

Background: How N-terminal acetylation affects the structure and function of α-syn remains unknown.
Results: N-terminally acetylated and WT α-syn are unfolded monomers and exhibit similar aggregation and cellular properties.
Conclusion: α-syn N-terminal acetylation does not dramatically affect its structure or oligomerization state in vitro and in intact cells.
Significance: Recombinant nonacetylated or Nα-acetylated α-syn remains suitable for α-syn biophysical studies.
Keywords: α-Synuclein, NMR, Parkinson disease, Protein Aggregation, Protein Conformation, N-terminal Acetylation, Membrane Binding, Subcellular Localization
Abstract
N-terminal acetylation is a very common post-translational modification, although its role in regulating protein physical properties and function remains poorly understood. α-Synuclein (α-syn), a protein that has been linked to the pathogenesis of Parkinson disease, is constitutively Nα-acetylated in vivo. Nevertheless, most of the biochemical and biophysical studies on the structure, aggregation, and function of α-syn in vitro utilize recombinant α-syn from Escherichia coli, which is not N-terminally acetylated. To elucidate the effect of Nα-acetylation on the biophysical and biological properties of α-syn, we produced Nα-acetylated α-syn first using a semisynthetic methodology based on expressed protein ligation (Berrade, L., and Camarero, J. A. (2009) Cell. Mol. Life Sci. 66, 3909–3922) and then a recombinant expression strategy, to compare its properties to unacetylated α-syn. We demonstrate that both WT and Nα-acetylated α-syn share a similar secondary structure and oligomeric state using both purified protein preparations and in-cell NMR on E. coli overexpressing Nα-acetylated α-syn. The two proteins have very close aggregation propensities as shown by thioflavin T binding and sedimentation assays. Furthermore, both Nα-acetylated and WT α-syn exhibited similar ability to bind synaptosomal membranes in vitro and in HeLa cells, where both internalized proteins exhibited prominent cytosolic subcellular distribution. We then determined the effect of attenuating Nα-acetylation in living cells, first by using a nonacetylable mutant and then by silencing the enzyme responsible for α-syn Nα-acetylation. Both approaches revealed similar subcellular distribution and membrane binding for both the nonacetylable mutant and WT α-syn, suggesting that N-terminal acetylation does not significantly affect its structure in vitro and in intact cells.
Introduction
α-Synuclein (α-syn)3 is a 140-residue natively disordered protein expressed mainly at presynaptic termini of neurons that has emerged as a key player in the pathogenesis of Parkinson disease (PD) and related synucleinopathies (2). Fibrillar and aggregated forms of α-syn are the major constituents of the intracellular protein inclusions called Lewy bodies (LBs), which are commonly found in surviving dopaminergic neurons of PD patients. Mutations in the protein sequence (A30P (3), E46K (4), and A53T (5)) or increased expression (due to gene duplication or triplication (6, 7) have been shown to cause early onset forms of PD and to enhance α-syn oligomerization and fibrillogenesis (8). Biochemical analysis of LBs and α-syn from PD patient brains has shown that α-syn undergoes diverse post-translational modifications (9) such as ubiquitination (10–12), phosphorylation (11, 13), truncation (14, 15), oxidation (16), and Nα-acetylation (11, 17). Whereas some of these post-translational modifications have been linked to pathogenic processes (9), it has recently become clear that the great majority, if not all, of native α-syn is N-terminally acetylated in both normal and diseased individuals (11, 17).
Nα-Acetylation is one of the most common post-translational (or more precisely, co-translational) modifications occurring in eukaryotic cells. It has been suggested that up to 80–90% of mammalian (18, 19) and 40–50% of yeast (20–22) proteins bear an Nα-acetyl group (23). Nevertheless, the role of this modification, which affects thousands of proteins, remains poorly understood (30). Nα-Acetylation is carried out by a family of three N-acetyltransferase enzymatic complexes that use acetyl-coenzyme A as a cofactor (24). Whereas these were initially identified in yeast (22, 23, 25) as NatA, -B, and -C, each of which composed of distinct regulatory and catalytic subunits, homologs of each of these complexes have been described in humans (26–28). In the case of α-syn, although the enzyme responsible for its Nα-acetylation has not yet been identified, the substrate specificity of NatB (which catalyzes Nα-acetylation of proteins retaining an initiator methionine followed by either Asp or Glu) suggests that it could be the enzyme mediating α-syn acetylation in human cells. As such, preliminary attempts to assess the effect of Nα-acetylation on α-syn behavior involved knocking down members of the NatB complex in yeast cells (29). Consistent with previous reports implicating Nα-acetylation with regulation of membrane targeting of some proteins (30), NatB silencing lead to the re-localization of an α-syn-GFP fusion construct from the plasma membrane to the cytosol. However, because that study did not assess the effect of NatB knockdown on α-syn Nα-acetylation, it remains unclear whether this effect was due to the specific loss of α-syn Nα-acetylation or due to the alteration of some other NatB-controlled pathway.
Despite the identification of quantitative Nα-acetylation of α-syn both as soluble protein and entrapped within Lewy bodies from human PD brains (11), all biophysical studies investigating α-syn structural, biochemical, and aggregation properties in vitro have been relying on recombinant α-syn from E. coli, which is the only system providing high protein yields required for such studies. However, even though prokaryotes do acetylate some of their proteins, this occurs via a distinct pathway that is not homologous to that mediated by yeast and human N-acetyltransferases (31); hence, recombinant α-syn purified from E. coli is not Nα-acetylated. We therefore sought to determine the effect of α-syn Nα-acetylation on its conformation, membrane interactions, aggregation properties, and finally subcellular localization in mammalian cells.
When we began this study, the lack of knowledge of a specific N-terminal acetyltransferase acting on α-syn, and the lack of chemical methods to specifically acetylate the N terminus of recombinant proteins, led us to rely on a protein semisynthesis approach based on expressed protein ligation (EPL) (1), which has been recently applied by our group to prepare N-terminal monoubiquitylated and C-terminal phosphorylated forms of α-syn (32, 33). Later, we applied a recombinant expression strategy based on co-expression of α-syn and NatB, which generated quantitatively acetylated α-syn based on mass spectroscopic analysis. We then utilized these approaches to generate Nα-acetylated α-syn in milligram quantities, and we determined the effect of Nα-acetylation on the structure, oligomerization, and aggregation of α-syn in vitro and in intact Escherichia coli using a battery of biophysical methods, including NMR, static light scattering, and sedimentation and thioflavin T fluorescence assays.
Together, our studies revealed that WT and Nα-acetylated α-syn exist predominantly as disordered proteins. NMR data in intact cells also ruled out the presence of stable or highly populated α-syn oligomers and confirmed the intrinsically disordered nature of the protein in E. coli regardless of its Nα-acetylation state or purification method. We also show that Nα-acetylation does not significantly influence α-syn fibrillization, membrane binding, or its subcellular localization in mammalian cells. These results and their implications for α-syn structure and function in health and disease are presented and discussed.
EXPERIMENTAL PROCEDURES
Chemicals
N-Methylpyrrolidone (extra-pure) was bought from Acros Organics (Geel, Belgium). The following were purchased from Sigma: ammonium iodide; benzyl mercaptan; t-butyl-mercaptan; 4-mercaptophenylacetic acid; 1,2-ethanedithiol; methyl 2-mercaptopropionate; 4-nitrophenylchloroformate; O-methylhydroxylamine hydrochloride; thioanisole; dimethyl sulfide; dichloromethane; N,N-diisopropylethylamine, N,N-dimethylformamide, tris(2-carboxyethyl)phosphine (TCEP); trifluoroacetic acid (TFA); thioflavin T (ThT), and triisopropylsilane. Guanidinium hydrochloride (GdnHCl) was from MP Biomedicals (Solon, OH). Fmoc-protected amino acids, di-Fmoc-3,4-diaminobenzoic acid, Fmoc-Rink-amide methylbenzhydrylamine resins (initial substitution 0.4 mmol/g), and O-benzotriazole-N,N,N′,N′-tetramethyluronium-hexafluorophosphate were from Anaspec (Fremont, CA). N-Acetylmethionine was purchased from Bachem (Bubendorf, Switzerland). Piperidine was bought from Merck. The water-soluble radical initiator 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) was purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan). Acetonitrile (HPLC gradient-grade) was obtained from VWR (West Chester, PA). Phenol crystals were from Carlo Erba reagents (Val de Reuil, France). Buffer solutions were from AppliChem (Darmstadt, Germany).
Semisynthesis of Nα-Acetylated α-syn
Peptide Synthesis, Thioester Formation, and Purification
The Ac-α-syn(1–10)-SR (R = CH2CH2SO3H) peptide was synthesized using Fmoc chemistry on a CS336X automated peptide synthesizer (CSBio, Menlo Park, CA), employing the in situ neutralization protocol. The peptide was assembled on a di-Fmoc-3,4-diaminobenzoic acid-loaded Rink-amide methylbenzhydrylamine resin to obtain a peptide thioester according to the method described by Blanco-Canosa and Dawson (34). After a first manual deprotection with 50% piperidine in N-methylpyrrolidone (two times for 10 min), the first residue was double-coupled manually (45-min coupling time each) before transferring the resin to the automated synthesizer. During automated synthesis, Fmoc groups were deprotected with 20% piperidine/N-methylpyrrolidone for 3 min and then repeated for 7 min. Amino acid coupling cycles were carried out with 5 eq of Fmoc-amino acids, 5 eq of O-benzotriazole-N,N,N′,N′-tetramethyluronium-hexafluorophosphate/HOBt, and 10 eq of N,N-diisopropylethylamine for 30 min. The N-terminal residue was coupled as N-acetylmethionine (Bachem), because on-resin acetylation with acetic anhydride would have also blocked the second amine functionality of the C-terminal diaminobenzoic acid linker, which must remain free to perform on-resin C-terminal activation (34). On-resin activation to obtain a C-terminal N-acyl-benzimidazolinone (Nbz) group was done by treatment with 5 eq of 4-nitrophenylchloroformate (10 mg/ml, 40 min, RT) in dry CH2Cl2, followed by 0.5 m N,N-diisopropylethylamine in N,N-dimethylformamide (15 min, RT). This procedure was repeated three times (three times with 15 ml of N,N-dimethylformamide and three times with 15 ml of dry CH2Cl2 wash cycles in between) to ensure quantitative conversion to the N-acyl-benzimidazolinone group. The peptide was finally cleaved from the resin and deprotected using 10 ml of Reagent H (35) to prevent methionine oxidation. Deprotection was done for 3–4 h at RT, followed by three consecutive precipitations in 10-fold excess of cold diethyl ether. Peptide precipitates were then lyophilized. Crude peptides were first treated with 0.45 m sodium mercaptoethanesulfonate at 37 °C for 30 min, before purification by reversed-phase HPLC (Waters 600 system) on a preparative C18 column (Cosmosil Protein-R, 20 × 250 mm, 5-μm particle size, and 300-Å pore size, at 10 ml/min using a linear gradient of solvent B in solvent A, from 25% B to 45% B over 40 min, where solvent A was 0.1% TFA in water and solvent B was 0.1% TFA in acetonitrile). Fractions were pooled according to purity as assessed by ESI-MS analysis (LTQ ion trap, Thermo Scientific), and final purity assessment was made by re-injection of pooled fractions on an analytical C18Cosmosil Protein-R column (4.6 × 250 mm, 5-μm particle size, and 300-Å pore size) connected to a Waters Alliance 27. Pure pooled fractions were evaporated to remove organic solvents, lyophilized, and stored at −20 °C under inert atmosphere.
