circles), S. pombe Mal3p (diamonds), or A. thaliana AtEB1A (triangles). When shown, error bars represent S.E. Symbols represent the measured data points; solid lines represent the fit to the data.
To determine the affinity of a non-labeled ligand that competes for the same EB1-binding site as MACFp1, a mixture of FC-MACFp1 (1.5 μm) and EB1 (5 μm) was titrated with increasing concentrations of unlabeled MACFp1 (up to 125 μm). A representative displacement binding isotherm at 30 °C is shown in Fig. 3C. The calculated Kd amounted to 3.5 ± 0.01 μm (Table 2), which is in excellent agreement with the ones determined by direct FP and ITC experiments (see above). To further test the specificity of our FP assay, we used a MACFp1 mutant in which the Ile and Pro residues of its SXIP motif were both mutated to asparagine (MACFp1-NN). This mutation was shown to abrogate EB1 binding and microtubule tip tracking of MACFp1 both in vitro and in cells (15). We further tested an EB1 mutant in which the SXIP-contacting residues Tyr-217 and Glu-225 were simultaneously mutated to alanine (EB1-Y217A/E225A). An analogous EB3 mutant failed to bind MCAK and recruit SXIP +TIPs to growing microtubule tips (17). As shown in Fig. 3C, binding of MACFp1-NN was reduced 70-fold compared with wild type MACFp1 in FP displacement experiments (Table 2). Similarly, binding of FC-MACFp1 to EB1-Y217A/E225A was strongly reduced compared with the binding to wild type EB1 (Fig. 3A), as expected from the loss of crucial hydrogen bond contacts between MACFp1 and EB1 (Fig. 1B). These results underpin the high specificity of our FP assay.
TABLE 2.
Dissociation constants obtained by FP displacement experiments and microtubule tip versus cytoplasm ratios of +TIP-derived polypeptide sequences
| Kd ± S.D. (monomer) | Tip/cytoplasma (monomer) | Tip/cytoplasma (dimer)e | |
|---|---|---|---|
| μm | |||
| MACFp1 | 3.5 ± 0.01 | NDb | ND |
| MACFp1-NN | mmc | No tip tracking | ND |
| APCp1 | 7.5 ± 0.1 | ND | ND |
| TrxMACF | 1.6 ± 0.5 | 1.6 ± 0.4 | 7.0 ± 3 |
| TrxMelan | 1.5 ± 0.4 | 1.5 ± 0.3 | 3.0 ± 1 |
| TrxAPC | 3.3 ± 0.5 | ND | ND |
| TrxMCAK | 10 ± 0.5 | 1.3 ± 0.1 | 2.2 ± 0.7 |
| TrxCLASP2 | 13 ± 0.8 | ND | ND |
| TrxIpl1-p1 | 15 ± 1 | ND | ND |
| TrxDDA3 | 28 ± 1 | 1.4 ± 0.3 | 2.7 ± 0.8 |
| TrxIpl1-p2 | 89 ± 3 | ND | ND |
| TrxFILIP | 126 ± 4 | No tip tracking | 1.2 ± 0.2 |
| TrxSTIM1 | 140 ± 3 | ND | ND |
| TrxSLAIN2 | mm | ND | ND |
| TrxNAV1 | mm | ND | ND |
| TrxTip150 | mm | ND | ND |
| Trxp140Cap | mm | No tip tracking | No tip tracking |
| TrxCLIP-170 | mm | ND | ND |
| TrxIpl1-p12 | 0.15 ± 0.01 | ND | ND |
| TrxMACF-GCN4 | <1 nmd | ND | ND |
a The experiments were performed using N-terminal GFP instead of Trx fusions.
b ND, not determined.
c mm, data could not be subjected to rigorous analysis; however, the shape of the binding isotherms suggested that the Kd values are in the millimolar range or higher.
d Data could not be subjected to rigorous analysis; however, the shape of the binding isotherms suggested that the Kd is below 1 nm.
e Dimerization was achieved by fusing GCN4 to the C-terminus of the indicated constructs.
Next, we tested the binding properties of MACFp1 to EBs from different species. As shown in Fig. 3D, FC-MACFp1 binds to the S. pombe and A. thaliana EB orthologues Mal3p and AtEB1A, respectively, although with slightly weaker affinities as compared with human EB1 (Kd = 10.3 ± 1.1 μm and 56 ± 1 μm for Mal3p and AtEB1A, respectively). These results are consistent with the conserved EBH domain of EBs acting as an SXIP recognition domain and with the observation that SXIP motifs embedded in disordered sequence regions enriched in basic, serine, and proline residues are ubiquitously found in +TIPs from different eukaryotic species. The limited differences in affinities between the EBs can be explained by their similar EBH domains and by the fact that their disordered C-terminal tails, which get ordered upon binding to SXIP motifs (15), vary in length and sequence (7). In line with these results, it has been shown that human EB1 can substitute for the complete loss of the mal3 gene in fission yeast (39).
