

Background: Hereditary startle disease is caused by genetic defects in inhibitory glycine receptor and transporter genes.
Results: Loss of function mutations in SLC6A5, with novel mechanisms of action, were identified in 17 individuals with startle disease.
Conclusion: Recessive mutations in SLC6A5 represent a second major cause of startle disease.
Significance: Genetic screening for startle disease should encompass both presynaptic and postsynaptic causes of disease.
Keywords: Electrophysiology, Glycine Receptors, Human Genetics, Neurological Diseases, Transporters, GlyT2, Glycine Transporter, Hyperekplexia, SLC6A5, Startle Disease
Abstract
Hereditary hyperekplexia or startle disease is characterized by an exaggerated startle response, evoked by tactile or auditory stimuli, leading to hypertonia and apnea episodes. Missense, nonsense, frameshift, splice site mutations, and large deletions in the human glycine receptor α1 subunit gene (GLRA1) are the major known cause of this disorder. However, mutations are also found in the genes encoding the glycine receptor β subunit (GLRB) and the presynaptic Na+/Cl−-dependent glycine transporter GlyT2 (SLC6A5). In this study, systematic DNA sequencing of SLC6A5 in 93 new unrelated human hyperekplexia patients revealed 20 sequence variants in 17 index cases presenting with homozygous or compound heterozygous recessive inheritance. Five apparently unrelated cases had the truncating mutation R439X. Genotype-phenotype analysis revealed a high rate of neonatal apneas and learning difficulties associated with SLC6A5 mutations. From the 20 SLC6A5 sequence variants, we investigated glycine uptake for 16 novel mutations, confirming that all were defective in glycine transport. Although the most common mechanism of disrupting GlyT2 function is protein truncation, new pathogenic mechanisms included splice site mutations and missense mutations affecting residues implicated in Cl− binding, conformational changes mediated by extracellular loop 4, and cation-π interactions. Detailed electrophysiology of mutation A275T revealed that this substitution results in a voltage-sensitive decrease in glycine transport caused by lower Na+ affinity. This study firmly establishes the combination of missense, nonsense, frameshift, and splice site mutations in the GlyT2 gene as the second major cause of startle disease.
Introduction
Startle disease is a rare disorder characterized by evoked episodes of hypertonia that can have serious consequences, including sudden infant death from apnea or aspiration pneumonia (1, 2). Although symptoms often diminish during the first year of life, the exaggerated startle response can persist into adulthood, leading to unprotected falls (3, 4). Fortunately, startle disease can be treated using clonazepam (4, 5), which potentiates inhibitory GABAA receptor function. An intervention called the Vigevano maneuver, involving flexing of the head and limbs toward the trunk, also dissipates and counteracts the effects of acute hypertonia and apnea episodes (2). Hyperekplexia is recognized and detected within specialist neurology and pediatric centers worldwide (6, 7). Key collaborations between clinicians and scientists have revealed that the primary cause of startle disease is defective inhibitory glycinergic transmission. Currently, the major known genetic cause of hyperekplexia is missense, nonsense, frameshift, or splice site mutations in the glycine receptor (GlyR)4 α1 gene (GLRA1) (8–10), although large GLRA1 deletions are also common in patients of Kurdish descent (11, 12). Rare mutations in the genes encoding the GlyR β subunit (GLRB) (13, 14) and the synaptic clustering proteins gephyrin (GPHN) (15) and collybistin (ARHGEF9) (16) have also been linked to hyperekplexia.
Because many patients do not harbor defects in these genes, some years ago we began to consider hyperekplexia as a synaptopathy. This led to the identification of missense, nonsense, and frameshift mutations in the GlyT2 gene (SLC6A5), encoding a Na+/Cl−-dependent neurotransmitter transporter that maintains a high presynaptic pool of glycine at glycinergic synapses (17, 18). Using detailed structure-function analyses, we demonstrated that GlyT2 mutations disrupted transporter membrane trafficking, Na+, or glycine-binding sites (17). Subsequently, unique GlyT2 mutations were discovered in cattle and dogs, causing startle disorders with early neonatal lethality (19, 20). Here, we present the outcomes from an international screening program targeting SLC6A5 in 93 unrelated hyperekplexia probands. This identified 20 recessive SLC6A5 mutations within 17 index cases, of which 19 are novel variants. These new mutations were characterized using [3H]glycine uptake assays and molecular modeling. Electrophysiological characterization of the GlyT2 mutation A275T revealed that this substitution results in a voltage-sensitive decrease in glycine transport, caused by lower Na+ binding at depolarized membrane potentials. Taken together with our recent study on GLRA1 mutations in startle disease (10), these results suggest that recessive modes of inheritance are far more common on a population basis than dominant mutations, explaining the apparent sporadic nature of this rare disorder.
EXPERIMENTAL PROCEDURES
Patients and Cases
Individuals of either sex with a clinical diagnosis of hyperekplexia were ascertained by referral from neurologists, pediatricians, or clinical geneticists from international centers. Informed consent and clinical data were obtained by the referring clinician (local research ethics committee, the South West Wales Research Ethics Committee). Ninety-three unrelated index patients were ascertained in this population study after evaluation by clinical criteria included a nonhabituating startle response (positive nose tap test), history of neonatal/infantile hypertonicity, negative results in GLRA1 gene screening by Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA), and the exclusion of phenocopies such as startle epilepsy (7).
Molecular Genetics
Exons and intron-exon boundaries of SLC6A5 were amplified from genomic DNA isolated from peripheral blood using established primers sets (17). To avoid allelic dropout, all of the primers were placed in intronic regions that were devoid of known single-nucleotide polymoryphisms found in the National Center for Biotechnology Information data base dbSNP. Following conventional PCR protocols with patient DNA, PCR products were purified with QIAquick purification kit (Qiagen) and directly sequenced using BigDye terminators and an ABI3100 automated sequencer (Applied Biosystems). Frequency of single-nucleotide polymoryphisms or mutations was assessed in 400 control alleles using restriction fragment length polymorphism, if a suitable restriction enzyme was available, or case:control contrast profiles using the Idaho LightScanner platform. In addition to Sanger sequencing, all DNA samples were screened for large deletions or insertions in SLC6A5 using a MLPA DNA detection kit (MRC-Holland).