Expression and Purification of the Recombinant Protein Fragment
Expression of recombinant α-syn Δ2–10 A11C (referred to as α-syn(A11C-140)) as performed as described previously (32). Briefly, BL21(DE3) cells transformed with a pT7-7 plasmid carrying the insert of interest were grown at 37 °C in LB medium induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at A600 = 0.6–0.7 and then incubated for 4 h. Cells were then harvested and lysed by sonication. The protein was then purified by anion-exchange followed by gel-filtration chromatography; dialyzed against water, and lyophilized. Because the N-terminal cysteine reacts with various endogenous aldehydes and ketones in E. coli, resulting in cyclic adducts that would not react during native chemical ligation (NCL), the lyophilized material was treated with 100 mm O-methylhydroxylamine and 30 eq of TCEP in 6 m GdnHCl at pH 4.0 for 1–2 h at 37 °C. The protein was then separated from the reagents and by-products by reversed-phase HPLC, lyophilized, and stored at −20 °C under inert atmosphere until use.
Native Chemical Ligation, Desulfurization, and Purification of the Semisynthetic Protein
For large scale ligations, α-syn(A11C-140) was dissolved in 1 ml of 6 m GdnHCl, 0.2 m sodium phosphate to a final concentration of 0.83 mm. 30 eq of TCEP were added to prevent disulfide formation. 1.2 eq of Ac-α-syn(1–10)SR were added, followed by 10 eq of 4-mercaptophenylacetic acid (with respect to the peptide fragment) as the NCL catalyst (36). The pH was then adjusted to 7.0 with aqueous NaOH, and the reaction was incubated at 37 °C under inert atmosphere for up to 4 h. Because 4-mercaptophenylacetic acid has been found to interfere with the radical-based desulfurization procedure, it was first removed by desalting the NCL reaction mixture using HiTrap desalting 5 ml (GE Healthcare) equilibrated with 200 mm of the volatile buffer ammonium bicarbonate. After lyophilization, the powder was redissolved in 800 μl of 6 m GdnHCl, 0.2 m sodium phosphate, pH 7.0. To this solution, 800 μl of 1.0 m TCEP in 6 m GdnHCl, pH 7.0, t-butyl mercaptan was added to a final concentration of 840 mm, and VA-044 to a final concentration of 5.4 mm. The reaction was complete within 3 h of incubation at 37 °C under argon. The ligation product was purified by reversed-phase HPLC on a semipreparative InertSil C8 column (7.6 × 250 mm; 5-μm particle size and 300-Å pore size, at 5 ml/min using a gradient from 25% B to 50% B over 30 min), evaporated, and lyophilized.
Recombinant Co-expression of Hyman α-syn and Yeast NatB in E. coli
To express recombinant Nα-acetylated human α-syn, an overnight pre-culture of BL21(DE3) cells carrying simultaneous plasmids encoding WT human α-syn and yeast NatB was used to inoculate 2 liters of LB medium containing 100 μg/ml ampicillin and 25 μg/ml chloramphenicol. Cells were grown at 37 °C with shaking until their A600 reached 0.6–0.8; the protein expression was then induced by adding isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 1 mm. At this point, the temperature of the incubator was reduced to 20 °C, and the cells were further incubated overnight. After harvesting the cells by centrifugation at 6000 × g for 2 min at 4 °C, bacterial pellets were resuspended in 40 ml of 20 mm Tris acetate, 5 mm EDTA, 1 mm PMSF, 1× protease inhibitor mixture (Sigma), pH 8.3, and lysed by ultrasonication. Cell lysates were cleared by high speed centrifugation at 48,000 × g for 20 min at 4 °C; supernatants were filtered (0.45 μm) and applied onto a HiPrep 16/10 Q FF anion-exchange column (GE Healthcare) equilibrated with 20 mm Tris, pH 8.0, at 1 ml/min. Note that the lysates were not boiled or chemically denatured at any point. Proteins were eluted with a 600-ml-long linear gradient of increasing NaCl concentration from 0 to 1 m at 2.5 ml/min on an Äkta Explorer 100 FPLC system (GE Healthcare). Collected fractions were analyzed by SDS-PAGE and Coomassie Blue staining. α-syn-positive fractions were concentrated using 10-kDa MWCO Amicon concentrators (Millipore), filtered (0.22 μm), and injected (0.5 ml per injection) into a HiLoad 16/60 Superdex 200 column (GE Healthcare) at 0.5 ml/min. Fractions collected around the elution volume of α-syn (∼90 ml) were analyzed by SDS-PAGE/Coomassie staining and further purified by hydrophobic interaction chromatography. Ammonium sulfate powder (Acros) was slowly added to a final concentration of 1 m to the pooled gel-filtration fractions kept on ice, which were then applied onto two HiTrap phenyl HP columns (GE Healthcare) connected in series and equilibrated with 50 mm sodium phosphate, pH 7.5, 1 m (NH4)2SO4 at 0.5 ml/min. Proteins were eluted with a 300-ml-long linear gradient of decreasing [(NH4)2SO4] from 1 to 0 m. Pure α-syn fractions were dialyzed twice against 20 mm sodium phosphate, pH 7.4, before analysis. Purification of recombinant Nα-acetylated α-syn in the presence of 10% glycerol and 0.1% n-octyl-β-d-glucopyranoside (BOG, purchased from Sigma) described by Trexler and Rhoades (37) was performed as mentioned above with the only differences being the buffers used during the first two chromatographic steps of the purification. The lysis buffer was replaced by 100 mm HEPES, 20 mm NaCl, 10% glycerol, 0.1% BOG, pH 7.4 (subsequently referred to as BOG buffer A), to which were added 1 mm PMSF and 1× protease inhibitor mixture (Sigma). Anion-exchange binding buffer was BOG buffer A. Anion-exchange elution buffer was BOG buffer A + 1 m NaCl. Gel-filtration chromatography buffer was BOG buffer A. Note that it is critical that BOG-containing buffers must be made fresh and stored at 4 °C.
Large Unilamellar Vesicle (LUV) Preparation
LUVs were prepared as described before (13), except that the buffer was changed to 20 mm sodium phosphate, pH 7.4, with the pH adjusted with phosphoric acid and not HCl to keep the buffer as UV-transparent as possible to allow accurate CD measurements in the low wavelength range. The appropriate volumes of LUV stock solution (10 mg/ml), buffer, and protein stock solutions were mixed to obtain protein/:lipid weight ratios as indicated in the corresponding figure.
Circular Dichroism
Lyophilized proteins were reconstituted in 20 mm sodium phosphate, pH 7.4, to a concentration of 10 μm. CD spectra were acquired in a Jasco J-815 spectrometer using a 1-mm-wide quartz cell. Data acquisition was carried out using the following parameters: wavelength range, 250 to 195 nm; data pitch, 0.2 nm; bandwidth, 1 nm; scanning speed, 50 nm/min (continuous mode); digital integration time, 2 s. Each spectrum was the average of 10 subspectra that were further smoothed using the Savitzky-Golay filter (convolution width, 25 points).
Gel-filtration Chromatography/Static Light Scattering
Analytical gel-filtration chromatography was done on a 1200 series isocratic HPLC pump running at 0.4 ml/min connected to a GE Healthcare Superdex 200 10/300 GL equilibrated with 50 mm Tris, pH 7.5, 150 mm NaCl, 50 ppm NaN3. Protein detection was done by monitoring absorbance at 275 nm (Agilent 1200 VWD). Absolute molecular weights were determined by stating light scattering using a Dawn Heleos II (Wyatt Corp.) multiangle light-scattering detector connected in series with the UV detector. The protein concentration data used to obtain molecular weights from light-scattering data (using the Zimm model) were derived from either refractive index measurements (Wyatt Optilab rEX, connected downstream of the LS detector) or UV measurements (Agilent 1200 VWD, upstream of the LS detector). Inter-detector delays and band broadening effects were corrected as appropriate in the Astra 5.3 analysis software (Wyatt Corp.).
Amyloid Formation and Fibrillization Assays
WT and Nα-acetylated α-syn were reconstituted in 50 mm Tris, pH 7.5, 150 mm NaCl and filtered through 0.22-μm nylon membranes. The final protein concentration was adjusted to 15 μm in a final volume of 450 μl. Fibrillization was done by incubating the protein at 37 °C with agitation on an orbital shaker (1200 rpm) for up to 5 days. Amyloid structure formation was monitored using the Thioflavin T binding assay as described before (38). Fibril morphology was assessed by negative-staining transmission electron microscopy. Briefly, for each time point, 5–10 μl of protein solution were spotted on a Formvar-coated carbon grid (Electron Microscopy Sciences). The grid was then dried and washed twice with 15 μl of water each time, dried again, and stained with 2% uranyl acetate. Images were acquired on a Philips CM-10 transmission electron microscope operated at 100 kV; micrographs were recorded with a slow scan CCD camera (Gatan Model 679). Fibrillization was also quantified by measurement of the decrease in soluble protein content over time. This was done by high speed centrifugation (20,000 × g for 10 min at 4 °C) of 17-μl aliquots. Fibrils were pelleted, and the supernatant represents the soluble protein fraction. 7 μl of the supernatant were then mixed with 2× Laemmli sample buffer before electrophoresis on homogeneous SDS-15% polyacrylamide gels and Coomassie Brilliant Blue R-450 staining.
