

Background: DR6-induced apoptosis mechanism is unknown.
Results: DR6-induced apoptosis is dependent on cytochrome c release and Bax translocation, but is independent of caspase-8 and Bid.
Conclusion: DR6-induced apoptosis is mediated by a unique pathway, different from type I and type II pathways.
Significance: This study will lead to a better understanding of the mechanism by which DR6 induces apoptosis.
Keywords: Apoptosis, Bcl-2 Family Proteins, Caspase, Cytochrome c, Mitochondria, Bax, Bid, DR6, Death Receptor
Abstract
Cells undergo apoptosis through two major pathways, the extrinsic pathway (death receptor pathway) and the intrinsic pathway (the mitochondrial pathway). These two pathways can be linked by caspase-8-activated truncated Bid formation. Very recently, death receptor 6 (DR6) was shown to be involved in the neurodegeneration observed in Alzheimer disease. DR6, also known as TNFRSF21, is a relatively new member of the death receptor family, and it was found that DR6 induces apoptosis when it is overexpressed. However, how the death signal mediated by DR6 is transduced intracellularly is not known. To this end, we have examined the roles of caspases, apoptogenic mitochondrial factor cytochrome c, and the Bcl-2 family proteins in DR6-induced apoptosis. Our data demonstrated that Bax translocation is absolutely required for DR6-induced apoptosis. On the other hand, inhibition of caspase-8 and knockdown of Bid have no effect on DR6-induced apoptosis. Our results strongly suggest that DR6-induced apoptosis occurs through a new pathway that is different from the type I and type II pathways through interacting with Bax.
Introduction
Apoptosis, also known as programmed cell death, plays a pivotal role in development, cancer, normal aging, and in neurological disorders such as Alzheimer disease and Parkinson disease (1). Cells undergo apoptosis through two major pathways: the extrinsic pathway (death receptor pathway) and the intrinsic pathway (mitochondrial pathway) (2). Both of these pathways have been implicated in the neuronal cell death found in Alzheimer disease. Upon triggering by specific stimuli, each of these pathways activates an initiator caspase, caspase-8 (death receptor pathway) and caspase-9 (mitochondrial pathway), respectively, which in turn activates the executor caspase-3 and eventually leads to apoptosis. In the intrinsic pathway, cytotoxic stimuli and proapoptotic signal-transducing molecules cause dysfunction of mitochondria and the release of cytochrome c from mitochondria into the cytosol, resulting in the formation of the cytochrome c/Apaf-1/caspase-9-containing apoptosome complex and the subsequent activation of procaspase-9, which itself activates caspase-3 and leads to apoptosis (for a review, see Ref. 3). On the other hand, the extrinsic signaling pathway leading to apoptosis involves transmembrane death receptors that are members of the tumor necrosis factor (TNF) receptor gene superfamily. Ligand-activated death receptors, such as Fas and TNF, recruit the Fas-associated death domain (FADD)2 protein, which in turn recruits the apoptosis-initiating protease caspase-8 through the death effector domain in both molecules to form the death-inducing signaling complex, resulting in the activation of caspase-8 (4, 5). Two signal transduction pathways transduce death receptor-mediated death signals: type I and type II. In the type I pathway, the amount of activated caspase-8 is sufficient to directly activate effector caspase-7, or caspase-3, leading to apoptosis. However, in the type II pathway, the limited amount of caspase-8 that is activated is not enough to directly activate the executor caspase-3 but is enough to cleave Bid, the BH3-domain-only protein of the B cell lymphoma (Bcl)-2 superfamily. Truncated Bid (tBid) then translocates to mitochondria and triggers cytochrome c release, resulting in mitochondria-dependent apoptosis (6).
Very recently, death receptor 6 (DR6) was shown to be involved in the neurodegeneration observed in Alzheimer disease (7). DR6, also known as TNFRSF21, is a relatively new member of the TNF receptor family with unknown biological function. DR6 was identified as a member of the TNF receptor-related death receptor family by searching the expressed sequence tag database using the protein sequence of the extracellular, cysteine-rich, ligand-binding domain of TNFR2, and it was found that DR6 induces apoptosis when it is overexpressed (8). However, the mechanism by which DR6 induces apoptosis is still unknown. Among these death receptors, DR6 is a unique member, as it was revealed that DR6-induced apoptosis was independent of the adaptor protein FADD (9), which is a key molecule-mediating death receptor-induced apoptotic signaling. This finding raised a question of how the death signal was transduced downstream from DR6. To this end, we have examined the roles of caspase-8, which is a key enzyme in mediating conventional death receptor-induced apoptosis (3), proapoptotic mitochondrial factor cytochrome c and the Bcl-2 family proteins in DR6-induced apoptosis. Our data strongly suggest that DR6 induced apoptosis through a pathway different from type I and type II pathways of Fas-mediated apoptosis, and instead, through a pathway that exclusively depends on the mitochondrial pathway and possibly through interacting with Bax.
EXPERIMENTAL PROCEDURES
Reagents
Lipofectamine 2000 transfection reagent, the T-REx System (tetracycline-regulated expression system), antibiotics blastindicin and Zeocin, and human DR6 cDNA clone were purchased from Invitrogen. Specific siRNAs and HiPerFect transfection reagent were purchased from Qiagen. Tet system-approved serum was purchased from Clontech Laboratories. Tetracycline and anti-β-actin antibody were purchased from Sigma. Complete Protease Inhibitor Mixture tablets were purchased from Roche Applied Science. The general caspase inhibitor, benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk), and caspase-8 inhibitor Z-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethylketone (Z-IETD-fmk) were purchased from Enzo Life Sciences. Anti-DR6 antibody was purchased from R&D Systems, and anti-cytochrome c was purchased from BD Biosciences. Antibodies against poly(ADP-ribose) polymerase (PARP), caspase-3, -6, -7, -8, -9, Bax, and Bid were purchased from Cell Signaling. Anti-COX I antibody was purchased from Santa Cruz Biotechnology. Horseradish peroxidase-linked anti-rabbit IgG antibody (from donkey), horseradish peroxidase-linked anti-mouse IgG antibody (from sheep), and the developing reagent ECL Plus were purchased from GE Healthcare. The plasmid pcDNA4/TO/LacZ-Myc-His, which expresses LacZ protein with a Myc tag after the addition of tetracycline, and the conventional plasmid pcDNA3.1/LacZ-Myc-His, which expresses Myc-tagged LacZ protein, were provided in the vector packages by the vendor (Invitrogen). Recombinant human tumor necrosis factor α (TNFα) and cycloheximide (CHX) were purchased from Millipore. Annexin V-enhanced green fluorescent protein apoptosis detection kit was purchased from GenScript. Trypsin without EDTA was purchased from Lonza.