Mutagenesis and [3H]Glycine Uptake Assays
GlyT2 mutations were introduced into the pRc/CMV-hGlyT2 expression construct (17) using the QuikChange site-directed mutagenesis kit (Stratagene), and the complete coding region was sequenced to verify that only the desired mutation had been introduced. For [3H]glycine uptake assays, HEK293 cells were grown in minimal essential medium (Earle's salts) supplemented with 10% (v/v) FCS, 2 mm l-glutamine, and 20 units/ml penicillin/streptomycin in 5% CO2, 95% air. The cells were plated on poly-d-lysine-coated 24-wells plates (Nunc), grown to 50% confluence, and transfected with 1 μg of total pRc/CMV-hGlyT2 or mutant DNAs using Lipofectamine LTX reagent (Invitrogen). After 24 h, the cells were washed twice with prewarmed buffer (118 mm NaCl, 1 mm NH2PO4, 26 mm NaHCO3, 1.5 mm MgSO4, 5 mm KCl, 1.3 mm CaCl2, 20 mm glucose) pre-equilibrated with 5% CO2, 95% air. After 2 min, the cells were incubated for 5 min in 0.1 μCi/ml [3H]glycine (60 Ci/mmol; PerkinElmer Life Sciences) at a final concentration of 300 μm. The cells were rinsed twice with ice-cold buffer pre-equilibrated with 5% CO2, 95% air and then digested in 0.1 m NaOH for 2 h. The samples were used for scintillation counting and for determination of protein concentration using the Bradford reagent (Bio-Rad). [3H]Glycine uptake was calculated as nmol/min/mg of protein and expressed as percentages of that in control cells transfected with the empty expression vector. All statistical comparisons used an unpaired Student's t test.
Molecular Modeling
Fold recognition of human GlyT2 was performed with GenTHREADER (21) and HHPRED (22). The structure of the bacterial leucine transporter (LeuT) (PDB: 2A65) (23) was identified as the best template (GenTHREADER p value: 2e-21; HHPred e-value: 0). An alignment between the human GlyT2 sequence and the LeuT structure was generated with the program T-coffee, resulting in 26% sequence identity. Based on this alignment, 50 homology models of human GlyT2 were calculated using MODELLER-9v7 and assessed with the DOPE statistical potential score (24). The model based on the lowest score (Normalized DOPE Z score: −1.162) was selected. Additional evaluation using the ProSA web server showed that the model quality (Z score = −2.07) fell within a range typically found for native proteins of similar size (25). Selected nonsynonymous substitutions were modeled into the GlyT2 homology model using the swapaa command in Chimera, using the Dunbrack backbone-dependent rotamer library (26) and taking into account the lowest clash score, the highest number of H-bonds, and the highest rotamer probability.
For modeling the GlyT2 chloride ion-binding site, Cl− was placed into our model at the coordinates of the carboxylate carbon of the LeuT Glu-290 side chain (27), based on the assumption that this position may be occupied by Cl− in homologous transporters (27–29). The residues involved in the coordination of Cl− ion are generally conserved within the SLC6 transporter family. In our model, these are: Tyr-233 in TM2, Gln-473 and Ser-477 in TM6, Asn-509 and Ser-513 in TM7, corresponding to Tyr-47, Gln-250, Thr-254, Asn-286, and Glu-290 in LeuT, respectively. Energy minimization was carried out on the model with the minimize structure option in Chimera (using the MMTK method) (30) and the AMBER 94 force field (31), allowing Cl− to fit more favorably in the binding pocket. In the resulting minimized structure, all distances between Cl− and the side chains of the interacting residues are below 4.5 Å.
Electrophysiological Analysis of GlyT2 Mutant A275T
Capped mRNAs were transcribed from linearized cDNAs using the T7 mMessage mMachine kit (Ambion, Austin, TX), and 50 ng of mRNAs were microinjected in Xenopus oocytes prepared and maintained as described (32). For nuclear injection experiments, 30 ng of each plasmid DNA was microinjected into the animal pole of multiple oocytes. 3–7 days later, whole cell currents were recorded in oocytes held at −40 mV with a two-electrode voltage clamp amplifier (Warner OC-725C; Harvard Apparatus, Holliston, MA) computer interfaced with a Digidata 1320A and pClamp 8 software (MDS Analytical Technologies). Steady-state currents were low pass filtered at 10 Hz and digitized at 100 Hz, whereas currents recorded using voltage step were filtered at 1 kHz and digitized at 5 kHz. Current and voltage electrodes were filled with 3 m KCl solution and had typical resistance of 1 MOhm. The oocytes were continuously bathed with a solution containing 100 mm NaCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm Hepes, pH 7.2, adjusted with KOH. Salt and glycine were from Sigma. The selective GlyT2 inhibitor ORG25543 was a generous gift from Organon.
RESULTS
Mutation Analysis of SLC6A5 in Hyperekplexia
A total of 93 index cases were screened for genetic variation in SLC6A5 coding exons and donor and acceptor splice junctions by Sanger DNA sequencing (17). Sequence variants were assigned as potential disease-causing mutations after exclusion from a panel of 400 human controls and cross-referencing with common SLC6A5 polymorphisms (17) and those found in dbSNP (supplemental Table S1). This revealed 20 sequence variants that were exclusive to 17 index cases, of which the vast majority (14 of 17) showed homozygous or compound heterozygous recessive inheritance (Fig. 1 and Table 1). These variants included four nonsense mutations (W151X, R191X, Y297X, and R439X), four frameshift mutations (P108L+fs25, L198R+fs123, S489F+fs39, and I665K+fs1), and three splice site mutations (IVS14+1ΔG, IVS13+1 G>T, and IVS8+1 G>A). Novel missense mutations affected residues in TM2 (L237P, P243T, and E248K), TM3 (A275T), TM7 (S513I), the extracellular TM7-TM8 loop (F547S), and the intracellular TM10-TM11 loop (Y656H and G657A). In three cases (case 12, W151X; case 13, R439X; and case 14, A275T), only a single defective SLC6A5 allele was detected. Notably, in case 13, the mutation detected (C1315T, resulting in R439X) is also found in the homozygous state in cases 4–6. This suggested that a second inactivating mutation might remain to be detected in these cases. However, we were unable to detect deletions of one or more SLC6A5 exons in these individuals using MLPA.
FIGURE 1.

Human GlyT2 mutations identified in individuals with startle disease. The amino acid sequence of human GlyT2 indicating the positions of putative transmembrane (TM) domains (colored boxes) and amino acid residues affected by hyperekplexia mutations; this study and Refs. 20–22). Blue and red triangles indicate residues in hGlyT2 that are likely to coordinate Na+ and Cl− ions, respectively, based on structure/function studies of the bacterial leucine transporter LeuT and other mammalian neurotransmitter transporters such as GAT-1 and SERT. However, GlyT2 binds three Na+ ions, whereas LeuT binds two, suggesting that other residues involved in Na+ coordination remain to be identified. Filled black circles indicate residues predicted to be involved in glycine binding.
TABLE 1.