Native and SDS-Gel Electrophoresis
Proteins in their native conformations were separated on custom-made native polyacrylamide gels using Bio-Rad gel-casting systems, with a separation section at 7.5% polyacrylamide buffered with 380 mm Tris buffer, pH 8.8, and a stacking section at 3% polyacrylamide buffered with 125 mm Tris, pH 6.8. Before application in the gel wells, samples were diluted in native sample buffer (310 mm Tris, pH 6.8, 50% glycerol, 0.05% bromphenol blue). Electrophoresis was carried out at 25 mA in constant current mode for ∼3 h on a Bio-Rad PowerPac 1000 supply. Gels were then stained with Coomassie Brilliant Blue or transferred on a nitrocellulose membrane (Whatman) using a semi-dry electrotransfer system (Bio-Rad) for Western blotting. For SDS-PAGE analysis, samples were diluted in loading buffer and separated on homogeneous 15% SDS-polyacrylamide gels (1.5 mm thickness). The electrophoresis and Western blot were conducted as described previously (39). Briefly, proteins were transferred to nitrocellulose membranes using the semidry blotting system (Bio-Rad) for 1 h. Membranes were then probed overnight with the primary antibody of interest after 30 min of blocking in Odyssey blocking buffer (Li-Cor Biosciences GmbH) diluted 1:3 in phosphate-buffered saline (Sigma). After four washes with PBST (phosphate-buffered saline, 0.01% (v/v) Tween 20 (Sigma)), membranes were incubated for 1 h with secondary antibodies (goat or rabbit Alexa Fluor 680 IgG) protected from light at room temperature. Immunoblots were finally washed four times with PBST and scanned using a Li-COR scanner at a wavelength of 700 nm.
NMR Sample Preparation
15N-Enriched α-syn for NMR studies was obtained by the media swap method (40). E. coli BL21(DE3) cells transfected with plasmid vectors encoding α-syn, NatB (generously provided by Dr. Daniel Mulvihill through Dr. Elizabeth Rhoades), or both were grown in 1 liter of LB medium at 37 °C until an A600 of 0.6, pelleted, washed in M9 minimal medium, pelleted again, and resuspended in 250 ml of M9 minimal medium supplemented with dextrose and 15N-labeled ammonium chloride. Cells were allowed to recover at 37 °C for 1 h before induction of protein expression by isopropyl 1-thio-β-d-galactopyranoside and harvested after 3 h. The resulting cell pellet (∼2 ml) was used immediately for NMR studies.
The pellet was resuspended by the addition of 500 μl of NMR buffer (100 mm NaCl, 10 mm NaPO4−, 10% D2O, pH 7.4). Approximately 500 μl of the resulting cell slurry, supplemented with 50 μl of D2O, was used as the in-cell NMR sample. The remaining slurry was diluted in half with NMR buffer and, after the addition of 1 mm EDTA and 1 mm PMSF (final concentrations), was subjected to sonication on ice with a micro-tip ultrasonication probe at low power for 6 min. The resulting lysate was centrifuged at 15,000 rpm for 20 min, and 550 μl of the supernatant was used as the lysate NMR sample.
Purified α-syn was obtained from E. coli BL21(DE3) cells transfected with plasmids encoding wild-type or E20C mutant α-syn, with and without NatB, grown in isotopically supplemented minimal media according to previous protocols (41). For paramagnetic relaxation enhancement (PRE) measurements, both unacetylated and Nα-acetylated 15N-labeled E20C α-syn were dissolved in NMR buffer and incubated with a 10-fold excess of the paramagnetic spin label S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate for 15 min. Unbound spin label was removed by buffer exchange, and the sample was divided in two. Dithiothreitol (DTT) was added (final concentration 2 mm) to one sample to reduce the spin label off the cysteine residue.
NMR Experiments
Spectra were collected at 10 °C on a Varian INOVA 600-MHz or a Bruker 800-MHz AVANCE spectrometer equipped with cryogenic probes. Because of the high concentration of α-syn in the cells, fairly short (∼20 min) HSQC experiments gave rise to high quality spectra. The in-cell sample proved refractory to shimming, so shimming was performed on the lysate sample, which was matched in volume and sample height to the in-cell sample.
PRE was measured as the intensity ratio of corresponding cross-peaks in matched HSQC spectra of diamagnetic and paramagnetic samples (single point method). Resonance assignments from unacetylated α-syn (41) were transferred to Nα-acetylated α-syn using an HNCA. Secondary Cα chemical shifts were calculated as reported previously (41) for a direct comparison. Changes in amide proton and nitrogen chemical shifts were calculated as shown in Equation 1,
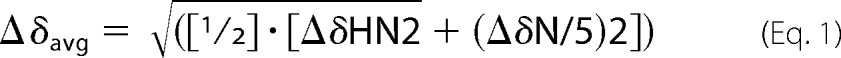 |
All spectra were processed with NMRpipe (42) and analyzed with NMRView (43).
Cell Culture and DNA Transfection
HeLa and HEK cells were grown at 37 °C, 5% CO2 in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and 1% penicillin/streptomycin. Transient transfection of HEK293T cells was performed using the standard calcium phosphate (CaPO4) transfection protocol. The amounts of transfected DNA were 0.5 μg when cells were plated on 24-well plate dishes for immunocytochemistry and 10 μg when cells were plated on 100-mm dishes for biochemical analysis. For studies involving NatB silencing, HEK cells were transfected with plasmids encoding scrambled shRNA or NatB shRNA4, and 24 h later, cells were either transfected with a plasmid encoding WT α-syn or not. 48 h later, cells were either harvested for biochemical analysis or fixed with (4% paraformaldehyde, 3% sucrose in PBS) for 20 min at RT and then washed with PBS for immunostaining. For D2P synuclein experiments, cells were analyzed 48 h post-transfection.
Proteofection of HeLa Cells
Approximately 500,000 HeLa cells plated in 6-well plates were proteofected at 50% confluency using the Chariot® reagent as per the manufacturer's instructions. Briefly, 4 μg of WT or Nα-acetylated (N-Ac) α-syn in PBS were incubated with 6 μl of Chariot® (diluted 1:10) in 100 μl of double distilled water for 30 min at room temperature (RT). Then the proteofection mixtures were added to the cells with 400 μl of serum-free media and incubated at 37 °C for 4 h. Cells were then trypsinized for 1 min at 37 °C and centrifuged for 3 min at 200 × g. The cell pellet was then resuspended in 1 ml of PBS to wash away un-internalized extracellular proteins. Cells were pelleted again by centrifugation at 200 × g (3 min at RT), resuspended in growth media, and then plated on poly-l-lysine-coated coverslips in 24-well plates. 24 h later, adherent cells were washed once with PBS, fixed with 4% paraformaldehyde, 3% sucrose in PBS for 20 min at RT, washed twice with PBS, and then stained with wheat germ agglutinin (WGA), Alexa 555-conjugated (5 μg/ml in PBS) for 10 min at RT. Cells were subsequently washed with PBS twice and then immunostained as detailed below.
Subcellular Localization Studies
After fixation with 4% paraformaldehyde, 3% sucrose in PBS for 20 min at RT, and then washing with PBS, cells were blocked with 0.1% Triton X-100, 3% bovine serum albumin (BSA) in PBS for 1 h at RT. Afterward, coverslips were incubated overnight at 4 °C with the primary mouse anti α-syn antibody (Syn-1, BD Transduction Laboratories) at a dilution of 1:1000 when analyzing overexpressed or proteofected synuclein and 1:500 when analyzing endogenous synuclein. Then cells were washed three times with PBS and incubated for 1 h at RT with donkey anti-mouse Alexa Fluor® 488 (1:1000) when analyzing proteofected synuclein or with donkey anti-mouse Alexa Fluor® 568 (1:1000) when analyzing endogenous or overexpressed synuclein. After three washes with PBS, the cells were stained with Hoechst 33342 (Invitrogen) for 15 min at RT, washed again twice with PBS, and then mounted on glass slides using DABCO mounting medium. Slides were then imaged using an LSM 700 (Zeiss) inverted confocal microscope. For the imaging of HEK cells transfected with NatB-shRNA (1–4), living cells in cell culture plates were imaged at ×10 magnification using the Leica DMI 4000 wide field fluorescent microscope.
Immunoprecipitation, In-gel Digestion, and Mass Spectrometric Analyses
HEK293T cells transfected with plasmids encoding WT or D2P α-syn, or with WT α-syn and NatB shRNA4, were harvested and lysed, and 5 μg of Syn-1 antibody (BD Transduction Laboratories) were added to the lysates. Following overnight incubation at 4 °C, 35 μl of protein G Fast Flow beads (GE Healthcare) were added to the lysates and incubated for 2 h at 4 °C. After washing the beads three times with 0.1% Triton X-100 in PBS, α-syn was eluted with 30 μl of 100 mm glycine, pH 2.5. Eluates were loaded on an SDS-15% polyacrylamide gel, and the bands corresponding to α-syn were excised after Coomassie Blue staining using Simply Blue Safe Stain (Invitrogen). Excised bands were destained with 50% ethanol in 100 mm ammonium bicarbonate, pH 8.5, followed by drying in an evacuated centrifuge, and the proteins were digested in-gel by covering gel pieces with 12 ng/μl proteomics-grade glutamyl C-terminal endopeptidase (Glu-C) (Sigma) and incubating overnight at 37 °C. Digested peptides were concentrated to dryness, resuspended in acetonitrile, and dried again. After repeating this step once more, dried peptides were resuspended in 20 μl of 20% formic acid, 2% acetonitrile, desalted on C18 OMIX tips (Agilent Technologies), and analyzed by MALDI-TOF-TOF tandem mass spectrometry operating in reflector positive mode on a 4800 series MALDI instrument (AB Sciex).