Construction of Expression Vector
The DR6 open reading frame was amplified by PCR from a DR6 cDNA clone from Invitrogen, with primer pair NotI-KDR6 (5′-GCGGCCGCCACCATGGGGACCTCTCCGAGCAGCAGCACCGCCC-3′) and XbaImyc DR6 (5′-TCTAGACAGCAGGTCAGGAAGATGGCTATAAACAG-3′). The DNA fragment generated by using this pair of primers contains a Kozak sequence, GCCACC (10), followed by an ATG codon and an XbaI site at the 3′ end, which allows insertion of the DNA fragment into the pcDNA4/TO/Myc-His-A (Invitrogen) and pcDNA3.1/Myc-His-A expression vector (Invitrogen) in-frame. The PCR product was first cloned into the TOPO TA cloning vector with pCR2.1-TOPO (Invitrogen), and the fidelity of the DNA sequence was confirmed by DNA sequencing. The inserted DNA was then digested with NotI/XbaI restriction enzymes and subcloned into pcDNA4/TO/Myc-His-A and pcDNA3.1/Myc-His-A expression vectors in-frame. Thus, the DR6 was expressed as a Myc-tagged polypeptide with the Myc-His tag fused at the C terminus.
Cell Culture and Transfection
Human cervical cancer HeLa cells, human embryonic kidney 293 (HEK293) cells, human neuroblastoma M17 cells, and human neuroglioma H4 cells were cultured in DMEM supplemented with 10% fetal bovine serum, 50 units/ml penicillin, and 50 μg/ml streptomycin. For transient transfection, cells were transfected with pcDNA 3.1 expressing DR6 or expressing LacZ using Lipofectamine 2000 following the manufacturer's instructions.
Establishing TRE-HeLa Stable Inducible Cell Line
HeLa cells were transfected with pcDNA6/TR plasmid DNA (Invitrogen) with Lipofectamine 2000 transfection reagent. The transfectants were cultured in DMEM (Lonza) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C, and the stable clones were selected by blasticidin (5 μg/ml). After testing for tetracycline-inducible gene expression by transient transfection with the positive control plasmid-expressing β-galactosidase (LacZ), the pcDNA6/TR stable clones were further transfected with pcDNA4/TO/Myc-His A-DR6 plasmid or with pcDNA4/TO/LacZ-Myc-His plasmid. The stable double transfectants were selected by the addition of blasticidin (5 μg/ml) and Zeocin (200 μg/ml).
Mitochondrial Preparation and Cytosolic Extracts
For examination of cytochrome c release, the cytosolic extracts and mitochondria-containing fractions were prepared by permeabilization of cells with streptolysin O using the method described previously by Mosser et al. (11) with slight modification (12). Briefly, cells (106) were washed with phosphate-buffered saline (PBS), collected by centrifugation, and resuspended in 10 μl of streptolysin O buffer (20 mm HEPES, pH 7.5, 250 mm sucrose, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm dithiothreitol, 0.1 mm phenylmethylsulfonyl fluoride, and 1× protease inhibitor mixture) containing 60 units of streptolysin O (Sigma). After incubation at 37 °C for 20 min, the permeabilization of cells was monitored by trypan blue staining. At the time when 95% cells were stained, permeabilized cells were pelleted by centrifugation at 16,000 × g for 15 min at 4 °C. The supernatant was collected as the cytosolic fraction, and the pellet was collected as a mitochondria-containing membrane fraction. Both the cytosolic and mitochondrial fractions were then subjected to SDS-polyacrylamide gel electrophoresis (PAGE) (10–14%) followed by Western blot analysis using antibodies against cytochrome c, Bax, and Bid.
siRNA Treatment
siRNA duplexes specific to Bax and Bid were generated by Qiagen. A control siRNA duplex that does not target any sequence in the genome (by BLAST search) was also purchased from Qiagen. Cells were transfected with these siRNAs three times at 2-day intervals using HiPerFect transfection reagent, following the instructions provided by the manufacturer. On day 5, cells were induced for DR6 expression by the addition of tetracycline. 48 h after induction, cells were harvested. Half of the cells were lysed and directly subjected to SDS-PAGE followed by Western blot analysis; the other half of the cells was used to prepare cytosolic and mitochondrial fractions.
SDS-PAGE, Western Blotting, and Immunoprecipitation
For routine Western blot analysis, cells and the mitochondria-containing membrane fraction were lysed by sonication for 20 s on ice in Western blot lysis buffer (50 mm Tris-HCl, pH 6.8, 8 m urea, 5% mercaptoethanol, 2% SDS, and protease inhibitor mixture). After the addition of 4 × SDS sample buffer and boiling at 100 °C for 7 min, samples were subjected to SDS-PAGE (PAGE, 8% for DR6 and LacZ, 10–14% for cytochrome c, Bax, and Bid) and transferred to a polyvinylidene fluoride membrane (Immobilon-P, Millipore). The membranes were probed with appropriate antibodies as described in the figure legends. For cytosolic fraction, samples were directly treated with 4 × SDS sample buffer as described previously (12).
For immunoprecipitation, cells were lysed in immunoprecipitation lysis buffer (1% CHAPS, 30 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm EDTA, and protease inhibitor mixture). After sonication for 20 s, the total cell lysates were centrifuged at 14,000 × g for 5 min at 4 °C to remove the cell debris, and the supernatants were incubated with anti-Bax, anti-DR6 (R&D Systems) or anti-Myc (Santa Cruz Biotechnology) together with protein A-Sepharose overnight at 4 °C. After washing five times with cold PBS, the immunoprecipitates were separated by SDS-PAGE and probed with the appropriate antibodies.
