

Background: CXCR7 is an atypical heptahelical receptor that functions as scavenger for the endogenous chemokine ligand but fails to signal through G-proteins.
Results: The CXCR7 C terminus causes uncoupling from G-protein, rapid receptor-mediated ligand uptake, and constitutive receptor degradation.
Conclusion: Atypical CXCR7 functions are encoded in C-terminal elements.
Significance: Identification of structural determinants regulating atypical and canonical functions of heptahelical receptors.
Keywords: Calcium, Chemokines, Cxcr4, G Protein-coupled Receptors (GPCR), Receptor Endocytosis, CXCL12, CXCR7, SDF-1, Decoy Receptor, Scavenger
Abstract
CXCL12 signaling through G protein-coupled CXCR4 regulates cell migration during ontogenesis and disease states including cancer and inflammation. The second CXCL12-receptor CXCR7 modulates the CXCL12/CXCR4 pathway by acting as a CXCL12 scavenger and exerts G protein-independent functions. Given the distinct properties of CXCR4 and CXCR7, we hypothesized that the distinct C-terminal domains differently regulate receptor trafficking and stability. Here, we examined epitope-tagged wild type and C-terminal mutant receptors in human embryonic kidney cells (HEK293) with respect to trafficking, stability, 125I-CXCL12 degradation, and G protein-coupling. The 24 CXCR7 C-terminal residues were sufficient to promote rapid spontaneous internalization. Replacement of the CXCR7 C terminus with that of CXCR4 (CXCR7–4tail mutant) abolished spontaneous internalization but permitted ligand-induced internalization and phosphorylation at the heterologous domain. The reverse tail-swap caused ligand-independent internalization of the resulting CXCR4–7tail mutant. Receptor-mediated 125I-CXCL12 uptake and release of 125I-CXCL12 degradation products were accelerated with receptors bearing the CXCR7 C terminus and impaired after conversion of CXCR7 C-terminal serine/threonine residues into alanines. C-terminal lysine residues were dispensable for plasma membrane targeting and the CXCL12 scavenger function but involved in constitutive degradation of CXCR7. Although the CXCR7 C terminus abolished G protein coupling in the CXCR4–7tail mutant, replacement of the CXCR7 C terminus, CXCR7 second intracellular loop, or both domains with the corresponding CXCR4 domain did not result in a G protein-coupled CXCR7 chimera. Taken together, we provide evidence that the CXCR7 C terminus influences the ligand-uptake/degradation rate, G protein coupling, and receptor stability. Regulatory pathways targeting CXCR7 C-terminal serine/threonine sites may control the CXCL12 scavenger activity of CXCR7.
Introduction
The chemokine stromal cell-derived factor-1 (CXCL12) is an indispensable regulator of cell migration processes that play a key role in a plethora of physiological and pathological conditions including embryogenesis, inflammation, and tumor metastasis (1–5). CXCR4 represents the classical CXCL12 receptor mediating CXCL12-dependent chemoattraction (6, 7). Recently, CXCR7 was identified as the second CXCL12 receptor (8). Although it is well established that CXCR7 plays a critical role in many CXCL12-dependent processes (1, 9, 10), close examination of CXCR7 revealed atypical features. In particular, identification of CXCR7-dependent G protein signaling and chemoattraction proved difficult in most tissues. Instead, CXCR7 was demonstrated to use a β-arrestin-dependent pathway resulting in MAP kinase activation (11–14). Recently, G protein coupling was demonstrated for CXCR7 in astrocyte cultures (15).
A distinctive feature of CXCR7 is its high affinity for CXCL12 and its ability to mediate efficient uptake and degradation of CXCL12 (16, 17). The decoy receptor function of CXCR7 exerts strong influence on CXCL12/CXCR4-dependent guidance of migratory cells. First, CXCR7 expressed by nonmigratory cells helps to shape chemokine gradients and prevents migration to nondesired locations, and second, CXCR7 expressed by migratory cells helps to sustain responsiveness of the CXCR4 pathway (10, 18).
Thus, the two CXCL12 receptors exert fundamentally different functions. CXCR4 represents a G protein-coupled chemoattractant receptor showing rapid phosphorylation at the C-terminal domain and β-arrestin-dependent endocytosis in response to CXCL12 stimulation (19, 20). Sustained stimulation causes CXCR4 degradation (21), a mechanism expected to desensitize the CXCL12/CXCR4 pathway once migratory cells reach CXCL12-rich environments. In contrast, CXCR7 is specialized on CXCL12 uptake and undergoes ligand-independent internalization (16, 17). Little is known about structural determinants that uncouple CXCR7 from G proteins, cause ligand-independent internalization, and permit CXCR7 to act as a decoy receptor. Furthermore, it has not been established how ligand-induced and ligand-independent CXCR7 internalization affect CXCR7 degradation.
In this study, we used wild type and tail-swap mutant receptors in an identical expression vector to directly compare how the CXCR4 and CXCR7 C-terminal domains influence receptor trafficking, stability, and function. Our findings indicate that the CXCR7 C terminus permits fast endocytosis of ligand-free and CXCL12-loaded receptors. Constitutively internalizing receptors bearing the CXCR7 C terminus were rapidly degraded in the absence of CXCL12. Although CXCL12 caused degradation of CXCR4, it did not accelerate degradation of CXCR7. G protein coupling could not be demonstrated for wild type CXCR7 and not be accomplished by replacing the CXCR7 C-terminal domain and CXCR7 second intracellular loop by the corresponding domains of CXCR4.
MATERIALS AND METHODS
Compounds and Antibodies
CXCL12 (PeproTech, Hamburg, Germany), 125I-CXCL12 (ARI0129, Hartmann Analytic, Braunschweig, Germany), AMD3100 (A5602, Sigma), cycloheximide (C4859, Sigma), λ-protein phosphatase (P0753, New England Biolabs GmbH, Frankfurt am Main, Germany), CCX733 and 11G8 monoclonal mouse anti-CXCR7 antibody (ChemoCentryx, Mountain View, CA) (22), anti-transferrin receptor antibody (13-6800; Invitrogen), and UMB-2 monoclonal rabbit anti-CXCR4 antibody (23). Polyclonal rabbit antisera against the hemagglutinin (HA) epitope MYPYDVPDYA were generated by injecting the peptide into rabbits 631–633 after coupling to keyhole limpet hemocyanin and emulsification with Freund's adjuvant. Anti-HA sera {631} and {632} were immunoaffinity purified and used for Western blot, ELISA, and immunocytochemistry including nontransfected cells as controls.
Plasmid Constructs
cDNAs for mouse wild type CXCR4 and CXCR7 were amplified by reverse transcription PCR from mouse spleen cDNA with NheI/NotI and NheI/ApaI extensions, respectively. Constructs received an N-terminal hemagglutinin epitope tag (MYPYDVPDYA) and were subcloned into pcDNATM 3.1(+) vector (V790-20, Invitrogen). The resulting constructs are referred to as CXCR4-WT and CXCR7-WT throughout the manuscript. cDNAs encoding C-terminal mutants were generated by gene synthesis (Eurofins MWG Operon, Ebersberg, Germany; see Table 1 for the protein sequences). Gene synthesis products were inserted into CXCR4-WT using the endogenous SphI site (bp 967) and the NotI extension. The CXCR7-WT construct received an XhoI site at bp 865 by introducing a silent mutation via site-directed mutagenesis. CXCR7 mutants were then generated using XhoI and ApaI. To replace the CXCR7 second cytosolic domain with the corresponding CXCR4 sequence, the desired sequence upstream of the XhoI site was synthesized and inserted using NheI and XhoI. Sequence identity of all constructs was verified by double strand sequencing. A CXCR7ΔC-term mutant lacking the C-terminal domain was provided by ChemoCentryx (22).
TABLE 1.
Amino acid sequences of wild type and mutant CXCR7 and CXCR4 receptors
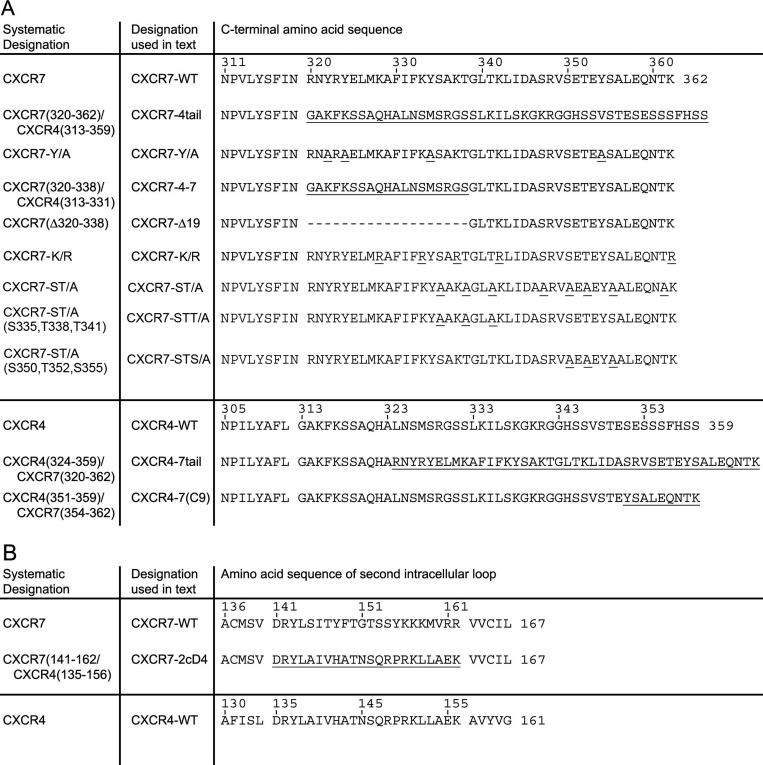
A, sequences of C-terminal mutants start with the conserved NPxxY motif at the end of the seventh transmembrane domain. B, sequences of CXCR7 chimeras in which the second cytosolic domain was replaced with the corresponding region of CXCR4 start with the end of the third transmembrane domain.