Details of SLC6A5 hyperekplexia mutations identified in this study
The modes of inheritance are as follows: NC, not confirmed; CH, compound heterozygosity; AR, autosomal recessive. Homozygous recessive inheritance, i.e., the same allele inherited from both parents, is indicated by (H). For compound heterozygosity, (P) indicates paternal allele, and (M) indicates maternal allele. Δ, deletion.
| Case | Mode | Exon | Genotype | Consequences | Class |
|---|---|---|---|---|---|
| 1 | AR | 14 | IVS14 + 1 ΔG (H) | Impact on splicing | Splice site |
| 2a | AR | 4 | C727A (H) | P243T | Missense |
| 3a | AR | 5 | C891A (H) | Y297X | Nonsense |
| 4 | AR | 8 | C1315T (H) | R439X | Nonsense |
| 5 | AR | 8 | C1315T (H) | R439X | Nonsense |
| 6 | AR | 8 | C1315T (H) | R439X | Nonsense |
| 7 | CH | 8 | C1315T (P) | R439X | Nonsense |
| 13 | IVS13 + 1 G>T (M) | Impact on splicing | Splice site | ||
| 8b | CH | 11 | T1640C (M) | F547S | Missense |
| 13 | T1966Cb | Y656H | Missense | ||
| 9a | CH | 4 | G742A (P) | E248K | Missense |
| 8 | IVS8 + 1 G>A (M) | Impact on splicing | Splice site | ||
| 10c | NC | 9 | ΔCT [1460–1467] | S489F + fs39 | Frameshift Δ |
| 14 | G1970C | G657A | Missense | ||
| 11a | CH | 3 | C571T (P) | R191X | Nonsense |
| 3 | ΔTG [593–594] (M) | L198R + fs123 | Frameshift Δ | ||
| 12 | NC | 2 | G452A (P) | W151X | Nonsense |
| 13 | NC | 8 | C1315T (P) | R439X | Nonsense |
| 14 | NC | 5 | G823A (P) | A275T | Missense |
| 15 | CH | 2 | ΔC [319–323] (P) | P108L + fs25 | Frameshift Δ |
| 14 | ΔT [1994] (M) | I665K + fs1 | Frameshift Δ | ||
| 16 | CH | 3 | ΔTG [593–594] (P) | L198R + fs123 | Frameshift Δ |
| 4 | T710C (M) | L237P | Missense | ||
| 17 | AR | 10 | G1538T (H) | S513I | Missense |
a Affected sibling with the same mutation(s).
b Only one parent was available to assess compound heterozygote status; here, the maternal allele did not carry the Y656H mutation.
c Parental samples were lost to contact, and long range PCR attempts were unsuccessful (22-kb target).
Truncating nonsense or frameshift mutations distributed throughout the GlyT2 coding region were found in 11 of 20 patients (Fig. 1 and Table 1). As well as loss of function via protein truncation, these mutations also have the potential to lead to nonsense-mediated RNA decay (33). We also identified a common nonsense mutation, C1315T, resulting in R439X, which was detected in several apparently unrelated cases from different ethnic backgrounds (Caucasian and Pakistani origins). Splice site mutations were identified in cases 1 (IVS14+1ΔG), 7 (IVS13+1 G>T), and 9 (IVS8+1 G>A), all affecting the +1 position (albeit in different introns). This means that the first nucleotide in the corresponding intron (i.e., part of the splice donor site consensus gt) is deleted or substituted. These changes are predicted to result in either mis-splicing or intron retention. Genotype-phenotype analysis revealed that all patients harboring mutations in SLC6A5 presented with classical hyperekplexia symptoms (supplemental Table S2) including neonatal muscle hypertonia and an exaggerated startle response to tactile or acoustic stimuli with preservation of consciousness. However, hyperekplexia caused by SLC6A5 mutations was also associated with high rates of recurrent neonatal apneas, learning difficulties, and developmental delay. There were frequent reports of speech delay in excess of motor delay, with subsequent catch-up in many. This seems unlikely to be due to the effect of benzodiazepines, because the vast majority of children received profound benefit from their administration.
Functional Analysis of GlyT2 Variants Using 3H[Glycine] Uptake
To confirm the effects of GlyT2 mutations on glycine uptake, we tested the capacity of recombinantly expressed human GlyT2 and selected mutants produced by site-directed mutagenesis to mediate the uptake of [3H]glycine. GlyT2 harboring nonsense (W151X, R191X, Y297X, and R439X), frameshift (P108L+fs25, L198R+fs123, S489F+fs39, and I665K+fs1) and missense (L237P, P243T, S513I, F547S, Y656H, and G657A) mutations revealed no significant [3H]glycine uptake in HEK293 cells compared with control values (Fig. 2a). By contrast, the A275T mutant was capable of [3H]glycine uptake, although at a lower level than wild-type GlyT2. Because mutant E248K caused substantial cell death in transfected HEK293 cells, we assessed the function of GlyT2 E248K by nuclear injection in Xenopus oocytes (Fig. 2b). Unlike wild-type GlyT2 and GlyT2 A275T, GlyT2 E248K was unable to direct the formation of functional glycine transporters in this system, confirming loss of function for this mutant. Taken together, these results provide strong evidence for a bi-allelic loss of GlyT2 function in individuals 1–11 and 15–17. However, the pathogenic mechanisms underlying the clinical phenotype in patients 12 (heterozygous for W151X), 13 (heterozygous for R439X), and 14 (heterozygous for A275T) remained ambiguous. We therefore tested whether co-transfection of the two alleles would provide evidence for dominant effects, because complex-glycosylated GlyT2 forms dimers located at the cell surface (34). However, mixing W151X, A275T, or R439X with wild-type hGlyT2 in a 1:1 ratio did not substantially alter [3H]glycine uptake (Fig. 2c), suggesting that W151X, A275T, and R439X do not function in a dominant-negative manner in vitro.
FIGURE 2.