Flow Cytometric Analysis
HeLa and HEK293 cells were transiently transfected with LacZ and DR6. 24 h after transfection, both floating and attached cells were collected. After washing twice with ice-cold PBS, cells were stained with Annexin V-enhanced green fluorescent protein/propidium iodide (PI), using an Annexin V-enhanced green fluorescent protein apoptosis detection kit and analyzed using an Epics XL-MCL Flow Cytometer (Beckman Instruments) following the manufacturer's protocol. Both PI- and Annexin V-negative cells (quadrant P3) were considered normal intact cells, PI-negative and Annexin V-positive cells were considered early apoptotic (quadrant P4) cells, cells that were both PI- and Annexin V-positive (quadrant P2) were considered late apoptotic cells, and cells that were PI-positive and Annexin V-negative were considered mechanically injured cells (quadrant P1) during the experiment.
Cells were exposed to carvacrol at concentrations of 0, 200 μm, 400 μm, or 600 μm for 24 h. Cells were harvested after incubation and washed in cold PBS. Cells were resuspended in 400 μl of solution with 10 mm HEPES, 140 mm NaC1, and 2.5 mm CaC12, pH 7.4. Alexa Fluor 488 Annexin V/PI staining solution (Probes Invitrogen) was added in the dark. After incubation for 15 min, the cells were collected and analyzed in a FACScan flow cytometry analyzer. Excitation wavelength was at 488 nm, and the emitted green fluorescence of Annexin V (FL1) and red fluorescence of PI (FL2) were collected using 530-nm and 575-nm band pass filters, respectively.
RESULTS
Overexpression of DR6-induced Apoptotic Cell Death in Various Types of Cells
To determine the apoptotic activity of DR6, we transfected M17 neuroblastoma cells, human H4 neuroglioma cells, human cervical cancer HeLa cells, and human embryonic kidney 293 (HEK293) cells with Myc-tagged DR6. As shown in Fig. 1A, transfection with DR6 resulted in apoptotic cleavage of PARP, an indicator of apoptosis (13), in all the transfected cells (lanes 2, 4, 6, and 8). As a control, no apoptotic sign was detected in cells transfected with LacZ protein (lanes 1, 3, 5, and 7). The multiple bands detected in the DR6-transfected cells are apparently a result of different glycosylation (14). To confirm further the apoptosis induced by DR6, an Annexin V/PI staining assay was employed to detect cells in early apoptotic and late apoptotic/necrotic stages. 24 h after transfection, cells were collected and stained with Annexin V/PI and analyzed in a FACScan flow cytometry analyzer as described under “Experimental Procedures.” As shown in Fig. 1B, the amounts of early apoptosis and late apoptosis/necrosis cells were about 40 and 5%, respectively, in DR6-transfected (right column), HEK293 (top panel), and HeLa (bottom panel) cells as determined by the Annexin V+/PI-(P4) or Annexin V+/PI+ (P2) cells. In the LacZ-transfected cells (left column), the basal levels of Annexin V+/PI-(P4) or Annexin V+/PI+ (P2) cells were <1 and 2%, respectively. Taken together, these results clearly indicate that overexpression of DR6 induces apoptosis in the different types of cells. Next, to determine the mechanism by which DR6 induces apoptosis, we have established a Tet-On system for inducible expression of DR6 in HeLa cells and performed a time-course experiment to analyze the apoptosis upon induction of DR6. At various time points, equal numbers of cells were harvested, and the expression of DR6, cleavage of PARP, activation of caspases, and the translocation of mitochondria proapoptotic proteins were analyzed by Western blotting. As shown in the top panel of Fig. 2A, at 6-h induction of DR6, the 85-kDa fragment of PARP, which is characteristic of apoptosis, became detectable (lane 2, second panel). Caspase activation was also detected at this time point and increased in a time-dependent manner (lanes 2–6, third to sixth panels). As shown in Fig. 2B, a similar time-dependent release of cytochrome c into the cytosol and translocation of Bax to the mitochondria were also detected. However, the formation of tBid became detectable in the mitochondria at a later time point, the 48-h point (lane 6, third panel of the right column). Therefore, the following experiments were carried out at 48-h time points.
FIGURE 1.
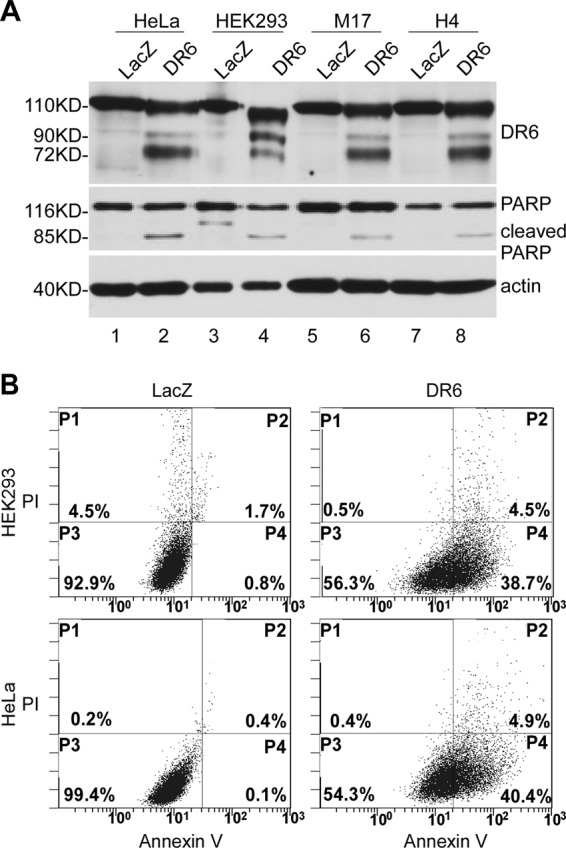
Overexpression of DR6-induced apoptosis in various types of cells. A, cells were transiently transfected with either the DR6-expressing construct pcDNA3.1/Myc-His/DR6 (DR6), which expresses a DR6-Myc fusion protein; or a control plasmid, pcDNA3.1/Myc-His/LacZ (LacZ), which expresses Myc-tagged LacZ protein. 24 h after transfection, cells were harvested, and the lysates were separated by SDS-PAGE, followed by Western blot analysis. Expression of DR6 and LacZ was detected using anti-Myc antibody (top panel). LacZ was detected as a 116-kDa peptide as expected (lanes 1, 3, 5, and 7). DR6 was detected as several bands within the molecular mass range 72–110 kDa, resulting from different glycosylation (14). PARP cleavage was detected in the DR6-expressing samples (lanes 2, 4, 6 and 8, middle panel) but not in the LacZ-transfected cells (lanes 1, 3, 5, and 7). This membrane was also reprobed with anti-actin antibody to indicate relative loading of samples (bottom panel). B, representative FACS scatter plots of three independent experiments are shown. DR6- (right column) or LacZ- (left column) transfected HeLa (bottom row) and HEK293 cells (top row) were double stained with Annexin-V-enhanced green fluorescent protein and PI. Fluorescence was detected using a fluorescence-activated Epics XL-MCL Flow Cytometer to analyze necrotic (PI-positive, quadrant P1), nonapoptotic (negative for both dyes, quadrant P3), early apoptotic (Annexin-positive/PI-negative, quadrant P4), and late apoptotic cells (positive for dyes, quadrant P2).