The enthalpy and entropy changes of the EB1-MACFp1 complex can be either directly assessed by ITC or extracted from the van't Hoff and Gibb's equations using the binding constants determined by our FP displacement assay at different temperatures (Fig. 4, A and B). The EB1-MACFp1 binding reaction was found to be exothermic (ΔHapp = −42.0 ± 0.1 kJ mol−1 and ΔSapp = −37.3 kJ mol−1 K−1, as determined by ITC, and ΔHapp = −20.9 ± 4.3 kJ mol−1 and ΔSapp = −39.5 kJ mol−1 K−1, as calculated from the van't Hoff plot of the FP data) and enthalpy-driven. The differences observed between ITC and FP are probably due to the different experimental conditions used. ITC was performed in PBS, whereas the Tris-based FP assay buffer is supplemented with 0.05% Tween 20 and 1 mg/ml BSA (for details, see “Experimental Procedures”). Despite this limitation, we found that the dissociation constants derived from the two methods do agree well (Kd = 3.5 ± 0.2 μm and 2.3 ± 0.2 μm, measured by FP and ITC, respectively). Moreover, we hypothesize that the highly unfavorable entropic contribution to the binding, derived from both methods, is most probably due to a conformational entropy penalty resulting from the ordering of residues in the C-terminal tail of EB1 upon peptide binding and of the peptide itself (15).
FIGURE 4.

Thermodynamic characterization of the EB1-MACFp1 interaction. A and B, Van't Hoff (A) and Gibb's (B) plots of the EB1-MACFp1 interaction. The binding constants were determined by FP measurements at different temperatures. Shown are the experimental data points with S.D. values (error bars) and the linear regressions used to derive ΔHapp, following the van't Hoff equation, ln Kb = −ΔH/RT + ΔS/R, and ΔSapp, following the Gibbs equation, ΔG = ΔH − TΔS. C, binding isotherms obtained at 30 °C for the EB1-MACFp1 interaction in the presence of different NaCl concentrations: 50 mm (circles), 150 mm (squares), 300 mm (diamonds), and 500 mm (triangles). Symbols represent the measured fractional saturation values calculated from the FP data using Equation 1. Error bars, S.E. values; solid lines, best fit to the data according to a simple ligand binding model described by Equation 2 (see “Experimental Procedures”).
Collectively, these results establish that our developed FP assay represents a fast, highly specific, economical (only protein quantities in the range of micrograms are required), and easy-to-perform method to quantitatively assess the EB-binding properties of SXIP-containing proteins and peptides.
Regulation of the SXIP-EB1 Interaction by Phosphorylation
We have previously hypothesized that electrostatic interactions between the positively charged residues flanking the SXIP site and the negatively charged residues of the C-terminal part of EBs contribute favorably to EB-SXIP complex formation (15). We tested this hypothesis by performing the FP assay under different ionic strength conditions. As shown in Fig. 4C, we observed a 15-fold decrease in EB1-MACFp1 binding affinity upon increasing the sodium chloride concentration of the solution from 50 to 500 mm. This result confirms the importance of electrostatic attractive interactions for stabilizing the EB1-SXIP complex.
To investigate the effect of phosphorylation in a canonical MtLS sequence, we performed a second SPOT analysis with a slightly shorter version of MACFp1 (residues 2–28). An array was generated consisting of this shorter MACFp1 peptide and all possible combinations of either phosphorylated serine or threonine residues in its sequence. The array was subsequently probed for EB1 binding, and the results were classified into EB binders and non-binders (Fig. 5). Single serine or threonine phosphorylation in the flanking region of the critical MACFp1 sequence 12PSKIPTPQRKSP23 (see above) has little influence on EB1 binding. In contrast, single phosphorylation of either the serine or threonine residue within 12PSKIPTPQRKSP23 significantly affected EB1 binding, suggesting a role of these residues for regulating the EB1-SXIP interaction. The effect of the negatively charged phosphate groups can be explained by long range electrostatic repulsive interactions with the negatively charged surface of the EBH domain (for phosphorylated residues in the SXIP flanking regions) or by a steric clash of the phosphoserine side chain with the surface of the EBH domain (for the phosphorylated serine residue of SXIP; Fig. 1). All but one sequence containing two phosphorylated serine or threonine residues did not bind to EB1 (supplemental Fig. 2). As an exception, the presence of two phosphoserines on the N-terminal side of 12PSKIPTPQRKSP23 does not drastically influence EB1 binding, in agreement with the substitution analysis showing that the N-terminal moiety of MACFp1 is less critical for the EB1-MACFp1 interaction.