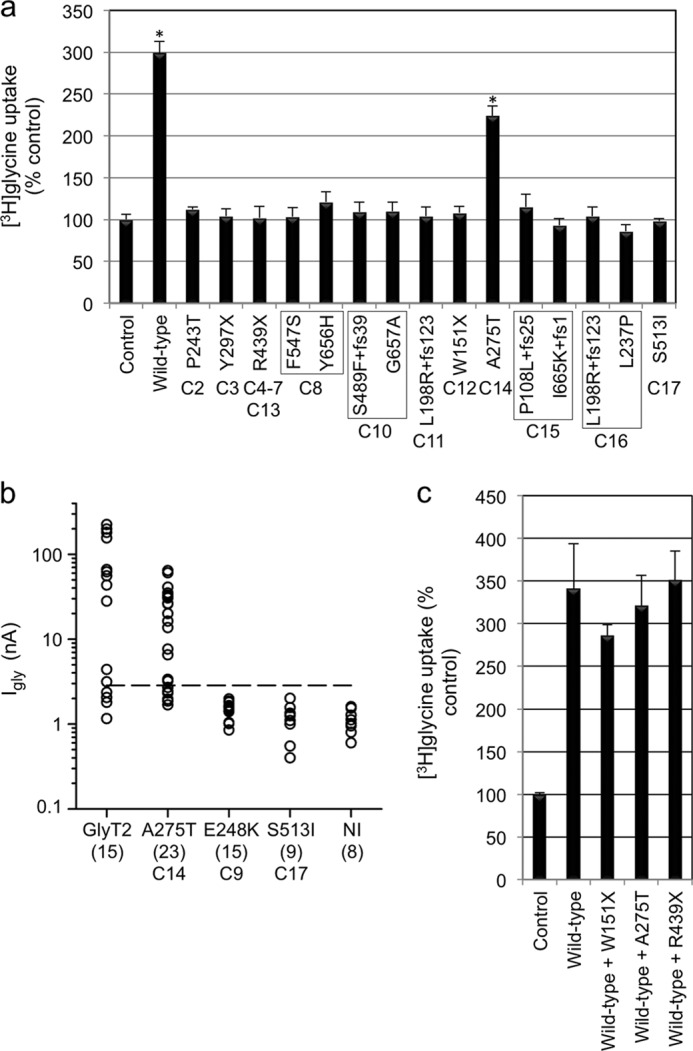
Functional activity of hGlyT2 and hyperekplexia mutants. a, glycine uptake in HEK293 cells transiently expressing hGlyT2 and single hyperekplexia mutants after 5 min of incubation with [3H]glycine (60 Ci/mmol; PerkinElmer Life Sciences) at a final concentration of 300 μm. Because low levels of glycine uptake are found in HEK293 cells (17), [3H]glycine uptake was calculated as nmol/min/mg of protein and then expressed as a percentage of the empty expression vector transfected control. Only wild-type GlyT2 and mutant A275T result in any detectable uptake above control values. b, steady-state amplitude of the current evoked by 1 mm glycine at −70 mV in Xenopus oocytes injected with selected GlyT2 expression cDNAs. Because 1 mm glycine evokes a small inward current of 1.1 ± 0.34 nA (n = 8) in non-injected (NI) oocytes (because of endogenous amino acid transporters), the threshold for heterologous expression of GlyT2 was set at +5 S.D. (∼2.8 nA, dashed line). Robust currents were observed for 10 of 14 oocytes injected with wild-type GlyT2 and 12 of 18 oocytes injected with the A275T mutant. By contrast, neither E248K or S513I mutants exhibited currents above this threshold (n = 15 and 9, respectively). c, selected [3H]glycine uptake experiments for mutations W151X, A275T, and R439X. We noted no significant effect of co-expressing these mutants with wild-type GlyT2, suggesting that these mutations do not show dominant-negative effects. The data are the means ± S.E. (n = 4–12). Statistical comparisons were made using an unpaired Students t test. For a, the asterisk indicates significantly different from empty vector control. p < 0.01.
Molecular Modeling of GlyT2 Mutations
To gain further insight into the precise molecular mechanisms underlying GlyT2 missense mutations, we examined each mutation individually and in the context of a homology model of human GlyT2 constructed using the crystal structure of the bacterial leucine transporter LeuT (Fig. 3a). Alignment of the Aquifex aeolicus LeuT with GlyT1 and GlyT2 also allowed residues potentially involved in coordinating glycine or Na+ and Cl− binding to be identified. Three new missense mutations result in substitution of residues in TM2 (L237P, P243T, and E248K). Introduction (L237P) or elimination (P243T) of proline residues in transmembrane helices is predicted to have a significant effect on local structural rigidity, destabilizing the native fold, which often results in accelerated degradation or aggregation of the mutant protein (35). By contrast, substitution E248K in TM2 results in the replacement of a negatively charged glutamate residue for a positively charged lysine, with a longer side chain that introduces potential clashes with Ala-480 and Ala-481 in TM6 (Fig. 3, b and c), which may affect the orientation of the key glycine-binding residue Trp-482.
FIGURE 3.

Molecular modeling of GlyT2 mutations. a, side view of the human GlyT2 monomer showing transmembrane helices as colored ribbons. Glycine and two (of three) sodium ions (purple spheres) are depicted. Note that the TM3-TM4 extracellular loop (EL2) was not modeled because of an insertion of residues 312–354 in GlyT2 relative to LeuTAA. b and c, substitution E248K in TM2 replaces a negatively charged glutamate for a positively charged lysine, with a longer side chain that introduces potential clashes (red lines) with Ala-480 and Ala-481 in TM6. This is likely to affect the orientation of the key glycine-binding residue Trp-482. d and e, substitution A275T in TM3 results in several predicted clashes with Ile-279 that are predicted to alter the orientation of the potential glycine-binding residue Tyr-287 and destabilize the unwound region of TM1, which contains several determinants of glycine and Na+ binding. f and g, substitution S513I in TM7 introduces a larger side chain at this position, resulting in predicted clashes with Asn-213 (TM1) and Asn-509 (TM7), both of which are involved in the binding of Na+. Note that Asn-509 and Ser-513 are also predicted to be involved in coordinating Cl− binding to GlyT2. h and i, substitution Y656H in the intracellular TM10-TM11 loop is predicted to abolish a cation-π interaction that exists between Tyr-656 and Arg-660 that may affect folding or stability or interfere with intracellular GlyT2 accessory protein interactions.
Substitution A275T in TM3 results in predicted clashes with Ile-279, which may alter the orientation of the predicted glycine-binding residue Tyr-287 further along TM3 (Fig. 3, d and e). The corresponding residue in LeuT (Tyr-108) not only anchors the leucine substrate but also stabilizes the unwound region of TM1 (36) by binding the amide nitrogen of Leu-25, equivalent to Leu-211 in GlyT2. Thus, A275T could destabilize the unwound region of TM1, which contains several determinants of glycine and Na+ binding. By contrast, substitution S513I in TM7 introduces a much larger side chain at this position, resulting in predicted clashes with Asn-213 (TM1) and Asn-509 (TM7), both of which are involved in Na+ binding (Fig. 3, f and g). It is also noteworthy that the recent discovery (27–29) of residues involved in Cl− binding in GAT-1 and SERT suggests that residues Tyr-233 (TM2), Gln-473, and Ser-477 (TM6) together with Asn-509 and Ser-513 (TM7) are likely to perform equivalent roles in GlyT2 (Fig. 4a). Substitution S513I in TM7 introduces a larger side chain (Fig. 4b), which lacks the hydroxyl necessary for the coordination of the Cl− ion and may prevent Cl− binding. Because a hydroxyl at this relative position is highly conserved in SLC6 transporters (27–29), the S513I substitution is predicted to interfere with glycine transport by disrupting the both Cl− (directly) and Na+ (indirectly) interactions.
FIGURE 4.