FIGURE 2.

Time course of DR6-induced apoptosis. After induction of DR6 expression by the addition of tetracycline (Tet), cells were harvested at various time points as indicated and divided into two portions. Half of the harvested cells were directly lysed and separated by SDS-PAGE, followed by Western blot analysis. The other half of the cells were used for preparation of cytosolic extracts and mitochondria-containing membrane fraction as described under “Experimental Procedures.” A, top panel shows induction of DR6 expression. Second panel shows PARP cleavage. Third, fourth, fifth, and sixth panels show activation of caspase-3, caspase-9, caspase-8, and caspase-7, respectively. The activation of caspases was determined by the formation of active forms of these caspases. This membrane was also reprobed with anti-actin antibody to indicate relative loading of samples (bottom panel). B, top panels show release of cytochrome c from mitochondria (right column) to the cytosol (left column). Second panels show translocation of Bax from cytosol (left panel) to mitochondria (right panel). Third panel show tBid formation and translocation to mitochondria. Fourth panel shows the reprobe of the second panels with anti-COX I antibody confirming the mitochondria remained intact during preparation.
Caspase-8 Is Not Required for DR6-induced Apoptosis and Cytochrome c Release and Bax Translocation
Next, we determined the effects of caspase inhibitors on DR6-induced apoptosis. To do so, cells were cultured in the presence or absence of the general caspase inhibitor Z-VAD (100 μm) or caspase-8 inhibitor Z-IETD (100 μm) for 30 min prior to induction of DR6 expression. As shown in Fig. 3A, upon induction of DR6 expression by the addition of tetracycline, PARP cleavage and caspase activation were detected in cells cultured in the absence of inhibitors (lane 2). Activation of caspase-3, caspase-7, caspase-8, and caspase-9 was expressed as the formation of the processed short active forms. Because the level of the active form of caspase-6 was too low to be detected, caspase-6 activation was expressed as the decrease in the level of procaspase-6. When the cells were cultured in the presence of general caspase inhibitor Z-VAD, neither PARP cleavage nor caspase activation was detected (lane 3), indicating that DR6-induced apoptosis depends on caspase activation. Interestingly, when the cells were cultured in the presence of caspase-8 inhibitor Z-IETD, DR6-induced caspase-8 activation was blocked (lane 4, third panel); however, PARP cleavage (lane 4, second panel) and the activation of other caspases were not affected (lane 4, panels 4–7).
FIGURE 3.
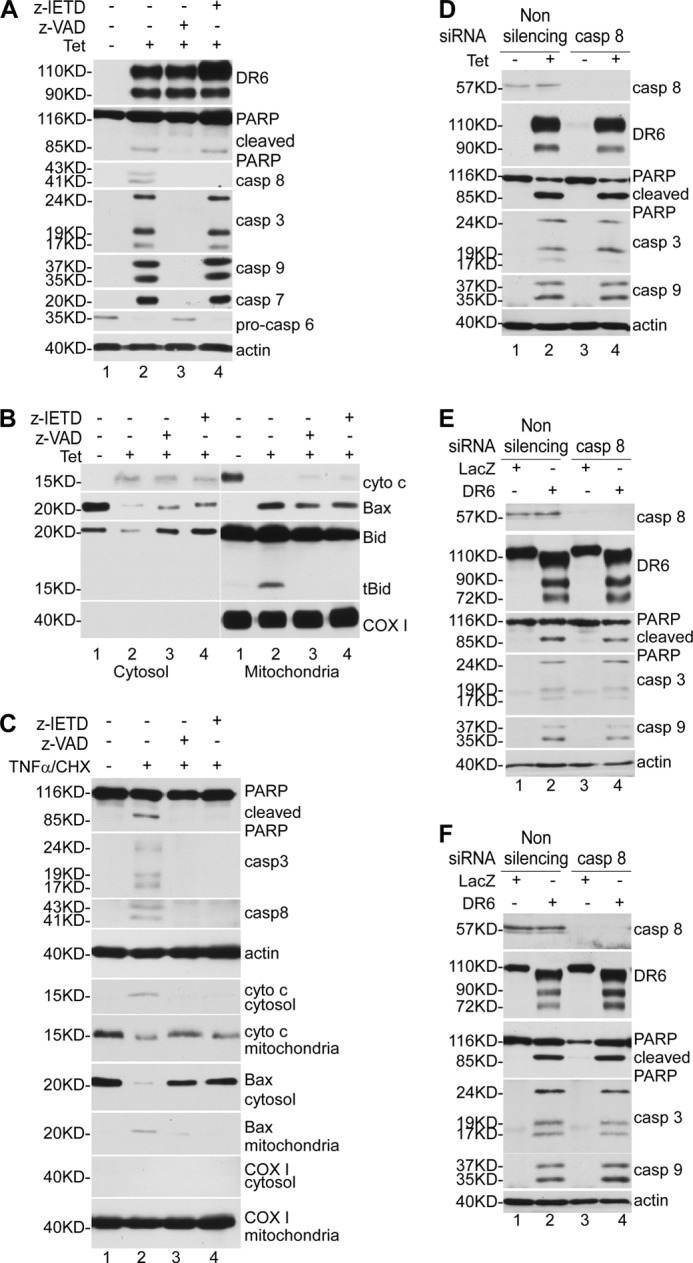
Caspase-8 is not required for DR6-induced apoptosis. A, T-RE HeLa-inducible cells were cultured in the presence or absence of caspase inhibitors and induced for DR6 expression for 48 h. Half of the cells were directly lysed and subjected to Western blot analysis, and the other half were used for preparation of cytosolic extracts and mitochondria-containing membrane fraction. DR6 expression, PARP cleavage, and caspase activation were determined as described in the legend to Fig. 2. Because the active form of caspase-6 is hardly detectable, caspase-6 activation was determined by the decrease in procaspase-6. The general caspase inhibitor Z-VAD was used at 100 μm (lane 3), and the caspase-8-specific inhibitor Z-IETD was used at 100 μm (lane 4). In lanes 1 and 2, cells were treated with vehicle only. The bottom panel is the reprobe of the membrane of the fifth panel with anti-actin antibody to indicate relative loading of samples. B, neither Z-VAD nor Z-IETD blocked cytochrome c release and Bax translocation induced by DR6. Cellular fractionation and Western blot analysis were performed as described under “Experimental Procedures.” The mitochondrial protein COX I was detected only in the membrane fraction (bottom panel), indicating that the mitochondria remained intact during the sample preparation. As shown by previous studies, in addition to tBid, a significant Bid proprotein was detected in the mitochondrial fraction (19, 20). C, caspase-8 inhibitor abrogates