FIGURE 5.

Effects of phosphorylation of MACFp1 on EB1 binding. The sequence 2–28 of MACFp1 is shown at the top. The serine and threonine residues that were systematically phosphorylated are underlined, and the corresponding residue number is shown below. The SKIP motif is highlighted by a line above the sequence. The bottom panel displays schematic representations of the phosphorylated MACFp1 variants tested. Phosphorylated residues are indicated. For simplicity, double numbering of sites (e.g. pS5/8) refers to single modified residues (i.e. phospho-Ser-5 or -8 (pS5 or pS8)). Phosphorylated MACFp1 variants are classified as EB1 binders (right) and non-binders (left). See also supplemental Fig. 2.
Collectively, these results demonstrate that long range electrostatic attractive interactions contribute to the stability of the EB-SXIP complex. They further suggest that this electrostatic mechanism can be exploited by cells to down-regulate the EB-binding activity of +TIPs through phosphorylation.
Binding of Different SXIP-containing +TIP Fragments to EBs and Their Localization to Growing Microtubule Ends
Next we sought to compare in vitro the properties of different SXIP-containing sequence regions from different +TIPs. For this purpose, we compiled a list of 14 +TIPs based on information from the literature and visual inspection of amino acid sequences (Fig. 6A). To test the EB1-binding properties of all these proteins, we fused ∼40 amino acid residue fragments encompassing SXIP motifs to the C terminus of oligohistidine-tagged thioredoxin (HisTrx). The validity of this approach was assessed by FP and ITC experiments; bacterially expressed and purified TrxMACF and TrxAPC proteins yielded consistent EB1-binding isotherms in both FP (Fig. 6B) and ITC experiments (Fig. 6C). As a control, no binding of HisTrx alone to EB1 was observed. The low micromolar Kd values obtained for the EB1-TrxMACF and EB1-TrxAPC interactions are in agreement with those measured from equivalent but untagged polypeptide versions of MACF and APC (Fig. 6B and Table 2), demonstrating that the presence of the HisTrx tag does not significantly influence the EB1-binding properties of peptides.
Using the approach described above, we next determined the affinities of the 14 +TIP sequences shown in Fig. 6A. The best EB1 binders displayed Kds between 1.5 and 3.3 μm. They are followed by seven +TIP sequences with affinities ranging from 10 to 140 μm (Table 2). Five +TIP sequences displayed Kd values in the millimolar range or higher. Based on this analysis, we classified the different sequences into EB1 binders (Kd values in the range of 1–140 μm) and non-binders (Kd values in the millimolar range; Fig. 6A). It is not straightforward to explain why some sequences bind and others do not bind to EB1 based on the present data. A priori, all tested +TIP fragments possess an SXIP motif, display similar physicochemical properties (net charge, hydrophobicity, hydrophilicity), and are all predicted to be intrinsically disordered in isolation. Based on our SPOT analysis (see above), a conspicuous notion is that all non-binders contain an unfavorable residue at the X position of the SXIP motifs as well as at least one additional unfavorable residue in the SXIP flanking regions (Table 1). The EB1 binders melanophilin and DDA3 (differential display and activated by p53) also contain an unfavorable residue at the x-position of SXIP (APC contains a glutamine, which is tolerated); however, their SXIP flanking regions are enriched in favorable ones. The SXIP motif of melanophilin is immediately flanked by favorable basic and hydrophobic residues, and DDA3 contains an additional SXIP-like motif (see Fig. 6A, underlined residues).
We cannot exclude at this point the possibility that additional factors like, for example, different conformational properties unfavorably correlated residue combinations that are not detected in the single-point substitution SPOT analysis or that additional long range nonspecific interactions could also contribute to the EB1-binding activity of different MtLS sequences. In fact, nuclear magnetic resonance spectroscopy on an APC fragment (15) demonstrated that a residue segment upstream of the SXIP motif also interacts, although weakly, with the C-terminal domain of EB1 (Fig. 6A). This result suggests that sequence context is a factor that can contribute to the function of an MtLS.