Murine Synaptosomal Binding Assays
The binding assays were conducted as described previously (44). Synaptosomes that were generated from the brain of a single knock-out human α-synuclein mouse were partially fractionated as described below. All steps were done on ice or at 4 °C. Briefly, whole brains, sans cerebella, were extracted from mice and homogenized with 10 strokes in a Dounce homogenizer rotating at 500 rpm. The homogenization buffer contained 320 mm sucrose, 1 mm EGTA, and 5 mm HEPES, pH 7.4. Homogenates were centrifuged at 1050 × g for 10 min, and the supernatants were centrifuged at 13,300 × g for 15 min. The pellets were gently resuspended in homogenization buffer and loaded onto a discontinuous 13.9 to 5% Ficoll gradient. Gradients were centrifuged at 86,808 × g on a Beckman SW41 swinging rotor, and intact synaptosomes were extracted from the 13 to 9% interface. The synaptosomes were gently resuspended in extracellular buffer (130 mm NaCl, 5 mm KCl, 20 mm Na HEPES, 5 mm NaHCO3, 1.2 mm Na2HPO4·7H2O, 1 mm MgCl2, 10 mm glucose, and 2.5 mm CaCl2, pH 7.4). A centrifugation at 13,300 relative centrifugal force for 15 min was performed to remove residual Ficoll. The synaptosomes were then hypotonically lysed with vigorous agitation in swelling buffer (10 mm HEPES and 18 mm potassium acetate, pH 7.2). After centrifugation, the resultant pellet was resuspended in intracellular replacement buffer (25 mm HEPES, 145 mm potassium acetate, and 2.5 mm MgCl2, pH 7.2) and divided into 10 equal aliquots of 100 μl each. Each aliquot was resuspended either in 1.5 mg/ml dialyzed cytosol that was derived from brains of α-synuclein knock-out mice, as described previously (45), or a corresponding volume of intracellular replacement buffer; and 0.5 and 1 μg of A30P, WT α-syn produced from E. coli, Nα-acetylated α-syn, or a corresponding volume of intracellular replacement buffer were added prior to 10 min of incubation in a 37 °C water bath. Each aliquot was then centrifuged for 5 min at 20,000 relative centrifugal force and at 4 °C. The resultant pellet was washed with intracellular replacement buffer and was resuspended in Tris lysis buffer (100 mm NaCl, 50 mm Tris, and 1 mm EDTA, pH 7.2) with 1% Triton X-100. This resuspension (bound) and the supernatant (unbound) were run on Western blot analyses. Protein samples were boiled briefly in loading buffer (10% glycerol, 0.05 m Tris, 2% SDS, bromophenol blue, and 2.5% mercaptoethanol) and separated by electrophoresis using 12% Tris-glycine polyacrylamide gels. Proteins were transferred to nitrocellulose and probed with Syn-1 (1:1000, BD Transduction Laboratories) followed by HRP-conjugated secondary antibody. The bands were quantified with ImageJ software. Statistical comparisons were calculated with GraphPad InStat software using Student's t test for comparisons between two groups or analysis of variance (Bonferroni test) for multiple comparisons.
RESULTS
Preparation of Nα-Acetylated α-syn
To produce α-syn acetylated specifically at the N terminus, we first utilized a semisynthetic methodology (1) that we recently developed to allow for site-specific introduction of post-translational modifications in the N-terminus of α-syn based on expressed protein ligation (EPL) (32). The present EPL-based method relies on the ligation of a synthetic Nα-acetylated α-syn fragment (residues 1–10) with a recombinant protein consisting of α-syn residues 11–140. Fig. 1A summarizes the strategy we utilized to prepare semisynthetic Nα-acetylated α-syn. The detailed semisynthesis procedure was described under “Experimental Procedures.” The progress of the native chemical ligation reaction was monitored by SDS-PAGE and MALDI-TOF MS (supplemental Fig. S1). The reaction was >80% complete after 2 h (supplemental Fig. S1A), and formation of the product was confirmed by the appearance of the expected ligation product (supplemental Fig. S1B). The protein was then desulfurized to obtain the native alanine residue at position 11 (supplemental Fig. S1C). Finally, the desulfurized ligation product Ac-α-syn(1–140) was separated from the excess peptide fragment and unreacted α-syn(11–140) by reversed-phase HPLC. The semisynthetic Nα-acetylated α-syn was confirmed to be pure by SDS-PAGE, MALDI-TOF MS (Fig. 1, C and D), and analytical reversed-phase ultra-HPLC (supplemental Fig. S1D). From 6.2 mg of α-syn(A11C-140), the combined synthesis and purification yield was 2.9 mg.
FIGURE 1.

Production of Nα-acetylated α-syn. A, semisynthesis and recombinant expression strategy schemes. For the EPL approach, the sequence of α-syn is split into the fragments Ac-α-syn(1–10)SR, which is chemically synthesized as an N-terminally acetylated peptide thioester, although the fragment α-syn(A11C-140) is produced as a recombinant protein in E. coli with the temporary mutation A11C to mediate the native chemical ligation with the Ac-α-syn(1–10)SR peptide. The N-terminal cysteine must be free of disulfide and thiazolidine ring adducts before the NCL reaction. Following the ligation, the full-length Nα-acetylated protein is desulfurized to regenerate the native alanine residue at position 11. For the recombinant expression approach, bacterial cells carrying plasmids encoding both full-length WT α-syn and yeast NatB are used. Protein expression is also controlled by the T7 promoter/polymerase system. B and C show MALDI-TOF MS analyses of purified recombinant (observed, 14,506 Da; theoretical, 14,503 Da) and semisynthetic (observed, 14,504 Da) Nα-acetylated α-syn, respectively. Stars indicate sinapinic acid matrix adducts. D, SDS-PAGE/Coomassie staining of Nα-acetylated semisynthetic (s.-s) and recombinant (rec) α-syn. 2 μg of proteins were loaded on homogeneous SDS-15% polyacrylamide gels. E, clear native PAGE analysis of WT and semisynthetic N-Ac α-syn. 2 μg of each protein were migrated on a homogeneous 7.5% native polyacrylamide gel and then stained with Coomassie Blue. The migration of the proteins was compared with a set of globular standards (NativeMark Unstained protein standard, Invitrogen).
We also used a recombinant protein production strategy to obtain Nα-acetylated α-syn. We took advantage of a previous study by Johnson et al. (45) who created a vector for recombinant expression of the yeast N-acetyltransferase NatB (which has similar target protein sequence determinants as its human homolog) in E. coli and showed that co-expressing several proteins with NatB successfully resulted in their Nα-acetylation. Accordingly, we transformed BL21(DE3) E. coli cells sequentially with two vectors, first a pT7-7 plasmid encoding at human α-syn and then the pNatB vector (a kind gift from Prof. Daniel Mulvihill), which encodes for both NatB subunits (Fig. 1A). Recombinant Nα-acetylated α-syn was expressed and purified as described under “Experimental Procedures” (see also supplemental Fig. S2 for a detailed purification flow chart), and quantitative N-terminal acetylation of α-syn was determined by MALDI-TOF mass spectrometry (Fig. 1B) and analytical reversed-phase ultra-HPLC (supplemental Fig. S1E). This strategy to prepare Nα-acetylated α-syn was also described in a recent report (37).
Biophysical Characterization of Nα-Acetylated α-syn
We first examined whether Nα-acetylation has any impact on the oligomeric state of the protein and its conformation. To this end, we performed analytical gel-filtration chromatography coupled to on-line determination of absolute molecular weight by static light scattering. WT and Nα-acetylated α-syn were dissolved at 30 μm in 50 mm Tris, pH 7.5, 150 mm NaCl, 50 ppm NaN3 and injected at 0.4 ml/min in a Superdex 200 10/300 GL size-exclusion column. Both WT and Nα-acetylated proteins eluted at 14 ml (Fig. 2A), suggesting that Nα-acetylation does not have a large influence on the quaternary structure of α-syn. Furthermore, the molecular weight of Nα-acetylated α-syn as estimated by light scattering (13055 ± 1383 Da) was consistent with a monomeric state and similar to that of the WT protein (13,814 ± 576 Da). Note that there is no difference between Nα-acetylated α-syn produced by semisynthesis or by recombinant production under nondenaturing purification (Fig. 2A). We also performed circular dichroism spectroscopy and showed that Nα-acetylation does not alter the predominantly disordered structure of α-syn (Fig. 2B). Again, both recombinant and semisynthetic Nα-acetylated proteins behaved very similarly, thus validating the use of either preparation method to study α-syn Nα-acetylation. Native PAGE with Coomassie Brilliant Blue staining also confirmed the similarity between recombinant WT and Nα-acetylated α-syn proteins. Both proteins migrated at an apparent molecular mass above 66 kDa when compared with the globular protein standards (Fig. 1E). This behavior is consistent with an unfolded character giving rise to a hydrodynamic radius similar to that of a larger globular protein, and therefore to a lower electrophoretic mobility on native PAGE, as determined previously for α-syn (46) and confirmed recently by our group (47).
FIGURE 2.

Biophysical characterization of Nα-acetylated α-syn. A, gel-filtration/light scattering analysis. Purified lyophilized WT and N-Ac α-syn were diluted to 30 μm in gel-filtration buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 0.05% w/v NaN3) and filtered (0.22 μm). Samples (100 μl) were run at 0.4 ml/min on a Superdex 200 10/300 GL column connected with UV multiangle light scattering and differential refractive index detectors. Molar masses were calculated based on concentrations determined on line by differential refractometry (dn/dc = 0.185 ml/g). The left ordinate axis shows the elution pattern of WT (continuous line), semisynthetic (s.-s., dashed line), and recombinant (rec., dotted line) N-Ac α-syn. The right ordinate axis shows calculated molecular weights for WT (open circles), semisynthetic (s.-s., closed circles), and recombinant (rec., squares) N-Ac α-syn. B, circular dichroism spectra of WT (continuous line), semisynthetic (s.-s., dashed line), and recombinant (rec., dotted line) N-Ac α-syn. Proteins were dissolved to 10 μm in 20 mm sodium phosphate, pH 7.4 (without lipids), and analyzed in a 1-mm quartz cell.