apoptosis induced by co-treatment with TNFα and CHX. Cells were pretreated with caspase inhibitors or vehicle only for 30 min and induced for apoptosis by TNFα and CHX for 6 h. PARP cleavage, caspase activation, cytochrome c release, and Bax translocation were determined as described by Western blot analysis. The fourth panel is the reprobe of the membrane of the third panel with anti-actin antibody to indicate relative loading of samples. The mitochondrial protein COX I was detected only in the membrane fraction (bottom panel), indicating that the mitochondria remained intact during sample preparation. Knockdown of caspase-8 had no effect on DR6-induced apoptosis in HeLa cells (D), HEK293 cells (E), and H4 cells (F). Top panel shows Western blot for caspase-8; second panel, Western blot for DR6; third panel, Western blot for PARP; fourth panel, Western blot for caspase-3; fifth panel, Western blot for caspase-9; bottom panel, membranes in the fifth panel were reprobed with anti-actin antibody to indicate relative loading of samples.
Next, we determined the effects of caspase inhibitors on the release of cytochrome c and the translocation of Bax and tBid induced by DR6. As shown in Fig. 3B, none of the caspase inhibitors had an effect on DR6-induced cytochrome c release and Bax translocation (lanes 3 and 4 of first and second panels of both left and right columns). These data strongly indicate that the release of cytochrome c from mitochondria to the cytosol and the translocation of Bax from cytosol to mitochondria are independent of caspase activity. On the other hand, it is notable that both the inhibitors blocked tBid formation (lanes 3 and 4, third panel of the right column), suggesting that DR6-induced tBid formation is dependent on caspase activation. The observation that caspase inhibitors blocked tBid formation but had no effect on Bax and cytochrome c translocation strongly suggests that Bax translocation and cytochrome c release are independent of tBid formation. This is in agreement with the observation that DR6-induced apoptosis is independent of caspase-8 activation, which cleaves Bid to generate tBid.
To validate our data that DR6-induced apoptosis being independent of caspase-8 is not an artificial result of our experimental conditions, as a control, we examined the effect of caspase-8 inhibitor on TNFα-induced apoptosis under the same experimental conditions. As shown in Fig. 3C, when HeLa cells were treated with TNFα (30 ng/ml) and CHX (3 μm), PARP cleavage (lane 2, top panel), caspase activation (lanes 2, second and third panels), and cytochrome c release and Bax translocation (lane 2, fifth to eighth panels) were detected. However, when the cells were pretreated with general caspase inhibitor Z-VAD (100 μm) and caspase-8-specific inhibitor Z-IETD (100 μm) for 30 min prior to the addition of TNFα and CHX, as shown in lanes 3 and 4, all the apoptotic changes, including PARP cleavage, caspase activation, cytochrome c release, and Bax translocation, were completely inhibited; this result was as expected because it is well known that caspase-8 is the key factor in mediating TNFα-induced apoptosis (15). This result indicates that our data that DR6-induced apoptosis is independent of caspase-8 are not artificial results of our experimental conditions.
Next, we employed the siRNA approach to determine further the effect of knockdown of caspase-8 on DR6-induced apoptosis. As shown in Fig. 3D, caspase-8 was completely knocked down by treatment with caspase-8-specific siRNA (lanes 3 and 4, top panel). However, DR6-induced PARP cleavage (third panel) and caspase-3 and caspase-9 activation were not affected by knockdown of caspase-8 (compare lane 4 with lane 2, third to fifth panels). To determine further whether the caspase-8-independent apoptosis induced by DR6 is a cell type-specific or a general mechanism, we examined the effect of knockdown of caspase-8 on DR6-induced apoptosis in HEK293 and H4 cells. As shown in Fig. 3, E and F, knockdown of caspase-8 had no effect on DR6-induced apoptosis in HEK293 and H4 cells. Taken together, these results clearly indicate that DR6-induced apoptosis is independent of caspase-8.