Several +TIPs contain multiple SXIP motifs either arranged in tandem on a single polypeptide chain and/or as a consequence of subunit oligomerization (4). We and others recently found that multiple SXIP motifs can act in concert to enhance microtubule tip localization in cells (15, 21, 40). To assess the effect of multiple SXIP motifs, we investigated the S. cerevisiae +TIP Ipl1p (24, 25), which contains two consecutive SXIP motifs (Fig. 6A), separated by 22 amino acid residues. Ipl1p-derived polypeptides encompassing the first (TrxIpl1-p1) and the second (TrxIpl1-p2) SXIP motif yielded Kd values of 15 and 90 μm, respectively (Table 2). In contrast, a Kd of 0.15 ± 0.01 μm was observed for a construct encompassing both SXIP motifs (TrxIpl1-p12), which represents an improvement in affinity of >100-fold over the single SXIP peptides (Fig. 6E and Table 2). To test the effect of an SXIP motif in an oligomerized context, we fused the sequence corresponding to MACFp1 to the C terminus of the two-stranded parallel leucine zipper coiled-coil domain of the yeast transcriptional activator GCN4 (TrxMACF-GCN4 (28)). As documented in Table 2, we found a >3-order of magnitude increase in EB1 binding affinity for TrxMACF-GCN4 compared with the corresponding, monomeric TrxMACF version (Fig. 6E). These results demonstrate that multiple SXIP motifs can cooperate to strongly increase the overall affinity of +TIPs for EBs.
The complexes formed in solution between EB1 and TrxIpl1-p12 or TrxMACF-GCN4 were further analyzed by multiangle light scattering experiments. EB1, TrxIpl1-p12, and TrxMACF-GCN4 alone yielded molecular masses of 60, 28, and 48 kDa, respectively (not shown). These values are consistent with EB1 and TrxMACF-GCN4 forming dimers (calculated molecular masses of the monomers: 29.9 and 22.3 kDa, respectively) and TrxIpl1-p12 staying monomeric in solution (calculated molecular mass of the monomer: 25.8 kDa). As shown in supplemental Fig. 3, the molecular masses obtained for EB1-TrxIpl1-p12 and EB1-TrxMACF-GCN4 mixtures amounted 100 and 82 kDa, respectively, consistent with the formation of 1:1 stoichiometric complexes between EB1 dimers and TrxMACF-GCN4 dimers (calculated molecular masses of the complex: 59.8 + 44.6 = 104.4 kDa) or EB1 dimers and TrxIpl1-p12 monomers (calculated molecular masses of the complex: 59.8 + 25.8 = 85.6 kDa). No higher oligomers were detected in both cases. These results suggest that two SXIP motifs either on the same polypeptide (TrxIpl1-p12) or in an oligomerized context (TrxMACF-GCN4) can simultaneously bind to the two SXIP-binding sites on the EBH domain of the EB1 dimer (6, 7).
To test the microtubule tip tracking activity of SXIP peptides, we fused selected representatives covering a wide range of EB1 binding affinities to the C terminus of GFP and transfected the resulting constructs into COS-7 cells (Fig. 7). As documented in Table 2, with the exception of p140Cap and FILIP, all tested monomeric SXIP sequence variants were able to track growing microtubule ends with a fluorescence tip-to-cytoplasm intensity ratio of about 1.5. When these peptides were tested as GCN4-mediated dimers, their accumulation at the microtubule tips improved significantly; the tip-to-cytoplasm ratio increased by a factor of 2–7. The dramatic increase in microtubule tip binding affinity of GCN4-MACF with a tip-to-cytoplasm ratio of 7 is consistent with its remarkably strong interaction with EB1 (see above). Notably, the FILIP-derived GFP-tagged peptide, which was not able to track microtubule ends as a monomer, could do so in its dimeric configuration. No microtubule tip accumulation, however, was detected for the p140Cap peptide either in its monomeric or in its dimeric form, in agreement with the very low affinity of this peptide for EB1. We conclude that other parts of the disordered regions of p140Cap confer the ability of interacting with EB1 and microtubule tip tracking in cells (23).
FIGURE 7.

Live cell imaging of monomeric and dimeric MtLS-containing peptides fused to GFP. Live images of COS-7 cells transiently transfected with the indicated GFP fusions are shown in two different ways. Panels on the left show single frames obtained by averaging of five consecutive images acquired with a 0.5-s interval (2.5 s in total). Panels on the right show maximum intensity projections of six consecutive and averaged frames, displayed in different colors. The first of the six frames is shown as grayscale (microtubule plus-ends are white), frames 2, 4, and 6 are shown in green (plus-ends appear green), and frames 3 and 5 are shown in red (plus-ends appear red). This representation facilitates the visualization of microtubule tip tracking events over time. Insets, enlargements of small parts of the images to illustrate the presence or absence of comet “tracks” reflecting +TIP accumulation at growing microtubule ends.
Taken together, these data suggest that single MtLS sequences display a wide range of affinities toward EB1. They further demonstrate that the acquisition of multiple SXIP motifs is a mechanism used by many +TIPs to enhance EB binding and consequently their microtubule tip localization activity in cells.