Molecular modeling of the GlyT2 chloride ion-binding site. a, exploded view of the human GlyT2 chloride ion binding site showing Cl− as a green sphere and coordination by Tyr-233 in TM2, Gln-473 and Ser-477 in TM6, and Asn-509 and Ser-513 in TM7. b, substitution S513I in TM7 introduces a larger side chain, which lacks the essential hydroxyl necessary for the coordination of Cl− and may occlude the binding site. Because a hydroxyl at this position is highly conserved in SLC6 transporters (27–29), this substitution is likely to disrupt glycine uptake by directly interfering with coordination of Cl−.
Substitution F547S affects a highly conserved residue found in extracellular TM7-TM8 loop 4B (EL4B). EL4B is composed of two short helices separated by an acute bend and participates in conformational mobility during the transport cycle in DAT (37), GAT-1 (38), GlyT1 (39), and SERT (40). Lastly, substitution Y656H in the intracellular TM10-TM11 loop is predicted to abolish a cation-π interaction that exists between Tyr-656 and Arg-660 (Fig. 3, h and i). This interaction is likely to have a significant role in the folding or stabilization of the protein in this unwound region or interfere with important intracellular GlyT2 accessory protein interactions, because a neighboring residue was substituted (G657A) in another individual.
Electrophysiological Analysis of the Single-hit GlyT2 Mutant A275T
To obtain further insights into the transport properties of the only partially functional GlyT2 mutant A275T, we performed a detailed electrophysiological characterization of the charge movement and steady-state kinetics of the A275T mutant expressed in Xenopus oocytes using two-electrode voltage clamp methodology (32, 41). In the absence of glycine, membrane expression of GlyT2 A275T induced Na+-dependent transient currents in response to voltage jumps. These transient currents were sensitive to glycine and the selective GlyT2 inhibitor ORG25543. The maximal charge movement was proportional to the maximal amplitude of glycine-evoked steady-state inward currents (Fig. 5, a and b). Current-voltage (I-V) relationships for different glycine concentrations showed no reversal potential, indicating that tight glycine coupling was preserved in A275T (Fig. 5c), although the dose-response curve showed a marked increase in voltage dependence of the glycine (EC50) and the maximal current (Imax; Fig. 5, d–f). Because glycine is electrically neutral, an increase in the EC50 voltage dependence of the A275T mutant suggests that, in line with our molecular modeling prediction, the allosteric interaction between Na+ and glycine binding may be compromised, particularly at depolarizing potentials. Indeed, the relative transport efficacy (Imax/EC50) of the A275T versus wild-type GlyT2 decreased from 101.3% at 100 mV to 21% at 0 mV (Fig. 5g). To further examine Na+ interactions for the A275T mutant, we compared the kinetics and charge distribution of wild-type GlyT2 and A275T transient currents isolated in the presence of 10 μm ORG25543 (Fig. 6a). On average, the membrane expression of A275T in different batches of oocytes was lower than for wild-type GlyT2 as suggested by the lower maximal charge distribution (Qmax) for A275T (10 ± 0.84 nC, n = 19) than for wild-type GlyT2 (20.6 ± 2.4 nC, n = 14). To assess Na+ binding to the A275T mutant, we measured zδ, a sensitive parameter of the Boltzmann equation used to fit the distribution of the charge movement associated with the transition between the Na+-bound and Na+-unbound conformations of the transporter (42). Although the equivalent charge (zδ) associated with the charge movement was unchanged in A275T (1.19 ± 0.02, n = 19, versus 1.21 ± 0.02 for wild-type GlyT2, n = 14), suggesting similar ionic coupling for wild-type GlyT2 and A275T, the half-distribution potential (V0.5) was shifted to a negative potential by −40 mV from −27.5 ± 1.8 mV (wild-type GlyT2, n = 14) to −67.4 ± 1.44 mV (A275T, n = 19; Fig. 6, b and c). This indicates a decrease in the apparent Na+ affinity for the A275T mutant. Further evidence supporting this finding was provided by the faster kinetics of the off transient currents for A275T during repolarization at −40mV (τw = 10.6 ± 0.5 ms, n = 15) than for wild-type GlyT2 (τw = 19.5 ± 1.45 ms, n = 10).
FIGURE 5.

Current-voltage relationships and dose-response curves for the GlyT2 mutant A275T. a, current traces evoked by 500-ms voltage steps from +50 mV to −140 mV by decrements of 10 mV in representative oocytes expressing wild-type GlyT2 (upper panels) and the GlyT2 A275T mutant (lower panels) in control solution (left traces) or in the presence of glycine (right traces). b, plots of current amplitude evoked by glycine (1 mm) at −40 mV and Qmax (see “Experimental Procedures” and Fig. 6) relationship for oocytes expressing wild-type GlyT2 (upper panel) or GlyT2 A275T (lower panel). The solid lines show the linear regression with a slope of 15.9 s−1 (r2 = 0.773, n = 20) and 24.1 s−1 (r2 = 0.774, n = 14) for wild-type GlyT2 and A275T, respectively. c, voltage dependence of steady-state currents evoked by the glycine concentrations indicated at left in a representative oocyte expressing GlyT2 A275T. d, dose-response curves of the glycine-evoked current at the membrane potentials indicated on right in the same oocytes as in c. The solid lines are the least square fit of the data to the Hill equation: Igly = Imax/(1 + EC50/[gly])n. e, effect of membrane potential on glycine EC50 for oocytes expressing GlyT2 A275T (red circles, n = 8) and wild-type GlyT2 (green circles, n = 7). The solid lines correspond to the fit of the data to the equation EC50 = A + BαuVm, where A = 23.7 and 29 μm, B = 62.1 and 379 μm, and α = 1.4 and 1.34 for wild-type GlyT2 and A275T, respectively, where u = F/RT (R is the universal gas constant, T is the absolute temperature, and F is Faraday's constant). f, voltage dependence of Imax for wild-type GlyT2 (n = 8) and A275T (n = 7). Imax values were normalized by the values at 0 mV. The solid lines correspond to the fit of the data to the equation Imax = −e −βuVm, with β = 0.3 and 0.22 for wild-type GlyT2 and A275T, respectively. g, relative efficacy (Imax/EC50) of A275T/wild-type GlyT2.
FIGURE 6.

Pre-steady-state kinetics of the GlyT2 mutant A275T. a, relaxation currents recorded in representative oocytes expressing wild-type GlyT2 (green) or GlyT2 A275T (red) during the repolarization to the holding potential (−40 mV) after 400-ms step pulses from +50 to −140 mV. Currents recorded in the presence of ORG25543 were subtracted. b, time integral of the relaxation currents for oocytes expressing wild-type GlyT2 (green circles, n = 14) or GlyT2-A275T (red circles, n = 19). The solid lines are the least square fit of the data to the Boltzmann equation: (QV − Qmin)/Qmax = 1/(1 + ezδu(V0.5−Vm)) c, box plots of Qmax, V0.5, zδ, and τ weighted distributions for oocytes expressing wild-type GlyT2 (green, n = 14) or GlyT2 A275T (red, n = 19). The box edges represent the 25th and 75th percentiles with the median line. The errors bars represent the 10th and 90th percentiles.