We then used 1H/15N HSQC NMR spectra as a more sensitive technique to determine whether α-syn Nα-acetylation could lead to conformational changes or ordering within the protein. To exclude potential effects of different purification protocols on the observed conformation of N-terminally acetylated α-syn, we collected proton-nitrogen correlation (1H/15N HSQC) spectra for α-syn overexpressed in intact E. coli cells (in-cell NMR (48–50), as well as corresponding spectra for cell lysates obtained by subjecting cells from the same pellet to sonication (note that a similar approach has been used to characterize unacetylated α-syn in intact cells).4 Fig. 3A shows an overlay of spectra obtained for lysed cells transfected with only α-syn (−NatB) or both α-syn and NatB (+NatB), as well as for purified recombinant unacetylated α-syn. Importantly, signals that are observed in cells transfected with NatB alone (data not shown) are boxed in Fig. 3A and do not originate from or report on α-syn. It is immediately evident that the spectrum of α-syn expressed in the presence of NatB is extremely similar to that expressed in the absence of NatB, indicating that there are no large scale conformational changes in the protein and that it remains highly disordered. Nevertheless, there are several resonances in the +NatB spectrum that show unambiguous changes in chemical shifts. Importantly, for these shifted peaks no remaining intensity was observed, down to the level of noise, at the original position observed in the −NatB spectrum. This implies that any modification of α-syn in the +NatB cells was quantitative and complete. Any remaining unmodified protein would give rise to signals at positions observed in the −NatB cells.
FIGURE 3.

NMR analyses of Nα-acetylated α-syn. A, proton-nitrogen correlation (HSQC) spectra for lysates from cells transfected with α-syn alone (black), α-syn and NatB (red), and purified recombinant α-syn (blue). Peaks present in spectra of lysates from cells transfected with NatB alone are indicated by boxes and do not originate from or report on synuclein. Resonances that change position upon co-transfection with NatB are indicated by arrows pointing from their position in the unmodified protein spectrum toward their position in the modified protein spectrum. Resonances for residues Met-1 and Asp-2, which are not typically observed for the unacetylated protein, are indicated. B, Cα secondary chemical shifts for Nα-acetylated versus unacetylated α-syn. Positive values indicate a preference for helical structure, and an increase in helicity is readily apparent for the N-terminal ∼10 residues of the acetylated protein. Shifts for the unacetylated protein are as originally reported (41). C, PRE for Nα-acetylated versus unacetylated α-syn spin-labeled at position 20. Long range transient contacts indicated by decreased resonance intensity in the C-terminal region of the protein are largely unaffected by acetylation. D, proton-nitrogen correlation (HSQC) spectra for intact cells transfected with α-syn alone (black), α-syn and NatB (red), and NatB alone (green). Cells were pelleted after growth and gently resuspended in buffer prior to loading into NMR tubes for data collection. Aliquots from the same resuspension were lysed by sonication to obtain the data in A. aS, α-syn.
NMR Demonstrates That N-terminal Acetylation Leads to Local Conformational Changes at the N Terminus of α-syn
To identify the new location of resonances shifted in the +NatB spectrum, we purified α-syn from +NatB cells and assigned its backbone NMR resonances using triple resonance spectroscopy (both the NMR data and mass spectrometry data confirm that protein purified from +NatB cells is indeed Nα-acetylated). A comparison of the NH shifts for the acetylated and unacetylated protein shows marked differences for residues 3–9 (Fig. 3A and data not shown). In addition, two new peaks in the +NatB spectrum were identified as belonging to Met-1 and Asp-2, which are not generally observed in the spectra of free recombinant unacetylated α-syn (although Asp-2 can be seen at lower pH). Although localized, the observed NH chemical shift changes extend further in sequence than is typically seen upon local covalent modifications of disordered proteins, for example by phosphorylation or mutagenesis, suggesting a possible structural change in the N-terminal region of the protein. To evaluate the structural consequences of α-syn acetylation, we analyzed both Cα chemical shifts, which are sensitive to secondary structure, and PRE data, which have been used to detect transient long range structure in unacetylated α-syn (51–53). The deviation of Cα chemical shifts from those expected for a random coil ensemble (Fig. 3B) indicates that there is a marked increase in helicity in the first ∼10 residues of α-syn upon N-terminal acetylation, providing an explanation for both the large amplitude of the observed NH chemical shift changes and of the range over which those changes extend. During the preparation of this manuscript, a report appeared analyzing the structural properties of purified Nα-acetylated α-syn by NMR (54). The results are entirely consistent with our own but include a further assertion that small but significant secondary structural changes are engendered further from the N terminus, including at positions corresponding to the three Parkinson-associated α-syn mutations, A30P, E46K, and A53T. Although small changes in these regions are apparent in our data as well, we cannot conclude whether they are significant in the absence of a more extensive analysis. In contrast to the localized secondary structural changes indicated by the chemical shift data, PRE measurements for α-syn spin-labeled at position 20 indicate that the transient long range contacts previously observed for unacetylated α-syn are not substantively altered in the Nα-acetylated protein (Fig. 3C).
In-cell NMR Demonstrates That NatB Modification Does Not Lead to Ordering or Large Scale Conformational Changes in α-syn
Fig. 3D shows an overlay of spectra obtained for intact cells transfected with only α-syn, only NatB, or both α-syn and NatB. Resonances that were observed in cells transfected with only NatB do not originate from or report on α-syn. The α-syn spectra with and without NatB were nearly identical. Interestingly, the N-terminal residues that were observed to shift positions upon NatB co-transfection in spectra of lysed cells are broadened beyond detection for both −NatB and +NatB cells. Nevertheless, the shifted positions of these resonances in lysates obtained from the same cell pellet establish that α-syn in the +NatB cells is indeed quantitatively modified. Importantly, the resonance intensities in the two spectra, normalized by the level of protein overexpression as estimated from SDS-PAGE, are comparable. The essentially identical spectra observed for unmodified and modified α-syn in intact cells have several implications for the effects of N-terminal acetylation on α-syn structure. First, the spectra demonstrate conclusively that when expressed in intact bacteria, with or without N-terminal acetylation and in the absence of any purification protocols, the majority of α-syn retains the intrinsically disordered character that was observed for the purified recombinant protein. Second, the spectra conclusively rule out any large scale conformational changes in α-syn upon N-terminal acetylation, such as the adoption of a highly helical structure. Such structural changes would necessarily lead to significant changes in chemical shifts. Third, the spectra conclusively rule out the presence of any highly populated or long lived oligomeric structure in N-terminally acetylated α-syn. Any significant fraction of such an oligomer would lead at a minimum to substantial signal reduction, if the oligomer were NMR-invisible, or to chemical shift changes if it were not. Neither of these effects was observed. The presence of a small population of any stable or dynamic oligomer(s) cannot be ruled out, but our data demonstrate that the majority of the protein in the cell does not populate such a structure.
Resonance broadening for the N-terminal residues of α-syn precludes direct observation of the effects of N-terminal acetylation on this region of the protein in intact cells. Such broadening could potentially be caused by inter-molecular interactions between α-syn and other cellular components, such as other proteins or possibly membranes. However, the interactions of α-syn with membranes in vitro do not recapitulate this localized broadening, suggesting that the responsible process is somehow different in nature. Whatever the process responsible, the fact that the same set of resonances is broadened for both unmodified and acetylated α-syn suggests that in this respect both proteins behave very similarly.
Effect of Nα-Acetylation on α-syn Aggregation
Having determined that the basic biophysical properties of N-Ac α-syn were similar to that of the WT protein, we wanted to examine whether Nα-acetylation could affect the aggregation propensity of α-syn in vitro, which is critical because aggregation of α-syn is recognized as a key event in the pathogenesis of synucleinopathies (55). Recombinant WT and N-Ac α-syn (15 or 50 μm, initial volume: 500 μl) were incubated in 1.5 ml of polypropylene-sealed screw-cap tubes at 37 °C for several days in 50 mm Tris, pH 7.5, 150 mm NaCl under agitation. Fibrillization kinetics were monitored by ThT binding assays. N-Ac and WT α-syn displayed similar ThT-binding propensities and kinetics of aggregation at 15 or 50 μm (Fig. 4, A and B). Amyloid fibril formation was further assessed by monitoring the decrease in soluble protein content by SDS-PAGE and Coomassie staining following high speed centrifugation. The loss of soluble protein was slightly slower with Nα-acetylated than with the unacetylated protein, which is more apparent at a protein concentration of 50 μm (Fig. 4, C and D). We also used transmission electron microscopy of negatively stained fibrillized samples to characterize the fibrils formed by WT and N-Ac α-syn. The acetylated protein formed fibrils indistinguishable from those obtained from the unacetylated protein (Fig. 4E), regardless of whether it was made by recombinant expression (Fig. 4F) or semisynthesis (see Fig. 5C), consistent with our observations that Nα-acetylation has no significant impact on α-syn aggregation. Similarly, no significant differences were observed when comparing the aggregation of WT and semisynthetic Nα-acetylated α-syn after 3 days of incubation at 37 °C. Interestingly, semisynthetic Nα-acetylated α-syn exhibited higher ThT signals at 3 days (Fig. 5A), suggesting that it forms fibrils faster than the WT protein. However, when we quantified fibrillization by monitoring the loss of soluble α-syn (Fig. 5B), we did not observe any differences between semisynthetic Nα-acetylated α-syn and the WT protein. These findings suggest that the differences in ThT signal at day 3 are likely due to differences in ThT binding, rather than the extent of fibrillization by both proteins (Fig. 5A).
FIGURE 4.

Fibrillization analysis of WT and N-Ac α-syn. A, thioflavin T binding assays of recombinant WT (white bars) and recombinant (rec.) Nα-acetylated (black bars) α-syn at 15 μm in 50 mm Tris, 150 mm NaCl, pH 7.5, at 37 °C and under orbital agitation at 1200 rpm. Aliquots were withdrawn at the mentioned time points and analyzed by ThT fluorescence. B, same experiment as in A was performed at a protein concentration of 50 μm. C, SDS-PAGE/Coomassie staining analysis of the soluble protein content. For gel analysis, samples were centrifuged (20,000 × g, 10 min, 4 °C) and supernatants (soluble fraction) were loaded on the gels. D, same experiment as in C was performed at a protein concentration of 50 μm. E and F, transmission electron micrographs of fibrillization samples of recombinant WT α-syn (E), recombinant N-Ac α-syn (F) stained with uranyl acetate. For this analysis, 5-μl aliquots were withdrawn after 24 h of incubation at 37 °C under orbital shaking at a protein concentration of 15 μm. Scale bars, 100 nm.