DISCUSSION
In this paper, we sought to investigate in detail the sequence determinants of a widespread microtubule tip localization signal, which we refer to as MtLS. The sequence features of MtLSs comprise a short and conserved motif, SXIP, which is embedded in unstructured regions of proteins. These properties are characteristic of short linear motifs, which are involved in an array of diverse protein-protein interactions and in the establishment of dynamic protein interaction networks in higher eukaryotes (41). In the case of MtLS, we observed an enrichment of serine, proline, and basic residues in the flanking region of the SXIP motif that contributes to MtLS function. Our SPOT analysis also shows that lysine and arginine are more frequently encountered at the X position of SXIP. Short linear motifs are typically fast evolving sequences that can easily be acquired during evolution (41), which suggests that the detailed knowledge of MtLS sequence determinants may enable the discovery of many more +TIPs in eukaryotic genomes. Our experiments with EBs and +TIPs from different species demonstrate that the mechanism of the MtLS is evolutionarily conserved throughout eukaryotes.
Previous structural work (15) and the biochemical/biophysical analysis presented here demonstrate that the SXIP motif of the MtLS is specifically recognized by the EBH domain of EBs. Collectively, the positively charged lysine and arginine residues flanking the SXIP site contribute favorably to the SXIP-EB complex through nonspecific long range attractive electrostatic interactions with the negatively charged and surface-exposed glutamate and aspartate residues of the EBH domain (15). Given the negatively charged outer surface of the microtubule lattice (42), we believe that the overall positively charged nature of the MtLS may in addition also interact with microtubule tips through similar nonspecific electrostatic attractive interactions.
The EB1 binding affinities of many isolated MtLS sequences that we have tested are in the low/medium micromolar range, the best binders displaying a dissociation constant around 1 μm. Such rather weak interactions are typical for short linear motifs that organize highly dynamic protein networks (43, 44), reminiscent to the ones orchestrated by EBs at growing microtubule tips (4, 12–14). Interestingly, we found a good correlation between EB binding affinity and microtubule tip tracking activity in cells, further underpinning the conclusion that the EB-SXIP interaction is the underlying mechanism used by many +TIPs to target the growing microtubule plus-end. Low affinity interactions are well suited to be fine tuned by, for example, posttranslational modifications like phosphorylation. Our past (15) and present results support the emerging concept that serine and threonine phosphorylation in the SXIP flanking regions is used in cells to destabilize the SXIP-EB complex and probably also the interaction with microtubules through an electrostatic repulsion effect. Prominent examples of +TIPs that are regulated by phosphorylation within their MtLS sequences include MCAK (15), CLASP (16), APC (7), and SLAIN2 (21).
Notably, we also found MtLS sequences that display affinities for EB1 in the high micromolar range. In this context, it is interesting to note that many +TIPs accumulate multiple MtLS either within single polypeptide chains or through subunit oligomerization (4, 12–14). Our data show that the presence of multiple MtLSs strongly enhances the affinity of proteins for EBs and, as a consequence, their microtubule tip localization activity in cells. Multiple MtLSs may strengthen their binding to EBs through an enforced proximity effect, a mechanism frequently exploited by short linear motifs (41).
In summary, our study defines the sequence determinants of a canonical MtLS. It thus provides a favorable basis to develop bioinformatics approaches to screen for novel +TIPs in entire genomes. Indeed, the data presented in this paper enabled us to discover and characterize 20 new +TIP candidates.5 Furthermore, the experimental pipeline established in this work, in particular the developed FP assay, enables high throughput screening of chemical libraries to find small molecule ligands that bind to the SXIP-binding site of EBs (supplemental Fig. 4). Such molecules could be employed as powerful tool compounds to study EB-mediated dynamic +TIP networks in vitro and in live cells. Moreover, because EBs are increasingly recognized to be implicated in human neoplastic processes (45–47), such compounds may also spur the development of novel therapeutic agents relevant to diseases such as cancer.
Supplementary Material
This work was supported by a Federation of European Biochemical Societies (FEBS) postdoctoral fellowship and a contract from the Juan de la Cierva program and a Marie Curie Career Integration grant (EB-SxIP; 293831) (to R. M. B.), by a Marie Curie IIF grant (MT-TIP Inhibitors; 253818) (to R. M.), by an EMBO grant (to D. G.), and by Swiss National Science Foundation Grants 31003A_122545 and 310030B_138659 (to M. O. S.).

This article contains supplemental Figs. 1–4.
D. Veprintsev, unpublished work.
K. Jiang, G. Toedt, S. Montenegro Gouveia, N. E. Davey, B. van der Vaart, I. Grigoriev, J. Larsen, L. Pedersen, K. Bezstarosti, J. Demmers, M. O. Steinmetz, T. J. Gibson, and A. Akhmanova, submitted for publication.