DISCUSSION
Our study represents the largest multicenter screening study for GlyT2 mutations in hyperekplexia to date, encompassing genotype-phenotype correlations, functional tests of GlyT2 mutants, molecular modeling, and electrophysiology. Our ongoing genetic screening program, which has accrued 230 index cases over the last 18 years (7), has identified an additional 20 mutations in the GlyT2 gene (SLC6A5) in 14 index cases showing homozygous or compound heterozygous recessive inheritance in 93 recently submitted samples (2006–2010). For the remaining three cases, we only identified a single defective allele but excluded allelic dropout and large deletions using MLPA. Although co-expression of the three mutants (W151X, A275T, and R439X) with wild-type GlyT2 did not reveal any dominant-negative effects in an in vitro [3H]glycine uptake assay, it is noteworthy that for the individual harboring the paternal A275T allele, startle was also observed in the father (supplemental Table S2), suggesting that a dominant effect may be present in vivo. However, inheritance of a single defective SLC6A5 allele (Y377X) has been observed in another study (18), so another possibility is that a second mutation in a promoter or regulatory element could explain cases 12 and 13 (Table 1). However, because the current minimal SLC6A5 promoter encompasses >90 kb upstream of exon 1 and regulatory motifs remain uncharacterized (43), it is not currently straightforward to address this possibility. Another plausible explanation is that these two cases may represent the first examples of digenic hyperekplexia, where single mutations in two unlinked genes are present in a single individual, and the combination of the two genetic lesions causes a disease phenotype that is not apparent in individuals harboring only one of these mutations.
Despite this caveat, our study effectively triples the number of known cases with SLC6A5 mutations, firmly establishing mutation in the GlyT2 gene as a second major cause of startle disease. Taken together with our recent study of mutations in the GlyR α1 subunit gene (GLRA1) (10), it is now apparent that recessive hyperekplexia is far more common on a population basis than dominant hyperekplexia, which received initial attention because of the characterization of extended families (8, 9) harboring dominant GLRA1 mutations such as R271 Q/L. In common with previous studies (17, 18), genotype-phenotype analysis revealed that GlyT2 mutations in humans are not as severe as those observed in mice, dogs, and cattle, where mutations in SLC6A5 cause extreme muscle rigidity, tremor, difficulty in breathing, and early mortality (19, 20, 44). However, it is noteworthy that therapeutic interventions are rarely applied in animals with startle disease, because this condition is often only recognized post mortem.
In humans, it is clear that SLC6A5 mutations result in more severe clinical signs than mutations in GLRA1. In particular, the majority (over 90%) of children with SLC6A5-linked hyperekplexia have recurrent neonatal apneas. This may explain the clinical and parental anxiety surrounding hyperekplexia and could underlie a small but discernible increase in mortality rate. Whether the increased rate of specific measures of developmental delay (such as late motor milestones) or learning disability is due to these apnea attacks is hard to prove. Because almost all children with SLC6A5-linked hyperekplexia have apneic attacks (and the number or severity of the attacks are not recorded), we cannot explain why nearly half the patients did not have any developmental or cognitive issues. Another potential explanation for these differences is that defective presynaptic glycine uptake is predicted to affect the function of multiple GlyR subtypes, i.e., α2β and α3β, as well as the predominant α1β isoform. In this regard, it is noteworthy that GlyR α3 knock-out mice were recently shown to have an irregular respiratory rhythm (45).
Our molecular modeling and [3H]glycine uptake studies have revealed the existence of several new pathogenic mechanisms that result in defective glycine transport. Previously known mechanisms included truncation of the GlyT2 polypeptide caused by nonsense and frameshift mutations (17, 18), to which we can now add splice site mutations that likely result in either exon skipping on intron retention (cases 1, 7, and 9; Table 1). Previous pathogenic mechanisms reported for missense mutations (17) that resulted in apparently normal membrane trafficking included defects in glycine (W482R in TM6) or Na+ binding (N509S in TM7). With the exception of S513I in TM7, the new missense substitutions reported in this study (L237P, P243T, E248K, A275T, F547S, Y656H, and G657A) do not directly affect predicted Na+-, Cl−-, or glycine-binding residues (Fig. 1). However, homology modeling of human GlyT2 based on the structure of the bacterial leucine transporter LeuT (23) and analysis of key structure-function studies of related transporters has allowed us to suggest several hypothetical pathogenic mechanisms for hyperekplexia mutations. These include mutations that are predicted to: (i) affect the conformation of TM2 (L237P, P243T); (ii) indirectly affect glycine (A275T and E248K) or Na+ (A275T and S513I) binding; (iii) directly affect Cl− binding (S513I); (iv) affect the conformational mobility of EL4 (F547S); and (v) affect cation-π or accessory protein interactions with the intracellular TM10-TM11 loop (Y656H and G657A). Although these mechanisms are all of intrinsic interest, our study confirms that the Cl−-binding residue Ser-513 in GlyT2 (equivalent to Ser-372 in SERT and Ser-331 in GAT-1) has a key functional role in glycine uptake. Replacement of Ser-372 in SERT with glutamate (S372E) or aspartate (S372D) results in Cl−-insensitive transporters with low uptake activity. However, consistent with our findings, substitution S372L completely abolished serotonin uptake (28), suggesting that either a Cl− ion or a carboxylate side chain as in LeuT is required in this area for the normal uptake cycle.
Lastly, our detailed electrophysiological characterization of the A275T mutation demonstrated that this mutation results in an increase in glycine EC50 at depolarizing potentials that reduces the ability of the transporter to pump glycine at low concentrations, a situation that prevails at the synaptic cleft. In addition, lower membrane expression of A275T may also reduce glycine uptake below the level required for supplying glycine in axons for maintaining adequate vesicle refilling (46). In line with our molecular modeling predictions, alterations in glycine uptake in A275T are primarily linked to a decrease in the affinity of Na+ binding with the apo-transporter as indicated by the leftward shift in the potential of half distribution. The Boltzmann distribution of charge movement indicated that the fraction of transporter bound with Na+ will decrease by a factor of ∼2 compared with wild-type GlyT2 at a negative potential of −60mV (0.42 and 0.82, respectively). Because glycine affinity is Na+-dependent (41), this difference in the fraction of Na+-bound transporter suggests that 50% of A275T may not be able to capture synaptically released glycine, thus reducing the local recycling that is required for the replenishment of vesicular transmitter levels (46).