FIGURE 5.

Fibrillization analysis of semisynthetic N-Ac α-syn. A, thioflavin T binding assays. Three independent 450-μl samples of WT α-syn (white bars) and Ac α-syn (black bars) each at 15 μm were incubated at 37 °C on an orbital shaker (1200 rpm). Aliquots (3 × 7 μl) were removed at several time points and diluted to a final volume of 70 μl of 50 mm glycine, pH 8.5, 0.1 mm ThT in a black, flat-bottom 384-well fluorescence assay plate. ThT signals were measured in a Tecan Infinite M200 using excitation at 450 nm and reading at 485 nm, with a manual gain factor of 80. Scale bars represent 500 nm. B, SDS-PAGE analysis of remaining α-syn-soluble protein content over the course of the fibrillization assays. The samples analyzed were the same as those shown in A. C, transmission electron micrograph semisynthetic N-Ac α-syn stained with uranyl acetate, prepared under the same conditions as in Fig. 4F.
Effect of Nα-Acetylation on Membrane Binding
Previous studies by Zabrocki et al. (29) in yeast suggested that Nα-acetylation could influence the membrane binding properties of α-syn in a yeast model of Parkinson disease. It has also been suggested that Nα-acetylation could enhance the stability of the membrane-bound α-syn by reinforcing the electrical dipole of the α-helical conformation adopted by α-syn upon interaction with negatively charged phospholipids (56). To determine whether the membrane binding properties of α-syn could be affected by Nα-acetylation, we performed an in vitro assay using phosphatidylglycerol (POPG) as a membrane model. Large POPG unilamellar vesicles with an average diameter of 100 nm were prepared at 1 mg/ml in 20 mm NaH2PO4, pH 7.6, by an extrusion procedure as described previously (38). WT or N-Ac α-syn (5 μm) were incubated at different POPG/protein mass ratios for 1 h at room temperature and then put on ice until the secondary structure content was assessed by CD spectroscopy (Fig. 6). Interestingly, we did not observe any difference in POPG binding affinity between the two proteins. As such, we investigated the binding of α-syn to LUVs composed of a 52.5:17.5:30 (mol %) phosphatidylcholine/phosphatidylserine/cholesterol, which were reported by Choi et al. (57) to be closer to the composition of synaptic vesicles. However, over the lipid concentration range we tested, which was the same as that used with pure POPG LUVs, we did not see any conformational shift toward the α-helical structure with both WT and N-Ac α-syn (data not shown). Accordingly, we then sought to further assess the effect of Nα-acetylation on α-syn interactions with biological membranes by using synaptosomal preparations obtained from α-syn-KO mice, as described before by Wislet-Gendebien et al. (44) (see “Experimental Procedures” for more details). Purified recombinant A30P α-syn, which has a lower membrane binding capacity (58, 59), was used as a control. Because a previous study established that cytosolic factors could partially rescue the membrane binding deficiency of A30P α-syn (44), we also sought to determine whether there could be an interplay between Nα-acetylation and murine brain cytosolic factors. Thus, cytosolic fractions were also collected and concentrated during synaptosomal extraction, and all binding assays were conducted both with or without preincubation with murine brain cytosolic factors (1.5 mg/ml). α-syn binding to synaptosomal membranes was measured from experiments where 0.5 μg of WT, A30P, or Nα-acetylated α-syn was incubated with synaptosomal fractions and was quantified by densitometry analysis from Western blots (Fig. 6, B and C). Although both recombinant WT and Nα-acetylated α-syn showed a higher binding propensity than A30P α-syn, we did not observe any significant differences between both proteins with respect to binding efficiency toward murine synaptosomes, with or without preincubation with cytosol (Fig. 6, B and C). Similar results were obtained when 1 μg of recombinant proteins was incubated with synaptosomes, although this higher amount saturated their α-syn binding capacity (not shown).
FIGURE 6.

In vitro membrane binding analyses. A, circular dichroism spectroscopy in the presence and absence of artificial POPG vesicles (100 nm average diameter). WT and N-Ac α-syn (10 μm each) were incubated in 20 mm sodium phosphate, pH 7.4, in the presence (0.5 or 5 mass equivalents) or absence of POPG liposomes for 30 min at RT before analysis by CD. Solid lines, WT α-syn. Dashed lines, N-Ac α-syn. Black lines, no POPG; dark gray lines, 5 eq of POPG; light gray lines, 5 eq of POPG. B, quantification of A30P, WT, and semisynthetic N-Ac α-syn binding to synaptosomal membranes isolated from α-syn-KO mice. 0.5 μg of each purified recombinant or semisynthetic protein was incubated with synaptosome-derived membranes at 37 °C for 10 min in absence or presence of dialyzed cytosol (1.5 mg/ml) before a centrifugation step (20,000 × g, 5 min) to separate bound (pellet) and unbound (supernatant) α-syn. Samples were then analyzed by SDS-PAGE/Western blot (primary antibody/Syn-1, 1:1000). C, representative Western blots that were used for densitometry-based quantification of α-syn binding (B).
Effect of Nα-Acetylation on α-syn Subcellular Localization and Membrane Binding in Mammalian Cells
To determine the effect of Nα-acetylation on the subcellular localization and membrane binding propensity of α-syn in mammalian cells, we used the proteofection reagent “Chariot®” to enhance the delivery of human recombinant WT and Nα-acetylated α-syn to HeLa cells. To minimize staining from noninternalized α-syn at the outer cell membrane, we introduced a trypsinization step followed by resuspending, washing, and then replating cells 4 h post-proteofection. Cells were then analyzed immunocytochemically 24 h later using a low concentration of the Syn-1 antibody to detect internalized proteins and not the minute levels of endogenous α-syn from HeLa cells, as well as using the WGA Alexa Fluor 555 conjugate that selectively binds N-acetylglucosamine and N-acetylneuraminic acid residues and hence selectively labels outer and inner cellular membranes allowing delineation of cellular morphology as reported previously (60, 61). Confocal imaging of proteofected cells revealed that both WT and N-Ac α-syn were internalized and that the subcellular localization of the internalized proteins is mostly cytosolic, with very little staining of the nuclear compartment (Fig. 7). Interestingly, a portion of the internalized proteins showed co-localization with inner membranes labeled with WGA, suggesting that both proteins are readily targeted to the endomembrane system upon internalization. However, both proteins showed similar subcellular co-localization indicating no significant effect of Nα-acetylation on the distribution or membrane binding properties of α-syn in vivo.
FIGURE 7.

Subcellular localization of WT versus N-Ac α-syn in HeLa cells. Confocal images of HeLa cells either proteofected with Chariot® alone or together with recombinant WT or semisynthetic Nα-acetylated α-syn (4 μg of protein were used in each case). Cells were immunostained using the Syn-1 antibody (1:1000, BD Transduction Laboratories) and 5 μg/ml of the membrane marker WGA. Nuclei are counterstained with Hoechst 33342.
Effect of Attenuating Nα-Acetylation on α-syn Subcellular Localization and Membrane Binding in Mammalian Cells
To further elucidate the biological role(s) of Nα-acetylation of α-syn, we set out to determine the effects of attenuating this modification in living cells. Doing so requires the knowledge of the N-terminal acetyltransferase responsible for α-syn Nα-acetylation in vivo. Given that the two N-terminal amino acids (including the initiator methionine) of nascent proteins determine which enzyme catalyzes their acetylation, the Met-Asp N-terminal sequence of α-syn strongly suggests that NatB could be mediating α-syn Nα-acetylation. Accordingly, we generated the D2P α-syn mutant having an altered N-terminal NatB recognition sequence and hence should not be acetylated (62). Note that the D2P mutation also causes removal of the initiator methionine by methionine aminopeptidases (30). After verifying by Western blot that D2P α-syn is overexpressed at a similar level and within the same time scale as WT α-syn (Fig. 8A), we analyzed transfected HEK293T lysates by SDS-PAGE after concentration of α-syn by immunoprecipitation, and the band corresponding to α-syn was digested with Glu-C. Extracted peptides were then analyzed by MALDI-TOF/TOF mass spectrometry with a focus on the N-terminal peptide generated by Glu-C proteolysis (MDVFMKGLSKAKE in the case of WT α-syn and PVFMKGLSKAKE for D2P α-syn). Fig. 8B shows that although overexpressed WT α-syn was found to be completely Nα-acetylated (as shown by the measured mass of 1525.71 Da for the α-syn N-terminal peptide and no signal at m/z = 1483.77 Da), the D2P mutant of α-syn is completely unacetylated, as only the nonacetylated peptide was detected (1334.68 Da). Indeed, no signal was detected for the acetylated D2P α-syn peptide (1376.76 Da), and D2P peptides with the first methionine retained were not observed. The sequences and acetylation states of both the WT and D2P N-terminal peptides were confirmed by tandem mass spectrometry as shown in Fig. 8C and supplemental Tables S1 and S2. After showing that the D2P α-syn mutant lacks Nα-acetylation, we sought to determine the biological effect of this loss on the subcellular localization and membrane binding abilities of α-syn in HEK cells (Fig. 8D). Interestingly, cells transfected with the D2P α-syn mutant showed similar distribution as that of WT α-syn, which is mostly cytosolic (Fig. 8D).
FIGURE 8.