Cell Culture and Transfection
Human embryonic kidney cells (HEK293 cells, DSMZ, Braunschweig, Germany) were cultivated in DMEM (PAA Laboratories, Pasching, Austria) supplemented with 10% defined fetal bovine serum (FBS GOLD, PAA), 2 mm l-glutamine, and 100 units/ml of penicillin/streptomycin (PAA). Chinese hamster ovary cells (CHO-K1 cells, DSMZ) were cultivated in Ham's F-12 (PAA) supplemented with 10% FBS, 2 mm l-glutamine, and 100 units/ml of penicillin/streptomycin. Transient transfection was accomplished using JetPEI reagent (PEQLAB Biotechnology, Erlangen, Germany). Jurkat T cells (ACC282, DSMZ) were cultivated in RPMI (PAA) supplemented with 10% defined FBS, 2 mm l-glutamine, and 100 units/ml of penicillin/streptomycin.
Radioligand Assays
HEK293 cells were seeded at a density of 150,000 cells/well in poly-l-lysine-coated 24-well plates, transfected with receptor-encoding constructs or empty vector 24 h after plating, and used for assay 24 h after transfection. 125I-CXCL12 (2200 Ci/mmol) was diluted with Ultra-MEM (Lonza Cologne GmbH, Cologne, Germany) containing 1% BSA (PAA) to a final concentration of 25 pmol/liter. In all radioligand assays, cultures received 300 μl of diluted 125I-CXCL12. The Cobra II γ-counter (Packard) was used to measure [125I].
Homologous competitive radioligand binding was performed by incubating cells for 2 h at 4 °C with 125I-CXCL12 containing increasing amounts of unlabeled CXCL12 (0.01–640 nm). Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and then lysed in 300 μl of 10 mm Tris buffer (pH 7.4). To estimate the IC50 (i.e. the equilibrium of bound 125I-CXCL12 and bound unlabeled CXCL12) the data were analyzed by nonlinear fitting using the “homologous competitive binding curve” option of GraphPad Prism 4.0a software.
Pulse-chase analyses of radioligand internalization were performed as described (24) by loading cells with 125I-CXCL12 for 2 h at 4 °C. Cultures were washed with ice-cold PBS and harvested either immediately (starting value) or transferred for 0, 5, 15, and 30 min to 37 °C to permit internalization before residual surface-bound 125I-CXCL12 was stripped by a double wash with acidic citrate buffer (50 mm sodium citrate, 90 mm NaCl, pH 4.5 (25)). Then, cells were lysed in 300 μl of 10 mm Tris buffer (pH 7.4). Counts of mock-transfected cultures were subtracted from counts of receptor-transfected cultures undergoing the same treatment. Percent internalization was calculated by dividing intracellular counts of acid-washed cultures by the starting value (initial bound 125I-CXCL12). To determine the effect of C-terminal point mutations (ST/A and K/R mutations) on CXCL12/CXCR7 internalization, percent of internalization in mutant receptor-transfected cultures was divided by percent of internalization in CXCR7-WT-transfected sister cultures.
For 125I-CXCL12 uptake and degradation experiments, HEK293 cells were transiently transfected using plasmid concentrations that produced similar signal intensities for the wild type and corresponding mutant receptors in anti-HA immunoblots. Thus, experimental conditions with similar total receptor levels were used. 125I-CXCL12 was applied to the transfectants at 37 °C for different time intervals. For radioligand uptake measurements, cells were subjected to acidic wash, lysed, and counted. To determine release of 125I-CXCL12 degradation products, cell supernatants were collected, precipitated with ice-cold 12.5% trichloroacetic acid (TCA) at 4 °C for 15 min, and pelleted at 21,000 × g at 4 °C for a further 15 min. Soluble counts in the supernatants were considered 125I-CXCL12 degradation products (26).
Transwell Migration Assay
HEK293 cells were seeded at a density of 700,0000 cells/well in 6-well plates. On the next day, the cells were transiently transfected with empty vector (mock) or receptor-encoding constructs using plasmid concentrations that produced similar expression levels of the different receptors when tested in immunoblots. The transfectants received Opti-MEM without phenol red supplemented with 20 nm CXCL12 for 16 h. The culture supernatants were collected, centrifuged, and used as chemoattractant for Jurkat T cells in a transwell migration assay (Costar number 3421). Opti-MEM without CXCL12 was used as control. Briefly, 250,000 Jurkat cells were seeded in 200 μl of Opti-MEM + 0.01% BSA in the upper chamber and permitted to migrate for 2 h at 37 °C. Cells in the lower chamber were stained with Calcein-AM (Sigma), harvested, centrifuged, resuspended in 100 μl of PBS, and transferred to a 96-well plate. Fluorescence was used as nonbiased readout for cell numbers in the lower chamber and recorded using a FlexStation 3 microplate reader.
Immunocytochemistry
150,000 cells/well were seeded in 12-well plates containing poly-l-lysine-coated coverslips. Cells were transfected after 24 h and used for immunocytochemistry 24 h thereafter. For pulse labeling of surface receptors, cells were transferred to 4 °C after the growth medium had been replaced by ice-cold Opti-MEM (Invitrogen) containing affinity purified rabbit anti-HA (0.25 μg/ml) or 11G8 antibody (4 μg/ml). After 60 min, cultures were washed with PBS, supplemented with Opti-MEM containing appropriate ligands or vehicle, and placed at 37 °C for various intervals to allow receptor internalization. Then, cells were fixed, permeabilized, and detected using Cy3-AffiniPure goat anti-rabbit IgG (number 111-165-003, Jackson ImmunoResearch Laboratories). Labeling was imaged using a LSM 510 Meta confocal laser scanning microscope (Carl Zeiss, Jena, Germany).
ELISA
Cultures were prepared as for the radioligand uptake assay. Surface receptors were labeled with anti-HA antibody and allowed to internalize as described in the previous paragraph before fixative was applied. Detection was performed in nonpermeabilized cultures using peroxidase-conjugated secondary antibody (number NA9340V, GE Healthcare) and One-StepTM Ultra TMB-ELISA reagent (number 34028, Thermo Fisher Scientific Inc.). Optical density was measured at 450 nm using the FlexStation3 microplate reader (Molecular Devices, Sunnyvale, CA). Parallel sets of mock-transfected cultures were prepared for background subtraction.
Immunoblotting
1,000,000 HEK293 cells were plated, transfected 24 h after plating, and used for assay 48 h after transfection. Treatment with cycloheximide (10 μg/ml) was performed at 37 °C. Cells were lysed in 1 ml of RIPA buffer and centrifuged for 30 min at 23,000 × g at 4 °C. Glycoproteins were enriched using wheat germ lectin-agarose beads as described (27). For dephosphorylation beads were incubated for 3 h at 37 °C with λ-protein phosphatase as described (10). Beads were washed with RIPA buffer before proteins were eluted for 20 min at 60 °C with SDS sample buffer. Samples were then subjected to 10% SDS-polyacrylamide gel electrophoresis and immunoblotted. Blots were immunodetected using anti-HA, anti-CXCR4 (UMB-2), anti-CXCR7 (11G8), or anti-transferrin receptor antibodies and appropriate peroxidase-conjugated secondary antibodies (number sc2004, Santa Cruz Biotechnology Inc.; number NXA931, GE Healthcare). Signals were imaged and analyzed using the Fusion-FX7 Chemiluminescence System and BIO-1D software (PEQLAB).
[Ca2+]i Measurements
Cells were batch transfected using JetPEI. A total of 50,000 HEK or 30,000 CHO cells per well were seeded onto poly-l-lysine-coated 96-well plates. G protein signaling was examined 24 h after plating by measuring increases in [Ca2+]i using the FlexStation3 microplate reader. For recordings, medium was replaced by probenecid-Ringer's solution (130 mm NaCl, 4 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 10 mm HEPES, pH 7.3, 20 mm glucose, 5 mm probenecid). Calcium loading dye (Calcium 4 assay kit, Molecular Devices) was added according to the manufacturer's instructions and measurements were started after a 1-h incubation at 37 °C. Baseline values were recorded for 30 s before CXCL12 application to a final concentration of 80 nm. Cell viability was confirmed by subsequent stimulation of a calcium response by the application of ATP at a final concentration of 20 μm. In antagonist assays 30 μm AMD3100 was added 15 min before start of measurements.
G Proteins
Murine Gα15, a promiscuous member of the Gq/11 G protein subfamily, was cotransfected with receptor constructs. Because chemokine receptors including CXCR4 signal through the Gi/o subfamily (28, 29), we also used chimeras that were engineered to link Gi/o-coupled receptors to Ca2+ mobilization: chimera qi5 consisting of murine Gαq with the 5 C-terminal amino acids changed from Gαq to Gαi2 (30) and chimera 16z44 consisting of human Gα16 and rat Gαz (31, 32).
Statistics
Statistical tests were done using Student's t test, one-way or two-way ANOVA3 including Bonferroni post tests. Levels of significance of p < 0.05, 0.01, and 0.001 were indicated by one, two, and three symbols, respectively.
RESULTS
Regulation of Ligand-independent Receptor Internalization by the CXCR7 C-terminal Domain
To study the influence of the CXCR7 C-terminal domain on CXCR7 regulation and function, we expressed wild type receptors (CXCR7-WT, CXCR4-WT) and tail-swap mutant receptors (CXCR7–4tail, CXCR4–7tail) with N-terminal HA-epitope tags in HEK293 cells (sequence information is detailed in Table 1). We first examined ligand-independent receptor trafficking by an immunocytochemical approach. Surface receptors were pulse-labeled at their extracellular N-terminal domain by applying anti-HA antibody to live cells at 4 °C (internalization-restrictive condition). Cells were washed and fixed after different ligand-free intervals (0, 5, 15, and 30 min) during which the cells were kept at 37 °C (internalization-permissive condition). To permit the detection of antibody-labeled receptors both at the cell surface and inside the cells, we permeabilized the cells before applying Cy3-coupled secondary antibody. Mock-transfected cells included as controls proved negative (not shown). Immediately after pulse labeling of surface receptors (0 min), prominent labeling was observed at the cell surface with each of the constructs (Fig. 1, A–D), indicating that the wild type and the chimeric receptors were targeted to the plasma membrane. Pulse-labeled CXCR7-WT and CXCR4–7tail receptors were partially internalized after 5 and 15 min (not shown) and appeared fully internalized after 30 min (Fig. 1, E and H). In contrast, only a minor proportion of the pulse-labeled CXCR4-WT and CXCR7–4tail receptors were internalized during the 30-min ligand-free interval (Fig. 1, F and G).