- +TIP
- microtubule plus-end-tracking protein
- EB
- end-binding protein
- MtLS
- microtubule tip localization signal
- EBH
- EB homology
- APC
- adenomatous poliposis coli
- MACF
- microtubule actin cross-linking factor
- MCAK
- mitotic centromere-associated kinesin
- CLIP
- cytoplasmic linker protein
- CLASP
- CLIP-associated protein
- STIM
- stromal interaction molecule
- SLAIN
- SLAIN motif-containing protein
- FILIP
- filamin A-interacting protein
- Trx
- thioredoxin
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- FC
- 5(6)-carboxyfluorescein
- FP
- fluorescence polarization
- ITC
- isothermal titration calorimetry
- MD
- molecular dynamics
- TIRF
- total internal reflection fluorescence
- EB1c
- EB1 C-terminal domain
- SI
- signal intensity
- HisTrx
- oligohistidine-tagged Trx
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Desai A., Mitchison T. J. (1997) Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 13, 83–117 [DOI] [PubMed] [Google Scholar]
- 2. Heald R., Nogales E. (2002) Microtubule dynamics. J. Cell Sci. 115, 3–4 [DOI] [PubMed] [Google Scholar]
- 3. Schuyler S. C., Pellman D. (2001) Microtubule “plus-end-tracking proteins.” The end is just the beginning. Cell 105, 421–424 [DOI] [PubMed] [Google Scholar]
- 4. Akhmanova A., Steinmetz M. O. (2008) Tracking the ends. A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 9, 309–322 [DOI] [PubMed] [Google Scholar]
- 5. Tirnauer J. S., Bierer B. E. (2000) EB1 proteins regulate microtubule dynamics, cell polarity, and chromosome stability. J. Cell Biol. 149, 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Slep K. C., Rogers S. L., Elliott S. L., Ohkura H., Kolodziej P. A., Vale R. D. (2005) Structural determinants for EB1-mediated recruitment of APC and spectraplakins to the microtubule plus end. J. Cell Biol. 168, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Honnappa S., John C. M., Kostrewa D., Winkler F. K., Steinmetz M. O. (2005) Structural insights into the EB1-APC interaction. EMBO J. 24, 261–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Groot C. O., Jelesarov I., Damberger F. F., Bjelić S., Schärer M. A., Bhavesh N. S., Grigoriev I., Buey R. M., Wüthrich K., Capitani G., Akhmanova A., Steinmetz M. O. (2010) Molecular insights into mammalian end-binding protein heterodimerization. J. Biol. Chem. 285, 5802–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buey R. M., Mohan R., Leslie K., Walzthoeni T., Missimer J. H., Menzel A., Bjelic S., Bargsten K., Grigoriev I., Smal I., Meijering E., Aebersold R., Akhmanova A., Steinmetz M. O. (2011) Insights into EB1 structure and the role of its C-terminal domain for discriminating microtubule tips from the lattice. Mol. Biol. Cell 22, 2912–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bieling P., Laan L., Schek H., Munteanu E. L., Sandblad L., Dogterom M., Brunner D., Surrey T. (2007) Reconstitution of a microtubule plus-end tracking system in vitro. Nature 450, 1100–1105 [DOI] [PubMed] [Google Scholar]
- 11. Komarova Y., De Groot C. O., Grigoriev I., Gouveia S. M., Munteanu E. L., Schober J. M., Honnappa S., Buey R. M., Hoogenraad C. C., Dogterom M., Borisy G. G., Steinmetz M. O., Akhmanova A. (2009) Mammalian end binding proteins control persistent microtubule growth. J. Cell Biol. 184, 691–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Akhmanova A., Steinmetz M. O. (2010) Microtubule +TIPs at a glance. J. Cell Sci. 123, 3415–3419 [DOI] [PubMed] [Google Scholar]
- 13. Galjart N. (2010) Plus-end-tracking proteins and their interactions at microtubule ends. Curr. Biol. 20, R528–537 [DOI] [PubMed] [Google Scholar]
- 14. Slep K. C. (2010) Structural and mechanistic insights into microtubule end-binding proteins. Curr. Opin. Cell Biol. 22, 88–95 [DOI] [PubMed] [Google Scholar]
- 15. Honnappa S., Gouveia S. M., Weisbrich A., Damberger F. F., Bhavesh N. S., Jawhari H., Grigoriev I., van Rijssel F. J., Buey R. M., Lawera A., Jelesarov I., Winkler F. K., Wüthrich K., Akhmanova A., Steinmetz M. O. (2009) An EB1-binding motif acts as a microtubule tip localization signal. Cell 138, 366–376 [DOI] [PubMed] [Google Scholar]