Effect of abolishing α-syn Nα-acetylation using the D2P mutant of α-syn. A, Western blot (WB) analysis of D2P α-syn expression in HEK cells. Cells grown in 6-well culture plates were transfected with 2 μg of plasmid encoding WT or D2P α-syn (or left untreated for the nontransfected control), and expression was assessed at 12, 24, and 48 h post-transfection. The membrane was probed with the Syn-1 clone (1:2000, overnight, 4 °C) for α-syn (lower panel) and with anti-actin (Abcam, 1:20,000, overnight, 4 °C). B, MALDI-TOF analysis of peptides obtained by Glu-C digestion of α-syn immunoprecipitated from HEK293T lysates obtained 48 h after transfection with either WT α-syn (top panel) or D2P α-syn (bottom panel). The analysis is focused on the N-terminal peptide of α-syn. Peaks corresponding to the N-terminal peptide of α-syn are labeled with the observed m/z (theoretical m/z of the Nα-acetylated WT α-syn peptide, 1525.78 Da; theoretical m/z of the nonacetylated D2P α-syn peptide, 1334.75 Da). Peaks labeled with an asterisk correspond to Glu-C autolysis peptides. C, MALDI-TOF/TOF tandem mass spectrometry of the peaks corresponding to the α-syn N-terminal peptides observed in B. Top panel, CID fragmentation spectrum of the 1525.71 Da parent ion in B (wt α-syn N-terminal peptide). Bottom panel, CID fragmentation spectrum of the 1334.68 Da parent ion in B (D2P α-syn N-terminal peptide). Peaks are labeled with their corresponding b or y ion. See supplemental Tables S1 and S2 for the observed and expected m/z of these ions. D, subcellular localization analysis of WT and D2P α-syn. Confocal images of HEK cells transfected with either WT or D2P α-syn. Cells were immuno-stained using the Syn-1 antibody (1:1000, BD Transduction Laboratories) and the WGA membrane marker (5 μg/ml). Nuclei were counterstained with Hoechst 33342.
As an alternative strategy to attenuate α-syn Nα-acetylation, we opted to silence NatB expression in HEK cells with four different plasmids encoding shRNA against NatB (as well as with a plasmid encoding scrambled shRNA as a control) to determine whether NatB actually acetylates α-syn in mammalian cells. Given that all of the used plasmids also encode for GFP, this served as a transfection marker that showed comparable efficiency of transfection across conditions by fluorescent microscopy (data not shown). Interestingly, one of the shRNAs (NatB-shRNA4) showed the most effective silencing of NatB 3 days post-transfection (Fig. 9A), without compromising the well being of the cells (data not shown). To determine whether NatB silencing in HEK cells attenuates α-syn acetylation, we immunoprecipitated overexpressed α-syn from cells transfected with scrambled shRNA or NatB-shRNA4, and we then determined the acetylation status of α-syn N-terminal peptides by LC-ESI-MS/MS, focusing on the N-terminal peptide generated by Glu-C proteolysis (MDVFMKGLSKAKE). After verifying α-syn expression (Fig. 9A) and immunoprecipitation of overexpressed α-syn by Coomassie staining and Western blotting (Fig. 9, B and C), mass spectrometry analysis showed that although only acetylated α-syn was detected in cells overexpressing scrambled shRNA (Fig. 9D and supplemental Fig. S3A), unacetylated α-syn became detectable in NatB-shRNA4-transfected cells (Fig. 9E and supplemental Fig. S3B). In line with our previous results, decreasing acetylation levels by NatB silencing had no major effect on the subcellular distribution of α-syn in HEK cells (Fig. 10). In fact, both overexpressed and endogenous α-syn showed similar cytosolic localization that was not altered upon pre-transfecting with NatB-shRNA4 or any of the other shRNA plasmids (data not shown).
FIGURE 9.

Expression of NatB shRNAs and analyses by immunoprecipitation and mass spectrometry. A, 24 h post-transfecting HEK cells with empty plasmids or plasmids encoding scrambled shRNA or NatB shRNA (1–4), cells were transfected with a plasmid encoding WT α-syn and then analyzed 48 h later. Western blotting using the NatB-specific antibody (Santa Cruz Biotechnology, catalog no. 68397) showed most efficient silencing of NatB using NatB-shRNA4, without affecting levels of overexpressed α-syn detected using the Syn-1 antibody. B, Coomassie staining. C, Western blotting using the Syn-1 antibody (BD Transduction Laboratories) revealed a band co-migrating with recombinant α-syn. Bands corresponding to the capture antibody light and heavy chains are labeled with L and H, respectively, on the right side of B and C. IP, immunoprecipitated. D, extracted ion chromatograms of α-syn N-terminal peptides obtained by Glu-C digestion of α-syn obtained from HEK cells transfected with a scrambled shRNA, showing the absence of signal (expected at 16.8 min) specific for the unacetylated and oxidized peptide (Ox-MDVFOx-MKGLSKAKE) and the signal for the acetylated and oxidized peptide (Ac-Ox-MDVFOx-MKGLSKAKE), which elutes at 25.98 min. E, extracted ion chromatograms of α-syn N-terminal peptides obtained by Glu-C digestion of α-syn obtained from HEK cells transfected with NatB-shRNA4, showing the detection of the unacetylated and oxidized peptide (Ox-MDVFOx-MKGLSKAKE), which elutes at 16.79 min, as well as the acetylated and oxidized peptide (Ac-Ox-MDVFOx-MKGLSKAKE), which elutes at 25.96 min, thus showing that NatB-shRNA4 silencing does not lead to complete loss of α-syn N-terminal acetylation. D and E, black lines show the extracted chromatograms of nonacetylated/oxidized α-syn N-terminal peptide (mass range, 758.3772–758.3848 Da; charge = 2); and the gray lines correspond to the acetylated/oxidized peptide (mass range, 779.3791–779.3869 Da; charge = 2). The mass ranges for the extracted ion chromatograms were chosen to be as narrow as possible to exclude nonspecific signals without too much decreasing the signal intensity. However, nonspecific peaks are still present and are labeled as such in D and E. Note that the nonoxidized peptides were not detected.
FIGURE 10.

NatB silencing does not affect the subcellular localization of endogenous or overexpressed α-syn in HEK cells. 24 h post-transfecting HEK cells with plasmids encoding scrambled shRNA or NatB shRNA4, cells were either transfected with a plasmid encoding WT α-syn (upper 2 panels) or not (lower 2 panels) and then immunostained using the Syn-1 antibody. Confocal fluorescent microscopy shows that cells transfected with scrambled or NatB shRNA4 constructs (expressing enhanced GFP) show similar subcellular localization of endogenously and exogenously expressed synuclein. Nuclei were counterstained with Hoechst 33342.
While this manuscript was in preparation, Trexler and Rhoades (37) reported that recombinant Nα-acetylated α-syn expressed, using the same method described above but purified in the presence of 10% glycerol and 0.1% of the detergent BOG, is α-helical and oligomeric. Furthermore, they suggested that both Nα-acetylation and glycerol/BOG during purification are required for obtaining a stable α-helical and oligomeric form of α-syn. In our hands, recombinant expression of Nα-acetylated α-syn in the presence or absence of BOG and using different purification protocols always yielded α-syn that is monomeric and disordered (supplemental Fig. S4). These findings are consistent with our NMR results demonstrating that Nα-acetylated α-syn in intact cells is disordered (Fig. 3).
DISCUSSION
Although the role of Nα-acetylation at the single protein level remains poorly understood, it has been proposed to regulate protein-protein interactions. Several studies have reported a stabilization of protein-protein interactions upon Nα-acetylation. For example, muscular α-tropomyosin requires N-terminal acetylation to functionally interact with actin as the unacetylated counterpart polymerizes abnormally and more importantly does not bind to fibrillar actin to form contraction-functional bundles (63). Another example is Tfs1p, a member of the stress-induced carboxypeptidase Y inhibitor and phosphatidylethanolamine-binding protein family, where N-terminal acetylation is essential for carboxypeptidase Y inhibition (64). In yeast, the assembly and stability of the Gag coat protein from the L-A double-stranded RNA virus is dependent on its N-terminal acetylation (65). However, Nα-acetylation does not always stabilize protein-protein interactions. For instance, N-terminal acetylation of the fetal hemoglobin γ-chain has been shown to weaken the hemoglobin tetramer. In this case, the loss of electrostatic stabilization in the acetylated protein (where N-terminal protonation stabilizes tetramer formation) was proposed to be the cause of the observed tetramer-weakening effect (66). At the cellular level, the roles of Nα-acetylation of proteins are diverse; it is required for cellular homeostasis, as knockdown of yeast NatA strongly perturbs protein synthesis (67), possibly because functional components of the 26 S proteasome regulatory particle require Nα-acetylation (68). In human cell lines, knockdown of NatB activity prevents normal cell cycle progression (27), whereas disruption of human NatC induces apoptosis in HeLa cells (28).
In this study, we report the semisynthesis and recombinant expression of Nα-acetylated α-syn and the effect of this co-translational modification on the structure, aggregation propensity, and membrane binding properties of the protein. N-terminal modification of α-syn was first reported by Jakes et al. (69) who measured an intact mass of 14,681 Da for α-syn extracted from human brains (whereas unmodified recombinant α-syn has a mass of 14,460 Da). Although the cause of this discrepancy was not investigated at the time, failure to sequence the protein by Edman degradation (69) suggested an N-terminal modification. To date, two research groups have reported Nα-acetylation of α-syn in vivo using mass spectrometry. The first was Anderson et al. (11) who reported that in human brains α-syn is quantitatively Nα-acetylated. Indeed, when they analyzed proteins extracted from cytosolic fractions and from Lewy bodies of human patients affected by dementia with Lewy bodies and from the brains of healthy controls using a “bottom-up” proteomics approach (11), the N-terminal tryptic peptide (MDVFMK) was detected with an m/z of 813 Da (+42 Da with respect to the theoretical mass of that peptide), which is consistent with a single acetylation event. MS/MS analysis of the peptide further confirmed that the modification occurred on the N-terminal amino acid. This finding was then recently corroborated by another study examining α-syn immunoprecipitates from human brain and cerebrospinal fluid samples obtained from either PD, dementia with LB patients, or healthy controls (17). Although the ubiquitous and irreversible (70) nature of α-syn Nα-acetylation complicates the identification of possible (if any) links to pathological processes, it remains crucial to understand the biophysical properties of Nα-acetylated α-syn. This is because despite the fact that the vast majority, if not all, of α-syn in human cells is subjected to Nα-acetylation, most studies on α-syn structural, aggregational, and functional properties have been performed using recombinant α-syn purified from E. coli, which is not Nα-acetylated.