Although sequential genetic analysis of our cohort of hyperekplexia DNAs has revealed many additional mutations in GLRA1 and SLC6A5, a high proportion of index cases (65%) remain mutation-negative for these major genes of effect. This suggests that other hyperekplexia/startle disease genes exist or that a number of different mutations remain to be characterized in regulatory elements at existing loci, e.g., upstream promoters. There are certainly precedents for extensive genetic heterogeneity in other neurological disorders. For example, mutations in both inhibitory GABAA receptor and voltage-gated Na+ channel genes are associated with generalized epilepsy with febrile seizures plus (GEFS+). Future challenges are to: (i) understand the regulatory elements controlling transcription of GLRA1, GLRB, and SLC6A5 and (ii) use alternative disease gene discovery techniques, such as array comparative genomic hybridization and exome/genome sequencing to search for pathogenic mutations in novel startle disease genes.
Supplementary Material
This work was supported by Medical Research Council Grant G0601585 (to R. J. H., K. H., and M. I. R.), Action Medical Research Grant 1966 (to R. J. H., R. H. T., and M. I. R.), a Bloomsbury Colleges studentship (to V. M. J., M. T., and R. J. H.), Career Development Award G0600084 (to M. T.), a National Institute of Social Care and Health Research grant (to M. I. R., R. H. T., and S. K. C.), and an Association Française contre les Myopathies grant (to S. S.).

This article contains supplemental Tables S1 and S2.
- GlyR
- glycine receptor
- MLPA
- multiplex ligation-dependent probe amplification
- LeuT
- leucine transporter.
REFERENCES
- 1. Nigro M. A., Lim H. C. (1992) Hyperekplexia and sudden neonatal death. Pediatr. Neurol. 8, 221–225 [DOI] [PubMed] [Google Scholar]
- 2. Vigevano F., Di Capua M., Dalla Bernardina B. (1989) Startle disease. An avoidable cause of sudden infant death. Lancet 1, 216. [DOI] [PubMed] [Google Scholar]
- 3. Bakker M. J., van Dijk J. G., van den Maagdenberg A. M., Tijssen M. A. (2006) Startle syndromes. Lancet Neurol. 5, 513–524 [DOI] [PubMed] [Google Scholar]
- 4. Thomas R. H., Stephenson J. B., Harvey R. J., Rees M. I. (2010) Hyperekplexia. Stiffness, startle and syncope. J. Ped. Neurol. 8, 11–14 [Google Scholar]
- 5. Bakker M. J., Peeters E. A., Tijssen M. A. (2009) Clonazepam is an effective treatment for hyperekplexia due to a SLC6A5 (GlyT2) mutation. Mov. Disord. 24, 1852–1854 [DOI] [PubMed] [Google Scholar]
- 6. Harvey R. J., Topf M., Harvey K., Rees M. I. (2008) The genetics of hyperekplexia. More than startle! Trends Genet. 24, 439–447 [DOI] [PubMed] [Google Scholar]
- 7. Davies J. S., Chung S. K., Thomas R. H., Robinson A., Hammond C. L., Mullins J. G., Carta E., Pearce B. R., Harvey K., Harvey R. J., Rees M. I. (2010) The glycinergic system in human startle disease. A genetic screening approach. Front. Mol. Neurosci. 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shiang R., Ryan S. G., Zhu Y. Z., Hahn A. F., O'Connell P., Wasmuth J. J. (1993) Mutations in the α1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat. Genet. 5, 351–358 [DOI] [PubMed] [Google Scholar]
- 9. Shiang R., Ryan S. G., Zhu Y. Z., Fielder T. J., Allen R. J., Fryer A., Yamashita S., O'Connell P., Wasmuth J. J. (1995) Mutational analysis of familial and sporadic hyperekplexia. Ann. Neurol. 38, 85–91 [DOI] [PubMed] [Google Scholar]
- 10. Chung S. K., Vanbellinghen J. F., Mullins J. G., Robinson A., Hantke J., Hammond C. L., Gilbert D. F., Freilinger M., Ryan M., Kruer M. C., Masri A., Gurses C., Ferrie C., Harvey K., Shiang R., Christodoulou J., Andermann F., Andermann E., Thomas R. H., Harvey R. J., Lynch J. W., Rees M. I. (2010) Pathophysiological mechanisms of dominant and recessive GLRA1 mutations in hyperekplexia. J. Neurosci. 30, 9612–9620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Becker K., Hohoff C., Schmitt B., Christen H. J., Neubauer B. A., Sandrieser T., Becker C. M. (2006) Identification of the microdeletion breakpoint in a GLRA1 null allele of Turkish hyperekplexia patients. Hum. Mutat. 27, 1061–1062 [DOI] [PubMed] [Google Scholar]
- 12. Sirén A., Legros B., Chahine L., Misson J. P., Pandolfo M. (2006) Hyperekplexia in Kurdish families. A possible GLRA1 founder mutation. Neurology 67, 137–139 [DOI] [PubMed] [Google Scholar]
- 13. Rees M. I., Lewis T. M., Kwok J. B., Mortier G. R., Govaert P., Snell R. G., Schofield P. R., Owen M. J. (2002) Hyperekplexia associated with compound heterozygote mutations in the β-subunit of the human inhibitory glycine receptor (GLRB). Hum. Mol. Genet. 11, 853–860 [DOI] [PubMed] [Google Scholar]
- 14. Al-Owain M., Colak D., Al-Bakheet A., Al-Hashmi N., Shuaib T., Al-Hemidan A., Aldhalaan H., Rahbeeni Z., Al-Sayed M., Al-Younes B., Ozand P. T., Kaya N. (2012) Novel mutation in GLRB in a large family with hereditary hyperekplexia. Clin. Genet. 81, 479–484 [DOI] [PubMed] [Google Scholar]
- 15. Rees M. I., Harvey K., Ward H., White J. H., Evans L., Duguid I. C., Hsu C. C., Coleman S. L., Miller J., Baer K., Waldvogel H. J., Gibbon F., Smart T. G., Owen M. J., Harvey R. J., Snell R. G. (2003) Isoform heterogeneity of the human gephyrin gene (GPHN), binding domains to the glycine receptor, and mutation analysis in hyperekplexia. J. Biol. Chem. 278, 24688–24696 [DOI] [PubMed] [Google Scholar]
- 16. Harvey K., Duguid I. C., Alldred M. J., Beatty S. E., Ward H., Keep N. H., Lingenfelter S. E., Pearce B. R., Lundgren J., Owen M. J., Smart T. G., Lüscher B., Rees M. I., Harvey R. J. (2004) The GDP-GTP exchange factor collybistin. An essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rees M. I., Harvey K., Pearce B. R., Chung S. K., Duguid I. C., Thomas P., Beatty S., Graham G. E., Armstrong L., Shiang R., Abbott K. J., Zuberi S. M., Stephenson J. B., Owen M. J., Tijssen M. A., van den Maagdenberg A. M., Smart T. G., Supplisson S., Harvey R. J. (2006) Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat. Genet. 38, 801–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eulenburg V., Becker K., Gomeza J., Schmitt B., Becker C. M., Betz H. (2006) Mutations within the human GlyT2 (SLC6A5) gene associated with hyperekplexia. Biochem. Biophys. Res. Commun. 348, 400–405 [DOI] [PubMed] [Google Scholar]