FIGURE 1.

Differences in ligand-independent internalization of wild type and tail-swap mutant CXCR4 and CXCR7 receptors. HEK293 cells were transiently transfected with constructs for HA-tagged receptors as indicated. A–H, surface receptors were pulse labeled with anti-HA antibody applied to live HEK293 cells at 4 °C. Confocal images show the subcellular localization of the labeled receptors after the anti-HA pulse (A–D) or after a 30-min ligand-free interval at 37 °C (E–H). I, quantitative analysis of receptor internalization by ELISA. Surface receptor levels after different ligand-free intervals are given as percentage of the surface receptor level immediately after pulse labeling (starting value). Data represent mean ± S.E. calculated from 4 independent experiments with 4 repeats each. Two-way ANOVA comparing wild type receptor and tail-swap mutant. Scale bar = 10 μm in A–H.
For quantitative analysis of receptor internalization, we performed an ELISA. Like in the immunocytochemical approach, surface receptors were labeled in live cells by anti-HA antibody at 4 °C and fixed either immediately (0 min) or after different ligand-free intervals at 37 °C. Surface receptors were then detected in nonpermeabilized cells by peroxidase-labeled secondary antibody and a peroxidase-driven color reaction. Signal intensities after the ligand-free intervals were expressed in percent of the signal at 0 min (Fig. 1I). The assay confirmed that CXCR7-WT and CXCR4–7tail receptors were rapidly depleted from the cell surface, whereas CXCR4-WT and CXCR7–4tail receptors showed little internalization. Together, the immunocytochemical and ELISA experiments demonstrate that replacement of the CXCR4 C-terminal domain with the CXCR7 C-terminal domain drives ligand-independent internalization of CXCR4. Conversely, replacement of the CXCR7 C-terminal domain with that of CXCR4 abolishes ligand-independent internalization of CXCR7.
Previous reports demonstrated that a CXCR7 mutant lacking the C-terminal domain (CXCR7ΔC-term) failed to internalize in the absence of ligand (Ref. 22; see also Fig. 5E). We now addressed the first 19 amino acids in the CXCR7 C-terminal domain (residues 320–338), which correspond to the putative helix eight-containing region. In one mutant (CXCR7–4-7), we replaced residues 320–338 of CXCR7 by the corresponding nonhelical domain of CXCR4 comprising residues 313–331 (33) and in another mutant (CXCR7-Δ19), we deleted residues 320–338. Analyses by immunocytochemistry and ELISA showed that both mutant receptors were efficiently internalized in the absence of ligand (Fig. 2, A, B, F, G, and K) suggesting that amino acids 339–362 are sufficient for ligand-independent internalization of CXCR7.
FIGURE 5.

Ligand-dependent activation and internalization of the CXCR7–4tail mutant. HEK293 cells were transiently transfected with CXCR7-WT, CXCR7–4tail, CXCR7ΔC-term, or CXCR4-WT as indicated. A–G, surface receptors were pulse labeled with anti-HA (A–D and G) or 11G8 anti-CXCR7 (E and F) antibody at 4 °C. A–F, confocal images show the subcellular receptor localization after 30 min at 37 °C in the absence (A, C, and E) or presence of CXCL12 (B, D, and F). CXCR7–4tail undergoes only ligand-dependent internalization (C and D), whereas CXCR7ΔC-term is internalization defective (E and F) and CXCR7-WT internalizes ligand independently (A and B). G, quantitative analysis of ligand-induced receptor internalization by ELISA. Cultures were incubated for 30 min at 37 °C with vehicle, CCX733 or CXCL12. Surface receptor levels are given as percentage of the surface receptor level immediately after pulse labeling (0 min). Results represent mean ± S.E. calculated from three to four independent experiments with 4 repeats each. Note that CCX733 and CXCL12 significantly increase receptor internalization. Asterisks indicate differences between the indicated groups (one-way ANOVA). H, immunoblots of wheat germ lectin-agarose (WGA)-purified cell lysates. Cultures were stimulated with CXCL12 and lysates were dephosphorylated with λ-PP as indicated. Upper panel, detection of CXCR4-WT and CXCR7–4tail protein using the anti-CXCR4 antibody UMB-2, which recognizes only the nonphosphorylated C-terminal CXCR4 epitope. Comparison of λ-PP-treated and λ-PP-untreated samples of nonstimulated cultures (lanes 1 and 2) indicates little constitutive phosphorylation of the UMB-2 epitope in CXCR4-WT but strong constitutive phosphorylation in CXCR7–4tail. CXCL12 treatment causes almost complete phosphorylation of the UMB-2 epitope both in CXCR4-WT and CXCR7–4tail (lanes 3 and 4). Middle and lower panels, aliquots of the samples shown in the upper panel were detected with anti-HA, 11G8, and anti-transferrin receptor (TFR) antibodies as indicated to confirm equal protein loading. H′, quantitative analyses of UMB-2 signal intensities in immunoblots from CXCR4-WT and CXCR7–4tail-transfected HEK293 cells using CCD camera-based densitometry. Relative amounts of nonphosphorylated (non-phospho) and total receptors were determined by measuring the UMB-2/HA ratios in λ-PP-untreated and λ-PP-treated samples, respectively. Results from 3 independent experiments were averaged and expressed as percentage of total receptor in cultures not receiving CXCL12 (*, p < 0.05, paired Student's t test).
FIGURE 2.

Motifs in the CXCR7 C-terminal domain regulating ligand-independent receptor internalization. HEK293 cells were transiently transfected with constructs for HA-tagged receptors carrying mutations in putative internalization regulating C-terminal motifs. In CXCR7–4-7 and CXCR7-Δ19 residues 320–338 of CXCR7 (putative helix eight-containing region) were replaced by the corresponding CXCR4 sequence or deleted. In CXCR4–7(C9) the last nine residues of CXCR4 were replaced by those of CXCR7. In CXCR7-Y/A and CXCR7-K/R all C-terminal tyrosine and lysine residues were replaced by alanines and arginines, respectively. A–J, surface receptors were pulse labeled by anti-HA antibody applied to live cells at 4 °C. Confocal images show the subcellular localization of label receptors immediately after the anti-HA pulse (0 min, A–E) or after a 30-min ligand-free interval at 37 °C (F–J). A, B, F, and G, CXCR7 residues 320–338 are not involved in ligand-independent CXCR7 internalization. C and H, the last nine CXCR7 C-terminal residues containing a YSAL motif are not sufficient for ligand-independent CXCR4 internalization. D, E, I, and J, C-terminal tyrosine and lysine residues are not critical for plasma membrane targeting and ligand-independent internalization of CXCR7. K, quantitative analysis of receptor internalization by ELISA. For each receptor, the surface receptor level after the 30-min ligand-free interval is expressed as percentage of its surface receptor level after 0 min (nonpermeabilized cells). L-O, fixed permeabilized and nonpermeabilized (nonperm.) transfectants of CXCR7-WT and CXCR7-K/R were immunostained with anti-HA antibody. Both receptors are present at the plasma membrane. Permeabilization reveals preponderant intracellular localization of CXCR7-WT and CXCR7-K/R. P, analysis of CXCR7-WT and CXCR7-K/R surface receptor expression in nonpermeabilized cells subjected to 3-h vehicle (veh.) or 3-h CHX treatments using ELISA. Student's t test comparing signals after 30 versus 0 min (*), vehicle versus CHX (§), and CXCR7-WT versus CXCR7-K/R ($). Data represent mean ± S.E. calculated from 3 independent experiments with 4 repeats each. Scale bar = 10 μm in A–J and L–O.
We then searched residues 339–362 for a structural motif triggering ligand-independent receptor internalization and identified a YSAL motif 9 residues before the C-terminal end of CXCR7. Recently a YXXL motif at the extreme end of the C terminus of CXCR3 has been shown to be involved in ligand-independent receptor endocytosis (34). We therefore hypothesized that the CXCR7 YSAL motif might exert a similar function. To test this assumption, we generated the CXCR4 mutant CXCR4–7(C9) with the last 9 residues replaced by those of CXCR7. Unlike CXCR4–7tail, in which the complete CXCR4 C-terminal domain was replaced by that of CXCR7, CXCR4–7(C9) failed to internalize in the absence of ligand (Fig. 2, C, H, and K). Thus, the extreme end of the CXCR7 C-terminal domain containing the YSAL motif is not sufficient to cause ligand-independent receptor internalization. The possibility that tyrosine residues in the C-terminal domain of CXCR7 are critical for ligand-independent receptor internalization was ruled out by the finding that a CXCR7-Y/A mutant in which all C-terminal tyrosine residues (positions 322, 324, 334, and 354) had been replaced with alanines showed efficient internalization (Fig. 2, D, I, and K). Together, these experiments indicate that residues 339–362 of CXCR7 are sufficient to trigger spontaneous internalization independently of a tyrosine-containing motif.