Overexpression of Bcl-2 and B Cell Lymphoma-extra Large (Bcl-xL) Inhibits DR6-induced Apoptosis
The above data clearly indicate that cytochrome c release and Bax translocation are events apparently upstream of caspase activation in DR6-induced apoptosis. Thus, to determine further the mechanism of DR6-induced apoptosis, we next determined the effects of antiapoptotic protein Bcl-2 and Bcl-xL on DR6-induced apoptosis. Cells were transiently transfected with Bcl-2 or Bcl-xL plasmids or a plasmid that expresses LacZ protein for 24 h and then induced for DR6 expression. As shown in Fig. 4, both Bcl-2 (A, lanes 3 and 4, top panel) and Bcl-xL (B, lanes 3 and 4, top panel) were expressed at significant levels. Endogenous Bcl-2 could be detected after longer exposure (data not shown). This may account for the susceptibility of these cells to DR6-induced apoptosis as suggested by a previous study (9). As shown in Fig. 4A, upon induction of DR6 expression, cytochrome c release from mitochondria to cytosol (lane 2, fifth and sixth panels) and Bax translocation from cytosol to mitochondria (lane 2, seventh and eighth panels) were detected in the LacZ-transfected cells, but these events were blocked in the Bcl-2-transfected cells (lane 4, panels 5–8). As shown in Fig. 4B, similarly, DR6-induced cytochrome c release from mitochondria to cytosol (lane 2, panels 5 and 6) and Bax translocation from cytosol to mitochondria (lane 2, panels 7 and 8) were detected in the LacZ-transfected cells, but these events were blocked in the Bcl-xL-transfected cells (lane 4, panels 5–8). As shown in Fig. 4C, upon induction of DR6, PARP cleavage (lane 2, top panel) and caspase activation (lane 2, panels 2–6) were observed in LacZ-transfected cells. However, in the Bcl-2- (lane 4) and Bcl-xL- (lane 6) transfected cells, DR6-induced PARP cleavage and caspase activation were completely blocked (lanes 4 and 6, panels 1–6). Furthermore, as shown in lane 2 of panels 8 and 9, DR6-induced tBid formation was detected in LacZ-transfected cells. However, this DR6-induced tBid formation was completely blocked by Bcl-2 and Bcl-xL (lanes 4 and 6, panels 8 and 9).
FIGURE 4.

Overexpression of antiapoptotic protein Bcl-2 and Bcl-xL inhibited DR6-induced apoptosis. Cell lysis and fractionation were performed as described previously. A and B, after 24-h transfection, a significant amount of Bcl-2 and Bcl-xL was detected (lanes 3 and 4, top panel). Because the recombinant Bcl-2 and Bcl-xL were expressed as a FLAG-tagged protein, the recombinant Bcl-2 and Bcl-xL were detected with a slower migration rate than endogenous Bcl-2 and Bcl-xL (compare lanes 3 and 4 with lanes 1 and 2). This membrane was also reprobed with anti-actin antibody to indicate relative loading of samples (fourth panel). Second and third panels show tetracycline (Tet)-induced DR6 expression, respectively; fifth and sixth panels, cytochrome c release from mitochondria; seventh and eighth panels, Bax translocation from cytosol to mitochondria. C, top panel shows Western blot for PARP; second to sixth panels, Western blots for caspases; seventh panel, Western blot for actin to indicate relative loading of lysate samples; eighth and ninth panels, Western blots for Bid; tenth and eleventh panels were stained for mitochondrial protein COX I.
Bax, but Not Bid, Is Required for DR6-induced Apoptosis
The data presented above strongly suggest that DR6-induced apoptosis is controlled by Bcl-2 family protein. To explore further the roles of Bax and Bid in DR6-induced apoptosis, we examined the effect of knockdown of Bax and Bid on DR6-induced apoptosis. As shown in lanes 3 and 4 of the top panels of Fig. 5, A and B, Bax and Bid were completely knocked down using the siRNA approach. As shown in Fig. 5A, upon induction of DR6 expression (lanes 2 and 4, second panel), PARP cleavage (lane 2, third panel), and tBid formation (lane 2, bottom panel) were observed in cells treated with nonsilencing control siRNA. However, when the cells were treated with Bax-specific siRNA, DR6-induced PARP cleavage and tBid formation were blocked (lane 4 of third and bottom panels). Interestingly, as shown in Fig. 5B, when the cells were treated with Bid-specific siRNA, DR6-induced PARP cleavage and Bax translocation were not disturbed (compare lane 4 with lane 2, of panels 3, 5, and 6). These results strongly indicate that DR6-induced Bax translocation is independent of tBid formation; however, tBid formation is dependent on Bax translocation. Moreover, as shown in Fig. 5C, as expected, in the nonsilencing control siRNA-treated cells, induction of DR6 expression resulted in caspase activation (lane 2 of panels 1–5), and cytochrome c release from mitochondria (lane 2 of panels 6 and 7). However, the DR6-induced caspase activation and cytochrome c release were completely blocked by knockdown of Bax (lane 4, panels 1–7). More interestingly, knockdown of Bid had no effect on DR6-induced caspase activation or cytochrome c release (lane 6, panels 1–7). Taken together, these results clearly indicate that tBid formation is not required for DR6-induced apoptosis.
FIGURE 5.

Knockdown of Bax blocked DR6-induced PARP cleavage, caspase activation, cytochrome c release, and tBid formation and translocation, and Bid is not required for DR6-induced apoptosis. A, top panel shows the Western blot for Bax; second panel, Western blot for DR6; third panel, Western blot for PARP; the membranes in the third panel were reprobed with anti-actin antibody to indicate relative loading of samples (fourth panel). Fifth and sixth panels show blots for Bid in cytosol and mitochondria, respectively. B, top panel shows the Western blot for Bid; second panel, Western blot for DR6; third panel, PARP; the membranes in the third panel were reprobed with anti-actin antibody to indicate relative loading of samples (fourth panel). Fifth and sixth panels show Western blots for Bax in cytosol and mitochondria, respectively. C, first to fifth panels show Western blots for caspases. Sixth and seventh panels show Western blots for cytochrome c in cytosolic and mitochondria-containing membrane fractions. Eighth and ninth panels were stained for mitochondrial protein COX I.