- 19. Charlier C., Coppieters W., Rollin F., Desmecht D., Agerholm J. S., Cambisano N., Carta E., Dardano S., Dive M., Fasquelle C., Frennet J. C., Hanset R., Hubin X., Jorgensen C., Karim L., Kent M., Harvey K., Pearce B. R., Simon P., Tama N., Nie H., Vandeputte S., Lien S., Longeri M., Fredholm M., Harvey R. J., Georges M. (2008) Highly effective SNP-based association mapping and management of recessive defects in livestock. Nat. Genet. 40, 449–454 [DOI] [PubMed] [Google Scholar]
- 20. Gill J. L., Capper D., Vanbellinghen J. F., Chung S. K., Higgins R. J., Rees M. I., Shelton G. D., Harvey R. J. (2011) Startle disease in Irish wolfhounds associated with a microdeletion in the glycine transporter GlyT2 gene. Neurobiol. Dis. 43, 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lobley A., Sadowski M. I., Jones D. T. (2009) pGenTHREADER and pDomTHREADER. New methods for improved protein fold recognition and superfamily discrimination. Bioinformatics 25, 1761–1767 [DOI] [PubMed] [Google Scholar]
- 22. Söding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 24. Shen M. Y., Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eswar N., John B., Mirkovic N., Fiser A., Ilyin V. A., Pieper U., Stuart A. C., Marti-Renom M. A., Madhusudhan M. S., Yerkovich B., Sali A. (2003) Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31, 3375–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dunbrack R. L., Jr. (2002) Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 12, 431–440 [DOI] [PubMed] [Google Scholar]
- 27. Zomot E., Bendahan A., Quick M., Zhao Y., Javitch J. A., Kanner B. I. (2007) Mechanism of chloride interaction with neurotransmitter:sodium symporters. Nature 449, 726–730 [DOI] [PubMed] [Google Scholar]
- 28. Forrest L. R., Tavoulari S., Zhang Y. W., Rudnick G., Honig B. (2007) Identification of a chloride ion binding site in Na+/Cl−-dependent transporters. Proc. Natl. Acad. Sci. U.S.A. 104, 12761–12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ben-Yona A., Bendahan A., Kanner B. I. (2011) A glutamine residue conserved in the neurotransmitter sodium, symporters is essential for the interaction of chloride with the GABA Transporter GAT-1. J. Biol. Chem. 286, 2826–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hinsen K. (2000) The molecular modeling toolkit. A new approach to molecular simulations. J. Comp. Chem. 21, 79–85 [Google Scholar]
- 31. Cornell W. D., Cieplak P., Bayly C. I., Gould I. R., Merz K. M., Jr., Ferguson D. M., Spellmeyer D. C., Fox T., Caldwell J. W., Kollman P. A. (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197 [Google Scholar]
- 32. Roux M. J., Martinez-Maza R., Le Goff A., Lopez-Corcuera B., Aragon C., Supplisson S. (2001) The glial and the neuronal glycine transporters differ in their reactivity to sulfhydryl reagents. J. Biol. Chem. 276, 17699–17705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stalder L., Mühlemann O. (2008) The meaning of nonsense. Trends Cell Biol. 18, 315–321 [DOI] [PubMed] [Google Scholar]
- 34. Bartholomäus I., Milan-Lobo L., Nicke A., Dutertre S., Hastrup H., Jha A., Gether U., Sitte H. H., Betz H., Eulenburg V. (2008) Glycine transporter dimers. Evidence for occurrence in the plasma membrane. J. Biol. Chem. 283, 10978–10991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bajaj K., Madhusudhan M. S., Adkar B. V., Chakrabarti P., Ramakrishnan C., Sali A., Varadarajan R. (2007) Stereochemical criteria for prediction of the effects of proline mutations on protein stability. PLoS Comput. Biol. 3, e241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Singh S. K., Piscitelli C. L., Yamashita A., Gouaux E. (2008) A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Norregaard L., Frederiksen D., Nielsen E. O., Gether U. (1998) Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J. 17, 4266–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zomot E., Kanner B. I. (2003) The interaction of the γ-aminobutyric acid transporter GAT-1 with the neurotransmitter is selectively impaired by sulfhydryl modification of a conformationally sensitive cysteine residue engineered into extracellular loop IV. J. Biol. Chem. 278, 42950–42958 [DOI] [PubMed] [Google Scholar]
- 39. Ju P., Aubrey K. R., Vandenberg R. J. (2004) Zn2+ inhibits glycine transport by glycine transporter subtype 1b. J. Biol. Chem. 279, 22983–22991 [DOI] [PubMed] [Google Scholar]
- 40. Mitchell S. M., Lee E., Garcia M. L., Stephan M. M. (2004) Structure and function of extracellular loop 4 of the serotonin transporter as revealed by cysteine-scanning mutagenesis. J. Biol. Chem. 279, 24089–24099 [DOI] [PubMed] [Google Scholar]
- 41. Roux M. J., Supplisson S. (2000) Neuronal and glial glycine transporters have different stoichiometries. Neuron 25, 373–383 [DOI] [PubMed] [Google Scholar]
- 42. Mager S., Cao Y., Lester H. A. (1998) Measurement of transient currents from neurotransmitter transporters expressed in Xenopus oocytes. Methods Enzymol. 296, 551–566 [DOI] [PubMed] [Google Scholar]
- 43. Zeilhofer H. U., Studler B., Arabadzisz D., Schweizer C., Ahmadi S., Layh B., Bösl M. R., Fritschy J. M. (2005) Glycinergic neurons expressing enhanced green fluorescent protein in bacterial artificial chromosome transgenic mice. J. Comp. Neurol. 482, 123–141 [DOI] [PubMed] [Google Scholar]
- 44. Gomeza J., Ohno K., Hülsmann S., Armsen W., Eulenburg V., Richter D. W., Laube B., Betz H. (2003) Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron 40, 797–806 [DOI] [PubMed] [Google Scholar]
- 45. Manzke T., Niebert M., Koch U. R., Caley A., Vogelgesang S., Hülsmann S., Ponimaskin E., Müller U., Smart T. G., Harvey R. J., Richter D. W. (2010) Serotonin receptor 1A-modulated phosphorylation of glycine receptor α3 controls breathing in mice. J. Clin. Invest. 120, 4118–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rousseau F., Aubrey K. R., Supplisson S. (2008) The glycine transporter GlyT2 controls the dynamics of synaptic vesicle refilling in inhibitory spinal cord neurons. J. Neurosci. 28, 9755–9768 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.