Ubiquitination at lysine residues plays an important role in G protein-coupled receptor regulation by affecting receptor trafficking and degradation (35). We therefore generated a CXCR7-K/R mutant in which all C-terminal lysine residues were replaced with arginines. Examination of anti-HA pulse-labeled surface receptors in the absence of CXCL12 using immunocytochemistry and ELISA demonstrated that CXCR7-K/R was targeted to the plasma membrane and internalized spontaneously (Fig. 2, E, J, and K). Consistently, immunostaining of CXCR7-WT and CXCR7-K/R in fixed cells with or without permeabilization prior to anti-HA antibody application showed exclusive plasma membrane staining for both receptors in nonpermeabilized cells (Fig. 2, L and N) as well as strong intracellular labeling and weak plasma membrane staining in permeabilized cells (Fig. 2, M and O). Plasma membrane targeting of CXCR7-K/R was confirmed by an anti-HA ELISA in nonpermeabilized cells showing that transfection with CXCR7-K/R caused a 5.2-fold signal increase as compared with mock transfection (p < 0.05; Student's t test; n = 3 with 4 repeats each).
To examine if the K/R mutations affect CXCR7 degradation, we applied the protein synthesis inhibitor cycloheximide (CHX; 10 μg/ml) or vehicle for 3 h prior to fixation and examined the receptor levels at the cell surface using the anti-HA ELISA. We observed a strong loss of CXCR7-WT and CXCR7-K/R at the cell surface in the presence of CHX (Fig. 2P). Comparison of CXCR7-WT and CXCR7-K/R showed that the loss of surface expression under CHX was attenuated with the K/R mutant.
Regulation of Receptor Stability by the CXCR7 C-terminal Domain
Because reduced surface receptor expression in the presence of CHX might be due to constitutive receptor degradation, we asked if the CXCR7 C terminus affects receptor stability. To test this assumption, we compared total receptor expression levels of the wild type and the tail-swap mutants in nontreated cultures and examined loss of receptor expression after CHX treatment using immunoblots of wheat germ lectin-agarose-purified cell lysates (Fig. 3). The anti-HA antibody recognized a single, nonspecific band at 80 kDa in lysates of nontreated mock-transfected and receptor-transfected cultures (Fig. 3, A and C). After transfection with CXCR7-WT and CXCR7–4tail, the antibody also detected a smear ranging from 50 to 70 kDa (Fig. 3A). A similar receptor-specific signal was obtained using the mouse monoclonal anti-CXCR7 antibody 11G8 (Fig. 5H). Analysis of the signal intensity in the 50–70-kDa area by densitometry showed that CXCR7–4tail was expressed at a 6.6-fold higher level than CXCR7-WT (Fig. 3B, Student's t test). CXCR4-WT and CXCR4–7tail migrated at 47 kDa with CXCR4-WT showing a 5-fold stronger signal than CXCR4–7tail (Fig. 3, C and D, Student's t test). We then treated transfected cultures for 60 or 180 min with 10 μg/ml of CHX and compared receptor expression levels after cycloheximide treatment with the level of nontreated sister cultures harvested at 0 min. Although cycloheximide had virtually no effect on CXCR4-WT, it caused an almost complete loss of CXCR7-WT and CXCR4–7tail within 180 min (Fig. 3, E–H). The expression level of the CXCR7–4tail and CXCR7-K/R mutants declined during cycloheximide treatment but at a slower rate than that of CXCR7-WT (Fig. 3, E and F). Collectively, these experiments indicate that the CXCR7 C-terminal domain regulates stability of CXCR7 in the absence of CXCL12 and that C-terminal lysine residues influence stability of CXCR7.
FIGURE 3.

The CXCR7 C-terminal domain regulates receptor degradation. A–J, immunoblots with wheat germ lectin agarose-purified lysates of HEK293 cells transiently transfected with empty vector (mock) or N-terminal HA-epitope-tagged wild type (7WT, 4WT), tail-swap mutant (7–4tail, 4–7tail) receptors, and CXCR7-K/R (7-K/R). The recombinantly expressed chemokine receptors were identified with an anti-HA antibody. The endogenous transferrin receptor (TFR) was labeled after stripping and served as loading control. For quantitative analysis of chemokine receptor expression levels, the HA/TFR-ratio was determined in at least 3 independent experiments using CCD camera-based densitometry and expressed as percentage of the group indicated in the figure. A, the anti HA-antibody recognizes the CXCR7-WT and CXCR7–4tail glycoproteins as a smear ranging from 50 to 70 kDa and produces a nonspecific signal at 80 kDa. B, the expression level of CXCR7–4tail is 6.6-fold higher than that of CXCR7-WT. C and D, CXCR4-WT and CXCR4–7tail migrate at 47 kDa with CXCR4-WT showing a 5-fold stronger signal than CXCR4–7tail. E-H, kinetics of receptor degradation in nonstimulated cultures receiving CHX for 0, 1, and 3 h to block protein synthesis. Blots with receptors carrying the CXCR7 C terminus were imaged with longer exposure times. Receptor degradation is attenuated in CXCR7-K/R and CXCR7–4tail as compared with CXCR7-WT and accelerated in CXCR4–7tail as compared with CXCR4-WT. I and J, influence of long term CXCL12 treatment (16 h) on receptor expression levels (CHX-free condition). CXCL12 treatment causes a 70% decrease of recombinant CXCR4 in CXCR4-WT transfectants. Levels of recombinant receptors are not affected by CXCL12 in CXCR7-WT, CXCR7–4tail, and CXCR4–7tail transfectants. *, Student's t test; #, two-way ANOVA; §, one-way ANOVA. Data represent mean ± S.E. calculated from three to five independent experiments.
We then wished to determine the effect of long term CXCL12 treatment on the expression level of the tail-swap mutants. To this end, we transfected cells with the different constructs and treated the cultures for 16 h with CXCL12 or vehicle. Wheat germ lectin-agarose-purified lysates of the four constructs were subjected to Western blotting and detected with anti-HA antibody (Fig. 3I; exposure times were optimized for each construct individually to account for unequal expression levels). After densitometry, we expressed the signal intensity of the CXCL12-treated group as percentage of the signal intensity of the vehicle-treated group. This showed that CXCL12 induced a 70% decline of CXCR4-WT and that CXCL12 had no significant effect on CXCR7-WT or on the chimeric receptors (Fig. 3J).
Chimeric CXCR7 Receptors Fail to Signal through G Proteins
In previous studies, G protein-dependent signaling could not be demonstrated for CXCR7 (14). Because chemokine receptor/G protein interactions occur via the second intracellular loop and are influenced by the C-terminal domain of G protein-coupled receptors, we examined G protein signaling in the tail-swap mutants as well as in CXCR7-WT and CXCR7–4tail receptors, in which the second intracellular loop had been replaced with that of CXCR4 (CXCR7–2cD4, CXCR7–2cD4–4tail). Before using the receptor chimeras in functional assays, we examined their affinity for CXCL12, subcellular targeting, and expression levels (Fig. 4, A–F). HEK293 cells were transfected with empty vector or with the receptor-encoding constructs and subjected to homologous competition binding experiments. Radiolabeled CXCL12 (25 pmol/liter) was competed for with increasing concentrations of unlabeled CXCL12 at internalization restrictive (4 °C) conditions. Because receptor-transfected cells bound about 5-fold more radioligand than mock-transfected cells, endogenous CXCL12 receptors had an insignificant effect under these experimental conditions. For the wild type and tail-swap mutants, we found the following rank order of IC50 values: CXCR7-WT (2.6 nm) < CXCR4–7tail (8.4 nm) < CXCR4-WT (23.8 nm) < CXCR7–4tail (25.2 nm). Replacement of the second intracellular loop in CXCR7-WT and CXCR7–4tail with the CXCR4 second intracellular loop had only a minor effect on the IC50 values (CXCR7–2cD4, 2.3 nm; CXCR7–2cD4–4tail, 11.3 nm). The IC50 values we obtained for CXCR4-WT and CXCR7-WT are somewhat higher than previously reported IC50 values (8, 11, 13), which might be caused by the preparation of unlabeled CXCL12 used in our experiments. However, our finding that CXCR7-WT has an ∼10 times higher potency than CXCR4-WT is in good agreement with the previous reports. In addition, the CXCR7–4tail receptor was ∼10 times less potent than the CXCR7-WT receptor, which corresponds to the 10 times lower potency of CXCR7ΔC-term compared with CXCR7 (22). Thus, the chimeric receptors display slightly altered CXCL12-binding properties but still function as high affinity receptors for CXCL12.
FIGURE 4.

G protein coupling is absent in wild type CXCR7 and CXCR7/CXCR4-chimeric receptors. A–H, HEK293 cells were transiently transfected with wild type receptors (CXCR7-WT, CXCR4-WT) and tail-swap mutants (CXCR7–4tail, CXCR4–7tail). A and B, homologous radioligand competition binding using 125I-CXCL12 (25 pm) and increasing concentrations of nonlabeled CXCL12. A and B, transfection with CXCR7-WT and CXCR7–4tail chimeras in which the second cytosolic domain (2cD) was replaced with the corresponding region of CXCR4 (mutants are referred to as CXCR7–2cD4 and CXCR7–2cD4–4tail). Results in A and B represent 4 independent experiments (3 repeats each) after normalization with the calculated top binding obtained for CXCR7-WT (A) and CXCR7–2cD4 (B). Potency of CXCR7-WT is 10-fold higher than potency of CXCR4-WT and CXCR7–4tail. Note that replacement of the 2cD does not affect potency of CXCR7-WT and CXCR7–4tail. C-F, CXCR7–2cD4 and CXCR7–2cD4–4tail transfectants were pulse-labeled with anti-HA antibody (4 °C). Cells were washed, fixed, permeabilized, and detected immediately after loading with the HA antibody (0 min) or after a 30-min ligand-free interval at 37 °C. C and E, both chimeras are present at the plasma membrane. D and F, pulse-labeled CXCR7–2cD4 surface receptors are rapidly internalized, whereas the CXCR7–2cD4–4tail receptors remain at the plasma membrane. G and H, analysis of G protein coupling. G, cells were transfected with the qi5 chimera alone or with CXCR4-WT in combination with qi5. Stimulation occurred at t = 30 s with 80 nm CXCL12. AMD3100 was applied 15 min prior to recording. Data were baseline corrected and traces were calculated as averaged traces from a single experiment with each trace representing data from three to four wells. H, mean maximum fluorescence amplitudes (± S.D.) were obtained. Experimental set up was as described for G, only that cells were transfected with qi5 in combination with different receptor constructs. Data were derived from two to three independent transfections with a minimum of three repeats (wells) per receptor. *, Student's t test.