- 16. Kumar P., Lyle K. S., Gierke S., Matov A., Danuser G., Wittmann T. (2009) GSK3β phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J. Cell Biol. 184, 895–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Montenegro Gouveia S., Leslie K., Kapitein L. C., Buey R. M., Grigoriev I., Wagenbach M., Smal I., Meijering E., Hoogenraad C. C., Wordeman L., Steinmetz M. O., Akhmanova A. (2010) In vitro reconstitution of the functional interplay between MCAK and EB3 at microtubule plus ends. Curr. Biol. 20, 1717–1722 [DOI] [PubMed] [Google Scholar]
- 18. Kumar P., Chimenti M. S., Pemble H., Schönichen A., Thompson O., Jacobson M. P., Wittmann T. (2012) Multisite phosphorylation disrupts arginine-glutamate salt bridge networks required for binding of cytoplasmic linker-associated protein 2 (CLASP2) to end-binding protein 1 (EB1). J. Biol. Chem. 287, 17050–17064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mimori-Kiyosue Y., Grigoriev I., Lansbergen G., Sasaki H., Matsui C., Severin F., Galjart N., Grosveld F., Vorobjev I., Tsukita S., Akhmanova A. (2005) CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J. Cell Biol. 168, 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grigoriev I., Gouveia S. M., van der Vaart B., Demmers J., Smyth J. T., Honnappa S., Splinter D., Steinmetz M. O., Putney J. W., Jr., Hoogenraad C. C., Akhmanova A. (2008) STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 18, 177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Vaart B., Manatschal C., Grigoriev I., Olieric V., Gouveia S. M., Bjelic S., Demmers J., Vorobjev I., Hoogenraad C. C., Steinmetz M. O., Akhmanova A. (2011) SLAIN2 links microtubule plus end-tracking proteins and controls microtubule growth in interphase. J. Cell Biol. 193, 1083–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li W., Miki T., Watanabe T., Kakeno M., Sugiyama I., Kaibuchi K., Goshima G. (2011) EB1 promotes microtubule dynamics by recruiting Sentin in Drosophila cells. J. Cell Biol. 193, 973–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jaworski J., Kapitein L. C., Gouveia S. M., Dortland B. R., Wulf P. S., Grigoriev I., Camera P., Spangler S. A., Di Stefano P., Demmers J., Krugers H., Defilippi P., Akhmanova A., Hoogenraad C. C. (2009) Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 61, 85–100 [DOI] [PubMed] [Google Scholar]
- 24. Zimniak T., Fitz V., Zhou H., Lampert F., Opravil S., Mechtler K., Stolt-Bergner P., Westermann S. (2012) Spatiotemporal regulation of Ipl1/Aurora activity by direct Cdk1 phosphorylation. Curr. Biol. 22, 787–793 [DOI] [PubMed] [Google Scholar]
- 25. Zimniak T., Stengl K., Mechtler K., Westermann S. (2009) Phosphoregulation of the budding yeast EB1 homologue Bim1p by Aurora/Ipl1p. J. Cell Biol. 186, 379–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 27. Olieric N., Kuchen M., Wagen S., Sauter M., Crone S., Edmondson S., Frey D., Ostermeier C., Steinmetz M. O., Jaussi R. (2010) Automated seamless DNA co-transformation cloning with direct expression vectors applying positive or negative insert selection. BMC Biotechnol. 10, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Shea E. K., Klemm J. D., Kim P. S., Alber T. (1991) X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 254, 539–544 [DOI] [PubMed] [Google Scholar]
- 29. Lu Q. (2005) Seamless cloning and gene fusion. Trends Biotechnol. 23, 199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 31. Zhang J. H., Chung T. D., Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 [DOI] [PubMed] [Google Scholar]
- 32. Wenschuh H., Volkmer-Engert R., Schmidt M., Schulz M., Schneider-Mergener J., Reineke U. (2000) Coherent membrane supports for parallel microsynthesis and screening of bioactive peptides. Biopolymers 55, 188–206 [DOI] [PubMed] [Google Scholar]
- 33. van der Spoel D., van Maaren P. J., Caleman C. (2012) GROMACS molecule and liquid database. Bioinformatics 28, 752–753 [DOI] [PubMed] [Google Scholar]
- 34. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM. The biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zoete V., Michielin O. (2007) Comparison between computational alanine scanning and per-residue binding free energy decomposition for protein-protein association using MM-GBSA. Application to the TCR-p-MHC complex. Proteins 67, 1026–1047 [DOI] [PubMed] [Google Scholar]
- 36. Frank R. (2002) The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports. Principles and applications. J. Immunol. Methods 267, 13–26 [DOI] [PubMed] [Google Scholar]
- 37. Volkmer R. (2009) Synthesis and application of peptide arrays. Quo vadis SPOT technology. Chembiochem 10, 1431–1442 [DOI] [PubMed] [Google Scholar]