DR6 Interacts with Bax
Because DR6-induced Bax translocation is independent of tBid formation, we sought to examine the possible interaction of DR6 with Bax by co-immunoprecipitation. After 48-h induction of DR6, cells were harvested, and cell lysates were subjected to immunoprecipitation as described under “Experimental Procedures.” As shown in Fig. 6, a high level of DR6 was detected in cells induced for Myc-tagged DR6 expression (lane 2, top panel), but the endogenous DR6 (lane 1, top panel) is hardly visible without prolonged exposure. A significant amount of Bax was detected in the cell lysates of both cells regardless of whether DR6 expression was induced (lanes 1 and 2, bottom panel). As shown in lane 4 of the top panel, a significant amount of DR6 was co-immunoprecipitated with Bax in cells induced for DR6 expression but not in cells without induction of DR6 expression (lane 3, top panel). Bax was immunoprecipitated by anti-Bax antibody regardless of whether DR6 expression was induced (lanes 3 and 4, bottom panel). However, when anti-Myc antibody was used, Bax was only co-immunoprecipitated in cells induced for Myc-tagged DR6 expression (compare lane 6 with lane 5, bottom panel). When nonrelated rabbit IgG was used, no Bax was immunoprecipitated from either cell lysate (lanes 7 and 8). These data suggest a possibility that DR6 induces Bax translocation via interacting with Bax.
FIGURE 6.
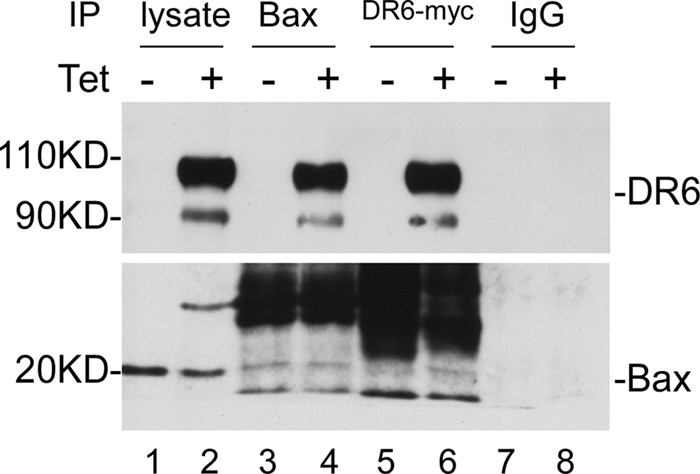
Co-immunoprecipitation of DR6 with Bax. 48 h after induction of DR6 expression, total cell lysates were immunoprecipitated with anti-Bax (lane 4) and with anti-Myc (lane 6) or with a control rabbit IgG (lane 8) and then probed with anti-DR6 (upper panel) and anti-Bax (lower panel). The endogenous DR6 (lane 1, top panel) is hardly visible without prolonged exposure.
DISCUSSION
DR6 is a relatively less characterized, new member of the TNF death receptor family of proteins. DR6 induces apoptosis when it is overexpressed either by transfection with a DR6-expressing plasmid (8) or by induction with TNFα (9). However, the mechanism by which DR6 induces apoptosis remains unknown. It has been shown that DR6 does not interact with the adaptor protein FADD, but interacts with TNF receptor-associated death domain (8). Furthermore, it was reported that FADD is not required for DR6-induced apoptosis, which leaves behind an unanswered question about how DR6 transduces apoptotic signals leading to the activation of caspases and apoptosis (9). A recent finding that implicates DR6 in the neurodegeneration observed in Alzheimer disease (7) prompted us to determine the mechanism of DR6-induced apoptosis. We first determined the effect of overexpression of DR6 on cell viability, and our data demonstrated that DR6 induced apoptosis in various types of cells including neuroblastoma and neuroglioma cells. To explore the mechanism by which DR6 induces apoptosis, we have established an inducible expression system. Using this system, our data demonstrated that DR6-induced apoptosis is completely inhibited by the general caspase inhibitor Z-VAD, indicating that DR6-induced apoptosis is caspase-dependent. On the other hand, DR6-induced apoptosis involves cytochrome c release and Bax translocation. Moreover, DR6-induced apoptotic events, including caspase activation and cytochrome c release, were completely inhibited by Bcl-2 and Bcl-xL and, specifically, by the knockdown of Bax. These findings clearly indicate that DR6-induced apoptosis is dependent on mitochondrial dysfunction.
With respect to the apoptosis mediated by death receptors, two major apoptosis signaling pathways have been established, type I and type II pathways, which are defined based on their level of mitochondrial dependence (16). According to the current models, in the type I pathway, upon activation, death receptors such as CD95 (APO-1/Fas) and TNF-related apoptosis-inducing ligand receptor recruit the adaptor protein FADD, which in turn recruits the initiator caspase-8 via the death effector domain to form the death-inducing signaling complex and results in the activation of a large amount of caspase-8. The activated caspase-8, in turn, directly activates the executor caspase-3 leading to apoptosis. On the other hand, in the type II pathway, only a limited amount of caspase-8 is activated at the death-inducing signaling complex. This low level of activated caspase-8 is not sufficient to activate caspase-3 directly but is enough to activate the mitochondria-based amplification loop by cleavage of Bid to tBid, followed by activation of Bax, leading to cytochrome c release and eventually apoptosis through activation of caspase-9 (5). Our observation that DR6-induced apoptosis is dependent on mitochondrial dysfunction suggests that DR6-induced apoptosis may be mediated by a type II pathway. This speculation is also supported by a previous finding that DR6-induced apoptosis was inhibited by caspase-8 inhibitor (9), which is a key enzyme in tBid formation. However, at the same time it was also shown that DR6-induced apoptosis was independent of FADD (9). Given the fact that either through direct interaction with FADD or indirect mediation by TNFR-associated death domain, FADD is necessary for receptor-induced caspase-8 activation (5, 17), the observation that caspase-8 inhibitor attenuated DR6-induced apoptosis is inconsistent with the finding that FADD is not required for DR6-induced apoptosis. To address this inconsistency, we reexamined the role of caspase-8 in DR6-induced apoptosis using our inducible system. Via this system, our data demonstrated that inhibition or knockdown of caspase-8 had no effect on the activation of other caspases and the apoptotic cleavage of PARP induced by DR6 in different types of cells (Fig. 3, A and D–F). This finding indicates that caspase-8 activation is not required for DR6-induced apoptosis. This discrepancy between our results and the previous finding may stem from different experimental conditions used in these two experiments. Nevertheless, the finding that caspase-8 is not required for DR6-induced apoptosis is in agreement with the fact that DR6 does not interact with FADD (8) and that DR6-induced apoptosis is independent of FADD (9). In addition, our data demonstrated that the general caspase inhibitor Z-VAD completely inhibited DR6-induced apoptosis but had no effect on cytochrome c release and Bax translocation. These results indicate that DR6-induced cytochrome c release and Bax translocation are independent of caspase activation. Taken together, the observations that DR6-induced apoptosis is independent of FADD and caspase-8 activation suggest that DR6-induced apoptosis is not mediated by the type II pathway. Moreover, tBid formation is required for bridging between caspase-8 activated by receptor complex and proapoptotic activation of mitochondria in the type II pathway (18). Thus, our observation that knockdown of Bid had no effect on DR6-induced Bax translocation, cytochrome c release, and apoptosis provided further strong support for our conclusion that DR6-induced apoptosis is not mediated by the type II pathway. Accordingly, DR6 induces apoptosis not through the known death receptor-mediated type I or type II pathways but via a novel apoptotic pathway that mediates the receptor apoptotic signal to mitochondrial dysfunction dependent on Bax translocation to mitochondria but independent of caspase-8 activation and tBid formation. To explore the mechanism of DR6-induced apoptosis, our data, which show that DR6 can be co-immunoprecipitated with Bax, strongly suggest a possibility that DR6 may mediate apoptotic signals through interaction with Bax.