Pulse labeling of surface receptors by anti-HA antibody and examination by immunocytochemistry demonstrated that the two CXCR7 chimeras containing the CXCR4 second intracellular loop were efficiently targeted to the plasma membrane (Fig. 4, C and E). Furthermore, the replacement did not influence ligand-independent receptor trafficking (Fig. 4, D and F): CXCR7–2cD4 internalized in the absence of ligand-like CXCR7-WT and CXCR7–2cD4–4tail stayed at the plasma membrane-like CXCR7–4tail. Signal intensities for CXCR7–2cD4 and CXCR7–2cD4–4tail in immunoblots tended to be higher than those for CXCR7-WT indicating that the mutants were efficiently expressed (n = 2, data not shown).
G protein signaling of the receptor chimeras was then examined by measuring [Ca2+]i transients, which is an established tool to study activation of G proteins of the Gq/11 subfamily or of chimeric Gi/o-Gq/11 G proteins (30, 32). Stimulation of naive HEK293 cells with CXCL12 produced a subtle AMD3100-sensitive Ca2+ signal (not shown), indicating that the cells expressed functional endogenous CXCR4 receptors. Transfection with CXCR4-WT alone did not enhance the CXCL12-induced Ca2+ response (not shown), suggesting that the amount of the endogenous G protein, which links CXCR4 to the calcium pathway, is limiting in HEK293 cells. Because CXCR4 is known to activate Gαi2 (28, 29) we next co-transfected the receptor-encoding constructs with qi5, a murine chimera consisting of Gαi2 and Gαq. Under these conditions, CXCL12 produced a marked Ca2+ response through CXCR4-WT but not through CXCR7-WT or through the chimeric receptors (Fig. 4, G and H).
Because the subtle Ca2+ response evoked by endogenous CXCR4 in HEK293 might mask weak signals by the chimeric receptors, we then used CHO-K1 cells, which do not express endogenous CXCL12 receptors. Using immunocytochemistry it was confirmed that the wild type and chimeric receptors were targeted to the plasma membrane of CHO-K1 cells (not shown). Stimulation with CXCL12 did not mobilize Ca2+ in CHO-K1 cells transfected with one of the wild type or chimeric receptors alone. Because Substance P produced a robust Ca2+ transient in cells transfected with the cognate Gq-coupled NK1 receptor (supplemental Fig. S1) it is likely that the chemokine receptors failed to activate the phospholipase C pathway through an endogenous Gq/11 protein in CHO-K1 cells. Also after co-transfection of the chemokine receptor-encoding constructs with Gα15, a murine promiscuous G protein of the Gq/11 subfamily, CXCL12-induced Ca2+ mobilization was not detected (not shown). After co-transfection of qi5, CXCL12 produced a marked Ca2+ response through CXCR4-WT but not through CXCR7-WT or the chimeric receptors (supplemental Fig. S1), which corresponds to our findings in HEK293 cells. It has been shown that the 16z44 chimera consisting of Gαz and Gα16 can improve the recognition of Gi-selective receptors (31). When co-transfected with one of our chemokine receptor constructs, 16z44 mediated a CXCL12-induced Ca2+ mobilization only with CXCR4-WT (supplemental Fig. S1).
To test whether the signaling defect of the CXCR4–7tail was due to low level expression, we then generated HEK293 cells and CHO-K1 cells stably expressing the mutant. Characterization of these cultures using the anti-HA antibody in immunocytochemistry and immunoblots showed that at least 80% of the cells expressed CXCR4–7tail and that the level of expression was comparable with that of CXCR4-WT after transient transfection. Still, stable CXCR4–7tail transfectants failed to produce a significant calcium response through qi5. Thus, the CXCR7 C terminus suppresses G protein coupling when fused to CXCR4. CXCR7 does not signal through G proteins and replacement of the CXCR7 second intracellular loop, the CXCR7 C terminus, or the CXCR7 C terminus and the second intracellular loop by the corresponding regions of CXCR4 is not sufficient to permit detectable G protein signaling through CXCR7.
Ligand-dependent Activation and Internalization of the CXCR7–4tail Mutant
Deletion of the CXCR7 C terminus abolishes internalization of CXCR7 (22). Because it is well established that the CXCR4 C-terminal domain mediates CXCL12-induced CXCR4 internalization through a GRK/phosphorylation-dependent mechanism (36), we tested whether the CXCR4 C terminus permits CXCL12-dependent phosphorylation and internalization when fused to CXCR7. In a first approach, we transfected HEK293 cells with CXCR7-WT, CXCR7ΔC-term lacking the C terminus, or CXCR7–4tail and examined receptor trafficking by immunocytochemistry (Fig. 5, A–F). Surface receptors were pulse labeled by applying antibody at 4 °C. Cells were washed and fixed after a 30-min interval at 37 °C during which they received vehicle or CXCL12. The experiment showed strong internalization of CXCR7-WT in the absence and presence of CXCL12 (Fig. 5, A and B). CXCR7ΔC-term was highly expressed at the plasma membrane and failed to internalize (Fig. 5, E and F). CXCR7–4tail showed prominent expression at the cell surface like CXCR7ΔC-term and only little spontaneous internalization (Fig. 5C). Unlike CXCR7ΔC-term, the CXCR7–4tail was capable of internalizing after CXCL12 exposure (Fig. 5D). Thus, the tail-swap largely abolished spontaneous internalization but preserved ligand-induced internalization.
We then performed quantitative analyses of receptor endocytosis by ELISA using CXCL12 and the CXCR7-selective compound CCX733 as ligands. This showed that CCX733 and CXCL12 accelerated internalization of CXCR7-WT and CXCR7–4tail (Fig. 5G).
We then addressed CXCL12-dependent phosphorylation of the CXCR7–4tail mutant using the rabbit monoclonal CXCR4 antibody UMB-2, which was raised against a synthetic peptide corresponding to the 22 C-terminal amino acids of CXCR4 (23). Recently, we established that binding of UMB-2 to CXCR4 is severely impaired after CXCL12 stimulation/phosphorylation of CXCR4 and that dephosphorylation of CXCR4 by λ-PP reinstates binding of UMB-2 to CXCR4 (Fig. 5, H, left panel, and H′ (10)). UMB-2 detected CXCR7–4tail in immunoblots as a smear between 50 and 70 kDa (Fig. 5H, first lane in upper right panel). A similar smear was seen when CXCR7–4tail was detected with the anti-HA antibody or with the N-terminal anti-CXCR7 antibody 11G8 (Fig. 5H, middle and right lower panels). Dephosphorylation of nonstimulated CXCR7–4tail lysates by λ-PP strongly enhanced the UMB-2 signal (Fig. 5, H, second lane in upper right panel, H′). In lysates of CXCL12-stimulated cultures, UMB-2 failed to recognize CXCR7–4tail but dephosphorylation fully restored UMB-2 binding (Fig. 5, H, third and fourth lanes in upper right panel, and H′). These experiments indicate that a large proportion of the CXCR7–4tail receptors is constitutively phosphorylated and that CXCL12 binding further enhances phosphorylation of the mutant.
The CXCR7 C-terminal Domain Accelerates Uptake and Degradation of CXCL12 as well as Release of CXCL12 Degradation Products
Next, we compared kinetics of CXCL12 uptake and degradation via the wild type, tail-swap mutant, and CXCR7ΔC-term receptors in transiently transfected HEK293 cells. For pulse-chase examination of receptor internalization, transfectants were loaded with radiolabeled CXCL12 at internalization-restrictive conditions (4 °C) and washed with PBS after loading. Some of the cultures were harvested immediately after the PBS wash to determine the starting value representing the initial amount of bound radioligand (comparison of the starting values between receptor-transfected cultures and mock-transfected cultures proved that all receptor-encoding constructs produced a >5-fold increase in radioligand binding; not shown). The remaining cultures were transferred to internalization permissive conditions at 37 °C. After various times (0, 5, 15, and 30 min), cells were washed with acidic buffer, which effectively removes surface-bound radioligand. To establish the internalization kinetics, the amount of residual internalized radioligand was counted and expressed as percentage of the starting value (Fig. 6, A and B). This showed that receptors bearing the CXCR7 C terminus mediated CXCL12 uptake considerably faster than the corresponding receptors bearing the CXCR4 C terminus. CXCR7ΔC-term failed to mediate any radioligand internalization (Fig. 6A).
FIGURE 6.