- 38. Weiser A. A., Or-Guil M., Tapia V., Leichsenring A., Schuchhardt J., Frömmel C., Volkmer-Engert R. (2005) SPOT synthesis. Reliability of array-based measurement of peptide binding affinity. Anal. Biochem. 342, 300–311 [DOI] [PubMed] [Google Scholar]
- 39. Beinhauer J. D., Hagan I. M., Hegemann J. H., Fleig U. (1997) Mal3, the fission yeast homologue of the human APC-interacting protein EB-1 is required for microtubule integrity and the maintenance of cell form. J. Cell Biol. 139, 717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Applewhite D. A., Grode K. D., Keller D., Zadeh A., Slep K. C., Rogers S. L. (2010) The spectraplakin Short stop is an actin-microtubule cross-linker that contributes to organization of the microtubule network. Mol. Biol. Cell 21, 1714–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davey N. E., Van Roey K., Weatheritt R. J., Toedt G., Uyar B., Altenberg B., Budd A., Diella F., Dinkel H., Gibson T. J. (2012) Attributes of short linear motifs. Mol. Biosyst. 8, 268–281 [DOI] [PubMed] [Google Scholar]
- 42. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems. Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Diella F., Haslam N., Chica C., Budd A., Michael S., Brown N. P., Trave G., Gibson T. J. (2008) Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front. Biosci. 13, 6580–6603 [DOI] [PubMed] [Google Scholar]
- 44. Dinkel H., Michael S., Weatheritt R. J., Davey N. E., Van Roey K., Altenberg B., Toedt G., Uyar B., Seiler M., Budd A., Jödicke L., Dammert M. A., Schroeter C., Hammer M., Schmidt T., Jehl P., McGuigan C., Dymecka M., Chica C., Luck K., Via A., Chatr-Aryamontri A., Haslam N., Grebnev G., Edwards R. J., Steinmetz M. O., Meiselbach H., Diella F., Gibson T. J. (2012) ELM. The database of eukaryotic linear motifs. Nucleic Acids Res. 40, D242–D251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu M., Yang S., Wang Y., Zhu H., Yan S., Zhang W., Quan L., Bai J., Xu N. (2009) EB1 acts as an oncogene via activating β-catenin/TCF pathway to promote cellular growth and inhibit apoptosis. Mol. Carcinog. 48, 212–219 [DOI] [PubMed] [Google Scholar]
- 46. Abiatari I., Gillen S., DeOliveira T., Klose T., Bo K., Giese N. A., Friess H., Kleeff J. (2009) The microtubule-associated protein MAPRE2 is involved in perineural invasion of pancreatic cancer cells. Int. J. Oncol. 35, 1111–1116 [DOI] [PubMed] [Google Scholar]
- 47. Dong X., Liu F., Sun L., Liu M., Li D., Su D., Zhu Z., Dong J. T., Fu L., Zhou J. (2010) Oncogenic function of microtubule end-binding protein 1 in breast cancer. J. Pathol. 220, 361–369 [DOI] [PubMed] [Google Scholar]
- 48. Wallace A. C., Laskowski R. A., Thornton J. M. (1995) LIGPLOT. A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 [DOI] [PubMed] [Google Scholar]
- 49. Askham J. M., Moncur P., Markham A. F., Morrison E. E. (2000) Regulation and function of the interaction between the APC tumor suppressor protein and EB1. Oncogene 19, 1950–1958 [DOI] [PubMed] [Google Scholar]
- 50. Wu X. S., Tsan G. L., Hammer J. A., 3rd (2005) Melanophilin and myosin Va track the microtubule plus end on EB1. J. Cell Biol. 171, 201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee T., Langford K. J., Askham J. M., Brüning-Richardson A., Morrison E. E. (2008) MCAK associates with EB1. Oncogene 27, 2494–2500 [DOI] [PubMed] [Google Scholar]
- 52. Hsieh P. C., Chang J. C., Sun W. T., Hsieh S. C., Wang M. C., Wang F. F. (2007) p53 downstream target DDA3 is a novel microtubule-associated protein that interacts with end-binding protein EB3 and activates β-catenin pathway. Oncogene 26, 4928–4940 [DOI] [PubMed] [Google Scholar]
- 53. Martínez-López M. J., Alcántara S., Mascaró C., Pérez-Branguli F., Ruiz-Lozano P., Maes T., Soriano E., Buesa C. (2005) Mouse neuron navigator 1, a novel microtubule-associated protein involved in neuronal migration. Mol. Cell Neurosci. 28, 599–612 [DOI] [PubMed] [Google Scholar]
- 54. Jiang K., Wang J., Liu J., Ward T., Wordeman L., Davidson A., Wang F., Yao X. (2009) TIP150 interacts with and targets MCAK at the microtubule plus ends. EMBO Rep. 10, 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Perez F., Diamantopoulos G. S., Stalder R., Kreis T. E. (1999) CLIP-170 highlights growing microtubule ends in vivo. Cell 96, 517–527 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.