Acknowledgment
We thank Misty R. Bailey for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R181741110 and R21AG039596 (to X. X.) and 0355339B (to M.-Z. C.). This work was also supported by an Alzheimer's Association grant and a grant from the American Health Assistance Foundation (to X. X.).
- FADD
- Fas-associated death domain
- tBid
- truncated Bid
- Bcl-2
- B cell lymphoma-2
- Bcl-xL
- B cell lymphoma-extra large
- DR6
- death receptor 6
- PARP
- poly(ADP-ribose) polymerase
- LacZ
- β-galactosidase
- Z-IETD-fmk
- benzyloxycarbonyl-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethylketone
- PI
- propidium iodide
- Z-VAD
- benzyloxycarbonyl-VAD
- CHX
- cycloheximide.
REFERENCES
- 1. Thompson C. B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267, 1456–1462 [DOI] [PubMed] [Google Scholar]
- 2. Youle R. J., Strasser A. (2008) The Bcl-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 3. Fulda S., Debatin K. M. (2006) Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811 [DOI] [PubMed] [Google Scholar]
- 4. Elmore S. (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilson N. S., Dixit V., Ashkenazi A. (2009) Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat. Immunol. 10, 348–355 [DOI] [PubMed] [Google Scholar]
- 6. Gonzalvez F., Ashkenazi A. (2010) New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 29, 4752–4765 [DOI] [PubMed] [Google Scholar]
- 7. Nikolaev A., McLaughlin T., O'Leary D. D., Tessier-Lavigne M. (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8. Pan G., Bauer J. H., Haridas V., Wang S., Liu D., Yu G., Vincenz C., Aggarwal B. B., Ni J., Dixit V. M. (1998) Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett. 431, 351–356 [DOI] [PubMed] [Google Scholar]
- 9. Kasof G. M., Lu J. J., Liu D., Speer B., Mongan K. N., Gomes B. C., Lorenzi M. V. (2001) Tumor necrosis factor α induces the expression of DR6, a member of the TNF receptor family, through activation of NF-κB. Oncogene 20, 7965–7975 [DOI] [PubMed] [Google Scholar]
- 10. Kozak M. (1991) Structural features in eukaryotic mRNAs that modulate the initiation of translation. J. Biol. Chem. 266, 19867–19870 [PubMed] [Google Scholar]
- 11. Mosser D. D., Caron A. W., Bourget L., Meriin A. B., Sherman M. Y., Morimoto R. I., Massie B. (2000) The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol. Cell. Biol. 20, 7146–7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu X., Shi Y. C., Gao W., Mao G., Zhao G., Agrawal S., Chisolm G. M., Sui D., Cui M. Z. (2002) The novel presenilin-1-associated protein is a proapoptotic mitochondrial protein. J. Biol. Chem. 277, 48913–48922 [DOI] [PubMed] [Google Scholar]
- 13. Dubrez L., Savoy I., Hamman A., Solary E. (1996) Pivotal role of a DEVD-sensitive step in etoposide-induced and Fas-mediated apoptotic pathways. EMBO J. 15, 5504–5512 [PMC free article] [PubMed] [Google Scholar]
- 14. Klíma M., Zájedová J., Doubravská L., Anděra L. (2009) Functional analysis of the posttranslational modifications of the death receptor 6. Biochim. Biophys. Acta 1793, 1579–1587 [DOI] [PubMed] [Google Scholar]
- 15. Ashkenazi A., Dixit V. M. (1998) Death receptors: signaling and modulation. Science 281, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 16. Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K. J., Debatin K. M., Krammer P. H., Peter M. E. (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17, 1675–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Micheau O., Tschopp J. (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 [DOI] [PubMed] [Google Scholar]
- 18. Kantari C., Walczak H. (2011) Caspase-8 and Bid: caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 1813, 558–563 [DOI] [PubMed] [Google Scholar]
- 19. Pucci B., Indelicato M., Paradisi V., Reali V., Pellegrini L., Aventaggiato M., Karpinich N. O., Fini M., Russo M. A., Farber J. L., Tafani M. (2009) ERK-1 MAP kinase prevents TNF-induced apoptosis through bad phosphorylation and inhibition of Bax translocation in HeLa Cells. J. Cell. Biochem. 108, 1166–1174 [DOI] [PubMed] [Google Scholar]
- 20. Schug Z. T., Gonzalvez F., Houtkooper R. H., Vaz F. M., Gottlieb E. (2011) Bid is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 18, 538–548 [DOI] [PMC free article] [PubMed] [Google Scholar]