Rapid endocytosis and degradation of 125I-CXCL12 depend on the CXCR7 C-terminal domain. HEK293 cells were transiently transfected with empty vector (mock), wild type receptors (CXCR7-WT, CXCR4-WT), tail-swap mutants (CXCR7–4tail, CXCR4–7tail), or CXCR7ΔC-term as indicated. A and B, internalization kinetics of receptor-radioligand complexes. Cells were loaded with radioligand at 4 °C (pulse), washed, and lysed immediately (starting value) or incubated for the indicated time intervals at 37 °C and lysed after an acidic wash (chase). Data show internalized 125I-CXCL12 of acid-washed groups as percent of the starting value. Receptors carrying the CXCR7 C-terminal domain mediate faster radioligand uptake than receptors carrying the CXCR4 C-terminal domain. CXCR7ΔC-term fails to internalize 125I-CXCL12. Results represent mean ± S.E. from 2 independent experiments with 5 repeats each. C and D, scavenging activity. Cells were incubated with 125I-CXCL12 for the indicated time intervals at 37 °C. Then, supernatants were subjected to TCA precipitation to separate degraded 125I-CXCL12 from precipitated 125I-CXCL12. Note faster accumulation of 125I-CXCL12 degradation products in culture supernatants of CXCR7-WT and CXCR4–7tail transfectants than in culture supernatants of CXCR7–4tail and CXCR4-WT transfectants, respectively. There is no significant increase in extracellular125I-CXCL12 degradation products in culture supernatants of CXCR7ΔC-term transfectants. E and F, intracellular accumulation of 125I-CXCL12. Transfectants were incubated with radioligand for the indicated time at 37 °C and subjected to an acidic wash to strip residual surface-bound 125I-CXCL12 before lysis and measurement of intracellular 125I. In cultures transfected with receptors carrying the CXCR7 C terminus, intracellular 125I-CXCL12 levels increase rapidly before decreasing (CXCR7-WT) or reaching a steady state (CXCR4–7tail). Cells overexpressing receptors with the CXCR4 C terminus continue to accumulate intracellular 125I-CXCL12 during the entire experimental period. CXCR7ΔC-term shows no intracellular accumulation of 125I-CXCL12. C–F, data are mean ± S.E. from 2 independent experiments with 4 repeats each. A–F, two-way ANOVA. Asterisks indicate significant differences between wild type receptors and corresponding tail-swap mutants. G and H, influence of receptor-mediated scavenger activity on functional CXCL12. CXCL12 (20 nm) was incubated overnight with HEK293 cells transfected with the indicated receptors. Supernatants were used to stimulate transwell migration of Jurkat T cells. G, graph shows fluorescence intensities of calcein-AM-stained migrated Jurkat T cells. CXCL12-supplemented medium incubated with mock-transfected cells causes a 9-fold increase in cell migration as compared with nonsupplemented medium. Preincubation with CXCR7-WT abolishes and preincubation with CXCR7ΔC-term reduces the migratory response. *, Student's t tests versus mock + CXCL12. H, migratory response with supernatants from CXCR7ΔC-term was used to normalize responses with supernatants from wild type and tail-swap mutant receptors. *, §, Student's t tests versus corresponding wild type receptor (*) or versus CXCR7ΔC-term (§). Data in G and H are 2–4 independent experiments with 2–6 repeats each.
Given that the CXCR7 C-terminal domain accelerates CXCL12 uptake, we examined how efficiently the receptors released degraded CXCL12 into the culture medium. To this end, we transfected HEK293 cells with amounts of plasmid that resulted in similar protein levels of the wild type and corresponding tail-swap mutant receptors (determined by immunoblotting). The transfectants were then incubated with radiolabeled CXCL12 for 5, 30, or 120 min at 37 °C before the culture medium was harvested and subjected to TCA precipitation. This procedure segregates nondegraded chemokine in the pellet from degraded chemokine in the supernatant. The amount of intracellularly accumulated radioligand was determined by harvesting the cells after washing with acidic buffer. The fastest increase of degraded CXCL12 was observed in the culture medium of CXCR7-WT and CXCR4–7tail transfectants. After 120 min, the medium of CXCR7-WT- and CXCR4–7tail-expressing cultures contained ∼2 times more degraded CXCL12 than the medium of CXCR7–4tail- and CXCR4-WT-expressing cultures, respectively (Fig. 6, C and D). Rapid release of degraded CXCL12 in cultures expressing the CXCR7 C terminus was associated with limited intracellular CXCL12 accumulation (Fig. 6, E and F). In contrast, CXCR4-WT- and CXCR7–4tail-expressing cultures continued to accumulate CXCL12 inside the cells over the 120-min incubation period. Together, these experiments indicate that rapid CXCL12 uptake and exocytosis of CXCL12 degradation products depend on the CXCR7 C- terminal domain.
Having shown that the CXCR7 C-terminal domain accelerates receptor-mediated sequestration and degradation of extracellular CXCL12, we wished to test the impact of this scavenger activity with an assay that depends on a biological response to functional ligand and chose CXCL12-induced transwell migration of Jurkat T cells. CXCL12 (20 nm) was incubated overnight with mock-transfected and receptor-transfected HEK293 cells (transfection conditions were adjusted to produce similar expression levels of wild type and mutant receptors). Then, the supernatants were harvested and used as chemoattractant to stimulate transwell migration of Jurkat T cells. CXCL12-supplemented medium preincubated with mock transfectants evoked a 9-fold increase in migration as compared with nonsupplemented medium (Fig. 6G). After preincubation with CXCR7-WT transfectants, the CXCL12-induced migratory response was almost eliminated, indicating that CXCR7-WT acts as a highly efficient CXCL12 scavenger under these conditions (Fig. 6G). Surprisingly, preincubation with CXCR7ΔC-term caused a 53% loss of functional CXCL12 (Fig. 6G). Because CXCR7ΔC-term does not internalize CXCL12, the observed loss of CXCL12 is attributable to mere receptor binding. To establish how internalizing receptors affect CXCL12 levels beyond mere binding, we normalized migratory responses triggered by supernatants from internalizing receptors to results obtained with CXCR7ΔC-term (Fig. 6H). This showed that all internalizing receptors depleted CXCL12 from supernatants more efficiently than CXCR7ΔC-term. The CXCR4 C terminus reduced the CXCL12 scavenger activity when fused to CXCR7 and the CXCR7 C terminus increased scavenger activity when fused to CXCR4. Collectively, these data provide evidence that the CXCR7 C-terminal domain plays a critical role in receptor-mediated CXCL12 degradation.
CXCR7 C-terminal Serine/Threonine Residues Influence the CXCL12 Scavenger Activity of CXCR7
Recently, it was reported that mutations in the CXCR7 C-terminal domain including truncation and conversion of all C-terminal lysine residues or serine/threonine residues into alanines affect CXCR7 trafficking (37, 38). We therefore examined the internalization rate and scavenger activity of our CXCR7-K/R mutant and of three mutants with C-terminal ST/A conversions: CXCR7-ST/A (conversion of all C-terminal Ser and Thr residues), CXCR7-STT/A (carrying S335A, T338A, and T341A), and CXCR7-STS/A (carrying S350A, T352A, and S355A). CXCL12 affinity of the 4 mutants as determined by homologous competitive radioligand binding ranged from 1.8 to 5.3 nm and was thus comparable with the IC50 of CXCR7-WT. We then studied internalization of CXCL12-receptor complexes using 125I-CXCL12 pulse/chase labeling of surface receptors in transiently transfected HEK293 cells. CXCR7-WT-transfected sister cultures were always examined in parallel and used for normalization (100% line in Fig. 7, A–C). Although 125I-CXCL12 internalization via CXCR7-K/R was virtually identical to that of CXCR7-WT, internalization via CXCR7-ST/A was significantly reduced at 5, 15, and 30 min (Fig. 7, A and B). Mutants with partial ST/A conversions showed significant differences only at the 5-min time point: 125I-CXCL12 internalization was slightly reduced with CXCR7-STT/A and strongly reduced with CXCR7-STS/A. Next, we examined kinetics of CXCL12 degradation and 125I-CXCL12 uptake after perpetuated 125I-CXCL12 exposure (Fig. 7, D–I). CXCR7-K/R transfectants released less 125I-CXCL12 degradation products and accumulated more intracellular 125I-CXCL12 than CXCR7-WT (Fig. 7, D and G). In CXCR7-ST/A transfectants and CXCR7-STS/A transfectants, both the release of 125I-CXCL12 degradation products and accumulation of intracellular 125I-CXCL12 were delayed (Fig. 7, E, F, H, and I). CXCR7-STT/A transfectants were similar to CXCR7-WT (Fig. 7, F and I). Finally, we preincubated medium containing 20 nm CXCL12 overnight with the transfectants and used the culture supernatants as chemoattractant for Jurkat T cells in Transwell migration assays. Supernatants from CXCR7ΔC-term transfectants were used as reference. Stronger chemotactic activity of the CXCR7-ST/A supernatants than of the CXCR7-WT supernatants indicated a reduced CXCL12 scavenger activity in the CXCR7-ST/A transfectants. In contrast, CXCR7-K/R was as effective as CXCR7-WT. Collectively these experiments show that C-terminal lysine residues are dispensable for efficient CXCR7-mediated CXCL12 internalization and degradation but involved in rapid release of 125I-CXCL12 degradation products. Serine and threonine residues in the C-terminal domain are required for rapid 125I-CXCL12 internalization and degradation.
FIGURE 7.

Influence of C-terminal lysine and serine/threonine residues on CXCR7-mediated endocytosis and degradation of CXCL12. HEK293 cells were transiently transfected with empty vector (mock), CXCR7-WT, CXCR7-K/R, a CXCR7-ST/A mutant lacking all C-terminal serine and threonine residues, CXCR7-STT/A with S335A, T338A, and T341A conversions, and CXCR7-STS/A with S350A, T352A, and S355A conversions. A–C, receptor-mediated internalization of 125I-CXCL12. Cells were loaded with radioligand at 4 °C (pulse), PBS washed, and lysed immediately (starting value) or incubated for the indicated time intervals at 37 °C and lysed after an acidic wash (chase). Percent internalization was calculated by dividing counts of internalized 125I-CXCL12 in the acid-washed groups by the starting value. Internalization data were then normalized to CXCR7-WT-transfected sister cultures (100% line). A, CXCR7-K/R and CXCR7-WT mediate similar 125I-CXCL12 internalization. B and C, as compared with CXCR7-WT, 125I-CXCL12 internalization via CXCR7-ST/A is reduced at 5, 15, and 30 min and 125I-CXCL12 internalization via CXCR7-STT/A and CXCR7-STS/A is reduced at 5 min after pulse labeling. D–F, release of degraded 125I-CXCL12. Cells were incubated with 125I-CXCL12 as indicated. Supernatants were subjected to TCA precipitation before degraded 125I-CXCL12 was counted. As compared with CXCR7-WT, CXCR7-K/R, CXCR7-ST/A, and CXCR7-STS/A cause slower accumulation of extracellular 125I-CXCL12 degradation products. G and H, intracellular accumulation of 125I-CXCL12. Cells were incubated with 125I-CXCL12 as indicated, subjected to acidic wash, and lysed. As compared with CXCR7-WT, CXCR7-K/R transfectants accumulate more, whereas CXCR7-ST/A and CXCR7-STS/A transfectants accumulate less 125I-CXCL12. A–I, two-way ANOVA. Asterisks indicate significant differences between wild type and mutant receptors. Data represent mean ± S.E. calculated from 2 independent experiments with 4 repeats each. J, influence of receptor-mediated scavenger activity on functional CXCL12. CXCL12 (20 nm) was incubated overnight with HEK293 cells transfected with the indicated receptors. Supernatants were used to stimulate transwell migration of Jurkat T cells. Migratory response with supernatants from CXCR7ΔC-term was used for normalization. * and §, Student's t tests versus CXCR7-WT (*) or versus CXCR7ΔC-term (§). Data are 3 independent experiments with 2–6 repeats each. K, schematic representation of the CXCR7 C terminus showing C-terminal serine (S) and threonine (T) residues and ST/A conversions in the mutant receptors.
DISCUSSION
Recent work established that CXCR4 and CXCR7 are dramatically different in their functions. Both receptors bind and internalize CXCL12 but only CXCR7 is an effective CXCL12 scavenger (8, 11, 18). CXCR7 is further distinct from CXCR4 in its failure to signal through heterotrimeric G proteins and in its property to undergo ligand-independent internalization (1, 12, 16, 17, 39). Little is known about structural elements underlying the different receptor functions (16, 22, 40). By generating tail-swap mutant CXCR4 and CXCR7 receptors, we provide evidence that replacement of the CXCR7 C terminus with the CXCR4 C terminus generates a CXCR7 chimera, which shows CXCL12-dependent internalization and CXCL12-dependent phosphorylation but lacks ligand-independent internalization. Furthermore, we provide evidence that C-terminal serine and threonine residues are involved in rapid CXCR7-dependent endocytosis and degradation of CXCL12. C-terminal lysine residues influence degradation of CXCR7 as well as rapid release of CXCL12 degradation products.
Spontaneous Internalization and G Protein Coupling of CXCR7
Our analyses of CXCR7 and CXCR4 mutant receptors showed that the CXCR7 C-terminal domain plays a critical role in ligand-independent receptor internalization: the deletion of this domain and its replacement by the corresponding CXCR4 C-terminal domain abrogate ligand-independent internalization of CXCR7. Conversely, replacement of the CXCR4 C-terminal domain with that of CXCR7 (CXCR4–7tail mutant) triggers rapid ligand-independent internalization of CXCR4. Currently, it is essentially unclear how this property of the CXCR7 C terminus influences CXCR7 function. The spontaneously internalizing CXCR4–7tail mutant provides a clue to this question: G protein signaling was fully abrogated whereas ligand uptake was accelerated. It therefore seems likely that the CXCR7 C terminus is sufficient to prevent efficient receptor/G protein coupling and to permit rapid receptor-mediated CXCL12 sequestration. However, the C terminus seems not to be the only CXCR7 domain that prevents effective G protein signaling, because the CXCR7–4tail mutant, which did not internalize in the absence of ligand, failed to signal through the G proteins tested. Because CXCR7 contains a DRYLSIT sequence in the second intracellular loop, which is distinct from DRY motifs found in typical chemokine receptors (14), we introduced the CXCR4 second intracellular loop into the CXCR7-WT and CXCR7–4tail receptors. Still, the resulting mutants failed to induce G protein signaling. Thus, although we cannot exclude that using more sensitive methods to monitor G protein activation might reveal such an activation, it is evident that simple replacements of cytosolic domains are insufficient to permit detection of a CXCR7/G protein pathway by FLIPR®-based technology.
Influence of the CXCR7 C-terminal Domain on Receptor Stability
Our immunoblot analyses showed that the expression levels of the CXCR7-WT and CXCR4–7tail receptors were considerably lower than those of the corresponding receptors bearing the CXCR4 C-terminal domain and that receptor degradation was much faster with receptors bearing the CXCR7 C-terminal domain than with receptors bearing the CXCR4 C-terminal domain. These results are consistent with a recent report demonstrating increased levels of the CXCR7ΔC-term mutant as compared with the wild type CXCR7 receptor (22). Thus, the CXCR7 C-terminal domain accelerates not only ligand-independent receptor internalization but also ligand-independent receptor degradation. Given that CXCR7 is ubiquitinated at lysine residues in the C terminus (37) and that the rapid loss of total and surface CXCR7 receptors during cycloheximide exposure were attenuated in our CXCR7-K/R mutant, it seems likely that C-terminal ubiquitination reduces stability of the CXCR7 protein.
Our analyses of the receptor expression levels in transfectants receiving long term CXCL12 treatment revealed another difference between CXCR4 and CXCR7: CXCL12 did not influence the CXCR7 expression level but induced the degradation of CXCR4. Thus, regulation of CXCR4 and CXCR7 protein levels by the respective C-terminal domains is fundamentally different. In contrast, regulation of CXCR3 and CXCR7 shows strong similarities because both receptors undergo rapid ligand-independent internalization and degradation (34).
Structural Elements in the CXCR7 C-terminal Domain Regulating Receptor Internalization and Scavenging Activity
Secondary structure predictions for CXCR7 (41, 42) indicate a helix VIII after the conserved NPXXY motif at the end of helix VII like in other Class A receptors. The recently reported crystal structure of human CXCR4 provided the surprising finding that CXCR4 lacks helix VIII (33). Using two C-terminal CXCR7 mutants, we addressed the question whether differences in the residues immediately after helix VII cause differential trafficking of CXCR4 and CXCR7: the domain of the putative helix VIII was deleted in one mutant and replaced with the corresponding nonhelical domain of CXCR4 in the other. Because neither mutant provided evidence for a key role of these residues in ligand-independent internalization of CXCR7, it seems likely that the last 24 residues of CXCR7 are sufficient for constitutive endosomal targeting. This part of CXCR7 contains a YXXL (YSAL) motif 9 residues before the C terminus. Although this kind of motif is involved in endosomal and lysosomal sorting of transmembrane proteins including G protein-coupled receptors (34, 43, 44), we found no evidence that a tyrosine-based motif in the CXCR7 C terminus is required for ligand-independent internalization. In the case of the atypical chemokine receptor D6, constitutive phosphorylation of a cluster of serine residues in the C terminus has been shown to be involved in β-arrestin recruitment, receptor trafficking, and receptor stability (24). Thus, the presence of potential phosphorylation sites in the C-terminal domain of CXCR7 suggests that a similar mechanism may be involved in trafficking and MAP kinase signaling of CXCR7 (11, 22, 40). Our finding that the CXCR4 C terminus shows strong constitutive phosphorylation when fused to CXCR7 points to the possibility that the CXCR7 protein prefers a conformation that attracts G protein-coupled receptor kinases. Indeed, conversion of all C-terminal serine and threonine residues into alanines eliminates β-arrestin recruitment and impairs CXCR7 trafficking (37). These findings are supported by our results showing that CXCR7-mediated CXCL12 uptake and CXCL12 degradation are slower with the CXCR7-ST/A mutant. Analyses of the CXCR7-STS and CXCR7-STT mutants suggests that the cluster of Ser-350, Thr-352, and Ser-355 is of particular relevance for internalization of CXCL12-CXCR7 complexes.
Direct comparison of the CXCR4 and CXCR7 wild type receptors with their tail-swap mutants under experimental conditions producing similar total protein levels clearly revealed that the CXCR7 C terminus is responsible for rapid receptor-mediated CXCL12 uptake and degradation. Given that CXCR4 becomes degraded after long term CXCL12 exposure and that the steady state expression of CXCR7 is not influenced by CXCL12, it is conceivable that CXCR7 is the more efficient CXCL12 scavenger under physiological conditions. As a consequence of its scavenger function, CXCR7 prevents CXCL12-induced CXCR4 degradation and balances the CXCL12/CXCR4 pathway (10).
Conclusion
We provide evidence that the CXCR7 C-terminal domain exerts a Janus-faced role in the regulation of CXCR7: it permits the highly efficient scavenger function and limits the protein expression level of CXCR7. The latter effect allows for tight control of CXCR7 functions by regulating CXCR7 expression or CXCR7 recycling processes presumably via ubiquitination and serine/threonine phosphorylation.
Acknowledgment
We thank Christine Anders for excellent technical assistance.
This work was supported by Federal State Sachsen-Anhalt with the European Fund for Regional Development Grant EFRE 2007–2013, Deutsche Forschungsgemeinschaft (DFG) Grant STU295/5-1, and Dr. Robert-Pfleger Stiftung (all to R. S.).

This article contains supplemental Fig. S1.
- ANOVA
- analysis of variance
- CHX
- cycloheximide
- λ-PP
- lambda protein phosphatase.
REFERENCES
- 1. Sierro F., Biben C., Martínez-Muñoz L., Mellado M., Ransohoff R. M., Li M., Woehl B., Leung H., Groom J., Batten M., Harvey R. P., Martínez-A C., Mackay C. R., Mackay F. (2007) Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. U.S.A. 104, 14759–14764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sun X., Cheng G., Hao M., Zheng J., Zhou X., Zhang J., Taichman R. S., Pienta K. J., Wang J. (2010) CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 29, 709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zou Y. R., Kottmann A. H., Kuroda M., Taniuchi I., Littman D. R. (1998) Function of the chemokine receptor CXCR4 in hematopoiesis and in cerebellar development. Nature 393, 595–599 [DOI] [PubMed] [Google Scholar]
- 4. Nagasawa T., Hirota S., Tachibana K., Takakura N., Nishikawa S., Kitamura Y., Yoshida N., Kikutani H., Kishimoto T. (1996) Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382, 635–638 [DOI] [PubMed] [Google Scholar]
- 5. Ma Q., Jones D., Borghesani P. R., Segal R. A., Nagasawa T., Kishimoto T., Bronson R. T., Springer T. A. (1998) Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 95, 9448–9453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oberlin E., Amara A., Bachelerie F., Bessia C., Virelizier J. L., Arenzana-Seisdedos F., Schwartz O., Heard J. M., Clark-Lewis I., Legler D. F., Loetscher M., Baggiolini M., Moser B. (1996) The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell line-adapted HIV-1. Nature 382, 833–835 [DOI] [PubMed] [Google Scholar]
- 7. Nagasawa T., Nakajima T., Tachibana K., Iizasa H., Bleul C. C., Yoshie O., Matsushima K., Yoshida N., Springer T. A., Kishimoto T. (1996) Molecular cloning and characterization of a murine pre-B-cell growth-stimulating factor/stromal cell-derived factor 1 receptor, a murine homolog of the human immunodeficiency virus 1 entry coreceptor fusin. Proc. Natl. Acad. Sci. U.S.A. 93, 14726–14729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Balabanian K., Lagane B., Infantino S., Chow K. Y., Harriague J., Moepps B., Arenzana-Seisdedos F., Thelen M., Bachelerie F. (2005) The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 280, 35760–35766 [DOI] [PubMed] [Google Scholar]
- 9. Maksym R. B., Tarnowski M., Grymula K., Tarnowska J., Wysoczynski M., Liu R., Czerny B., Ratajczak J., Kucia M., Ratajczak M. Z. (2009) The role of stromal-derived factor-1–CXCR7 axis in development and cancer. Eur. J. Pharmacol. 625, 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sánchez-Alcañiz J. A., Haege S., Mueller W., Pla R., Mackay F., Schulz S., López-Bendito G., Stumm R., Marín O. (2011) CXCR7 controls neuronal migration by regulating chemokine responsiveness. Neuron 69, 77–90 [DOI] [PubMed] [Google Scholar]
- 11. Burns J. M., Summers B. C., Wang Y., Melikian A., Berahovich R., Miao Z., Penfold M. E., Sunshine M. J., Littman D. R., Kuo C. J., Wei K., McMaster B. E., Wright K., Howard M. C., Schall T. J. (2006) A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 203, 2201–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rajagopal S., Kim J., Ahn S., Craig S., Lam C. M., Gerard N. P., Gerard C., Lefkowitz R. J. (2010) β-Arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. U.S.A. 107, 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kalatskaya I., Berchiche Y. A., Gravel S., Limberg B. J., Rosenbaum J. S., Heveker N. (2009) AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol. Pharmacol. 75, 1240–1247 [DOI] [PubMed] [Google Scholar]
- 14. Thelen M., Thelen S. (2008) CXCR7, CXCR4, and CXCL12. An eccentric trio? J. Neuroimmunol. 198, 9–13 [DOI] [PubMed] [Google Scholar]
- 15. Odemis V., Lipfert J., Kraft R., Hajek P., Abraham G., Hattermann K., Mentlein R., Engele J. (2012) The presumed atypical chemokine receptor CXCR7 signals through Gi/o proteins in primary rodent astrocytes and human glioma cells. Glia 60, 372–381 [DOI] [PubMed] [Google Scholar]
- 16. Naumann U., Cameroni E., Pruenster M., Mahabaleshwar H., Raz E., Zerwes H. G., Rot A., Thelen M. (2010) CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS One 5, e9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luker K. E., Steele J. M., Mihalko L. A., Ray P., Luker G. D. (2010) Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene 29, 4599–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boldajipour B., Mahabaleshwar H., Kardash E., Reichman-Fried M., Blaser H., Minina S., Wilson D., Xu Q., Raz E. (2008) Control of chemokine-guided cell migration by ligand sequestration. Cell 132, 463–473 [DOI] [PubMed] [Google Scholar]
- 19. Orsini M. J., Parent J. L., Mundell S. J., Marchese A., Benovic J. L. (1999) Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the C-terminal tail that mediate receptor internalization. J. Biol. Chem. 274, 31076–31086 [DOI] [PubMed] [Google Scholar]
- 20. Busillo J. M., Armando S., Sengupta R., Meucci O., Bouvier M., Benovic J. L. (2010) Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J. Biol. Chem. 285, 7805–7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marchese A., Benovic J. L. (2001) Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276, 45509–45512 [DOI] [PubMed] [Google Scholar]
- 22. Zabel B. A., Wang Y., Lewén S., Berahovich R. D., Penfold M. E., Zhang P., Powers J., Summers B. C., Miao Z., Zhao B., Jalili A., Janowska-Wieczorek A., Jaen J. C., Schall T. J. (2009) Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J. Immunol. 183, 3204–3211 [DOI] [PubMed] [Google Scholar]
- 23. Fischer T., Nagel F., Jacobs S., Stumm R., Schulz S. (2008) Reassessment of CXCR4 chemokine receptor expression in human normal and neoplastic tissues using the novel rabbit monoclonal antibody UMB-2. PLoS One 3, e4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCulloch C. V., Morrow V., Milasta S., Comerford I., Milligan G., Graham G. J., Isaacs N. W., Nibbs R. J. (2008) Multiple roles for the C-terminal tail of the chemokine scavenger D6. J. Biol. Chem. 283, 7972–7982 [DOI] [PubMed] [Google Scholar]
- 25. Kohout T. A., Nicholas S. L., Perry S. J., Reinhart G., Junger S., Struthers R. S. (2004) Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J. Biol. Chem. 279, 23214–23222 [DOI] [PubMed] [Google Scholar]
- 26. Fra A. M., Locati M., Otero K., Sironi M., Signorelli P., Massardi M. L., Gobbi M., Vecchi A., Sozzani S., Mantovani A. (2003) Cutting edge. Scavenging of inflammatory CC chemokines by the promiscuous putatively silent chemokine receptor D6. J. Immunol. 170, 2279–2282 [DOI] [PubMed] [Google Scholar]
- 27. Stumm R. K., Rummel J., Junker V., Culmsee C., Pfeiffer M., Krieglstein J., Höllt V., Schulz S. (2002) A dual role for the SDF-1/CXCR4 chemokine receptor system in adult brain. Isoform-selective regulation of SDF-1 expression modulates CXCR4-dependent neuronal plasticity and cerebral leukocyte recruitment after focal ischemia. J. Neurosci. 22, 5865–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kleemann P., Papa D., Vigil-Cruz S., Seifert R. (2008) Functional reconstitution of the human chemokine receptor CXCR4 with Gi/Go proteins in Sf9 insect cells. Naunyn Schmiedebergs Arch. Pharmacol. 378, 261–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moepps B., Frodl R., Rodewald H. R., Baggiolini M., Gierschik P. (1997) Two murine homologues of the human chemokine receptor CXCR4 mediating stromal cell-derived factor 1α activation of Gi2 are differentially expressed in vivo. Eur. J. Immunol. 27, 2102–2112 [DOI] [PubMed] [Google Scholar]
- 30. Conklin B. R., Farfel Z., Lustig K. D., Julius D., Bourne H. R. (1993) Substitution of three amino acids switches receptor specificity of Gαq to that of Gαi. Nature 363, 274–276 [DOI] [PubMed] [Google Scholar]
- 31. Mody S. M., Ho M. K., Joshi S. A., Wong Y. H. (2000) Incorporation of Gαz-specific sequence at the carboxyl terminus increases the promiscuity of Gα16 toward Gi-coupled receptors. Mol. Pharmacol. 57, 13–23 [PubMed] [Google Scholar]
- 32. Liu A. M., Ho M. K., Wong C. S., Chan J. H., Pau A. H., Wong Y. H. (2003) Gα16/z chimeras efficiently link a wide range of G protein-coupled receptors to calcium mobilization. J. Biomol. Screen. 8, 39–49 [DOI] [PubMed] [Google Scholar]
- 33. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meiser A., Mueller A., Wise E. L., McDonagh E. M., Petit S. J., Saran N., Clark P. C., Williams T. J., Pease J. E. (2008) The chemokine receptor CXCR3 is degraded following internalization and is replenished at the cell surface by de novo synthesis of receptor. J. Immunol. 180, 6713–6724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shenoy S. K. (2007) Seven-transmembrane receptors and ubiquitination. Circ. Res. 100, 1142–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barker B. L., Benovic J. L. (2011) G protein-coupled receptor kinase 5 phosphorylation of hip regulates internalization of the chemokine receptor CXCR4. Biochemistry 50, 6933–6941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Canals M., Scholten D. J., de Munnik S., Han M. K., Smit M. J., Leurs R. (2012) Ubiquitination of CXCR7 controls receptor trafficking. PLoS One 7, e34192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ray P., Mihalko L. A., Coggins N. L., Moudgil P., Ehrlich A., Luker K. E., Luker G. D. (2012) Carboxyl terminus of CXCR7 regulates receptor localization and function. Int. J. Biochem. Cell Biol. 44, 669–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Levoye A., Balabanian K., Baleux F., Bachelerie F., Lagane B. (2009) CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 113, 6085–6093 [DOI] [PubMed] [Google Scholar]
- 40. Gravel S., Malouf C., Boulais P. E., Berchiche Y. A., Oishi S., Fujii N., Leduc R., Sinnett D., Heveker N. (2010) The peptidomimetic CXCR4 antagonist TC14012 recruits β-arrestin to CXCR7. Roles of receptor domains. J. Biol. Chem. 285, 37939–37943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bryson K., McGuffin L. J., Marsden R. L., Ward J. J., Sodhi J. S., Jones D. T. (2005) Protein structure prediction servers at University College London. Nucleic Acids Res. 33, W36–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 43. Bonifacino J. S., Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 44. Paing M. M., Temple B. R., Trejo J. (2004) A tyrosine-based sorting signal regulates intracellular trafficking of protease-activated receptor-1. Multiple regulatory mechanisms for agonist-induced G protein-coupled receptor internalization. J. Biol. Chem. 279, 21938–21947 [DOI] [PubMed] [Google Scholar]