To study α-syn Nα-acetylation, one needs to use approaches that allow selective and quantitative acetylation of the N-terminal amine. This is not yet possible using chemical modification of the full-length protein, due to the small pKa difference between the N terminus and amino groups on the side chain of lysine residues. At the time we began this study, the enzyme responsible for α-syn Nα-acetylation in vivo was not known; therefore, we chose an EPL strategy for the initial characterization of Nα-acetylated α-syn. Later, we applied a recombinant expression strategy based on co-expression of α-syn and NatB, which generated quantitatively acetylated α-syn. We then utilized these approaches to generate Nα-acetylated α-syn in milligram quantities and showed that Nα-acetylated α-syn is monomeric and unstructured like its recombinant nonacetylated counterpart. In addition, the kinetics of Nα-acetylated α-syn oligomerization and fibrillogenesis were virtually identical to that of the WT nonacetylated protein.
We then assessed whether Nα-acetylation affects the membrane affinity of α-syn. Previous genetic screen studies of α-syn-GFP fusion proteins in yeast suggested that Nα-acetylation could stabilize α-syn-membrane interactions (29). In WT S. cerevisiae cells, α-syn-GFP has a mostly membranous localization, although in yeast cells lacking the NatB N-acetyltransferase, the fusion protein loses most of its membranous localization and becomes cytoplasmic. A recent study by Vamvaca et al. (56) speculated that Nα-acetylation may favor the α-helical conformation of α-syn by neutralizing the N-terminal positive charge and stabilizing the α-helix electrical dipole (71). Using in vitro liposome preparations and CD spectroscopy to investigate α-syn membrane binding, we did not observe such a dramatic effect. In fact, there was no significant difference in POPG liposome association between WT and semisynthetic Nα-acetylated α-syn. However, the use of vesicles comprised of a single lipid species such as POPG may not be an appropriate model, because cellular membranes display a very different composition (57, 72, 73). Furthermore, the high negative charge density at the surface of POPG-only vesicles may introduce an artificially large electrostatic component to the binding of the overall positively charged N-terminal domain of α-syn, which could mask putative differences between WT and N-Ac α-syn. Indeed, according to Cotman et al. (72), negatively charged lipids represent only about 12% (molar fraction) of the lipids present in rat synaptic plasma membranes. As such, to assess membrane binding under more physiologically relevant conditions, we studied membrane binding properties of exogenous semisynthetic α-syn (WT and N-Ac) using (α-syn-deficient) synaptosome-derived membranes. Again, we found no difference in membrane association between WT and N-Ac α-syn, both in presence and absence of brain cytosolic factors. This indicates that these components (such as ATP and cytosolic lipids), which were found to modulate WT and mutant α-syn binding to synaptosomes (44), do not act through nor in synergy with N-terminal acetylation of α-syn. In addition, we obtained similar results when we tested the effect of distribution of Nα-acetylation on the subcellular localization of α-syn in HeLa cells, as both WT and N-Ac α-syn showed a similar distribution that was mostly cytosolic with co-localization with inner membrane structures. These results suggest that Nα-acetylation does not significantly influence α-syn subcellular localization and membrane binding abilities in living cells. However, it remains possible that the intracellular distribution of the uptaken exogenously added α-syn differs from that of endogenous or overexpressed protein (which is quantitatively Nα-acetylated); furthermore, exogenously added recombinant α-syn could become Nα-acetylated when internalized in mammalian cells. We thus designed a nonacetylable α-syn mutant (D2P) based on the following: 1) the reported sequence determinants of protein Nα-acetylation (30); 2) the “XPX” rule reported by Goetze et al. (62), which states that having a proline as the second residue results in no Nα-acetylation in eukaryotic cells; and 3) recent studies suggesting that the human homolog of the yeast NatB N-acetyltransferase complex could be acetylating α-syn in vivo (29, 56). After verifying that this mutant is indeed not acetylated by mass spectrometry, we showed by immunocytochemistry that D2P α-syn and overexpressed WT (acetylated) α-syn exhibit similar subcellular distributions, therefore suggesting that Nα-acetylation does not influence α-syn membrane association, in agreement with all our previous findings.
As an alternative strategy to study the effect of attenuating α-syn Nα-acetylation on its subcellular localization, we opted to silence NatB in HEK cells using shRNA. Interestingly, although mass spectrometry analysis showed that some of the overexpressed α-syn in NatB-silenced cells is indeed unacetylated, hence suggesting that NatB does acetylate α-syn in vivo, deacetylation of α-syn was not complete. This could be due to the following: 1) only small amounts of remaining unsilenced NatB are sufficient to acetylate the rest of overexpressed synuclein, or 2) other enzymes are involved in acetylating α-syn. As such, the fact that silencing NatB did not have a prominent effect on the subcellular localization or membrane binding of endogenous or overexpressed synuclein in HEK cells is consistent with our observation that recombinant Nα-acetylated α-syn exhibited identical subcellular localization to nonacetylated WT α-syn.
Because of the recent report that recombinant Nα-acetylated α-syn purified in the presence of glycerol and the mild detergent BOG could form α-helically folded oligomers (37), we sought to clarify whether Nα-acetylated α-syn could exist as a structured oligomer in bacterial cells. To do so, we employed in-cell NMR to assess the structural consequences of N-terminal acetylation of α-syn in a manner that is independent of any purification protocol. Because NatB-mediated acetylation of α-syn leads to localized chemical shift changes (Fig. 3A), we could use NMR of cell lysates to document the extent of modification in intact cells obtained from the same cell pellet. Our data indicate that within the sensitivity of the NMR experiment, α-syn is quantitatively and completely acetylated in our cells (>95%), as any unmodified protein present at 5% or greater fraction would give rise to detectable resonances at positions distinct from those associated with the modified protein. Having established that our NatB co-transfected cells contain quantitatively acetylated α-syn, we collected NMR spectra in intact cells. α-syn spectra obtained with and without NatB co-transfection are essentially identical. The similarity between the spectra rules out any large scale ordering or structuring of α-syn upon N-terminal acetylation and proves that N-terminally acetylated α-syn expressed in bacteria is intrinsically disordered, independent of the purification protocol. The spectra also rule out any substantial (>10%) population of stable oligomeric protein, as this would lead to both decreases in resonance intensity and changes in chemical shifts. The data cannot rule out minor populations of a structured oligomer below the level of detection or the presence of some type of highly dynamic oligomers that fully preserve the intrinsically disordered structure of the protein. Such a dynamic oligomer, however, would be largely unprecedented and would be incompatible with the α-helical structure reported for recently proposed α-syn tetramers (74, 75). Our data also address the structural consequences of α-syn Nα-acetylation, demonstrating a localized increase in helicity in the N-terminal region of the protein, but no large scale conformational changes in, or ordering of, the protein. Importantly, we probed the overall conformational state of the protein in the absence of any potential artifact due to purification procedures, especially the introduction of detergents such as those used in recent studies of N-terminally modified α-syn (37, 74). We note that the interactions of Nα-acetylated α-syn with such detergents may be different from those of the unmodified protein, which may underlie the helical structure reported in those studies. Indeed, together with our failure to reproduce the data from Trexler and Rhoades (37) when using their purification method, followed by a hydrophobic interaction chromatography step, the NMR measurements strongly suggest that the glycerol/BOG used during their purification of Nα-acetylated α-syn may be responsible for the reported folding and apparent tetramerization of α-syn.
In conclusion, our findings suggest that Nα-acetylation does not have prominent effects on the biophysical and membrane binding properties of α-syn, in vitro and in vivo. In fact, Nα-acetylated synuclein (produced either semisynthetically or recombinantly from E. coli) shared similar secondary structure, oligomeric state, aggregation propensities, and ability to bind synaptosomal membranes in vitro, as well as in HeLa cells. Moreover, attenuating Nα-acetylation in living cells, by using a nonacetylable mutant or by silencing the enzyme responsible for α-syn Nα-acetylation, did not affect the subcellular distribution and membrane binding of α-syn. Nevertheless, we suggest that recombinant Nα-acetylated α-syn expression should become a standard for in vitro experiments, because it better mimics the real state of the protein in vivo. Although it is very likely that other groups will use recombinant expression instead of protein semisynthesis, it should be kept in mind that the EPL approach we used here retains unique advantages as it enables future studies on cross-talk between Nα-acetylation and other α-syn N-terminal modifications.
Supplementary Material
Acknowledgments
We thank Nathalie Jordan, Trudy Ramlall, and John Perrin for their outstanding technical support, Dr. Marc Moniatte and Diego Chiappe (EPFL Proteomics Core Facility) for LC-ESI-MS/MS analyses, and Prof. Daniel P. Mulvihill (University of Kent, United Kingdom) for kindly providing the pNatB plasmid.
Note Added in Proof
While this manuscript was under review, a report appeared describing another NMR study on the effects of N-terminal acetylation on α-syn structure (Maltsev, A. S., Ying, J., and Bax, A. (2012) Impact of N-terminal acetylation of α-synuclein on its Random coil and lipid binding properties. Biochemistry 51, 5004–5013) with results entirely consistent with those presented herein, except that N-terminal acetylation was found to increase lipid binding by α-syn. This discrepancy may result from differences in lipid vesicle composition and/or preparation. In addition, this study found that acetylation leads to a very slight increase in the hydrodynamic radius of α-syn, suggesting that the trend towards slightly decreased long-range contacts in our PRE data may be significant.
This work was supported, in whole or in part, by National Institutes of Health Grant AG019391 (to D. E.). This work was also supported by the Swiss Federal Institute of Technology, Lausanne (to H. A. L.), and European Research Council Starting Grant 243182 (to H. A. L. and B. F.).

This article contains supplemental Figs. S1–S4, Tables S1 and S2.
P. Selenko, personal communication.
- α-syn
- α-synuclein
- n-Ac
- Nα-acetylated
- BOG
- n-octyl-β-d-glucopyranoside
- PD
- Parkinson disease
- LB
- Lewy body
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- TCEP
- tris(2-carboxyethyl)phosphine
- ThT
- thioflavin T
- LUV
- large unilamellar vesicle
- PRE
- paramagnetic relaxation enhancement
- GdnHCl
- guanidinium hydrochloride
- POPG
- phosphatidylglycerol
- WGA
- wheat germ agglutinin
- NCL
- native chemical ligation
- EPL
- expressed protein ligation.
REFERENCES
- 1. Berrade L., Camarero J. A. (2009) Expressed protein ligation. A resourceful tool to study protein structure and function. Cell. Mol. Life Sci. 66, 3909–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cookson M. R., van der Brug M. (2008) Cell systems and the toxic mechanism(s) of α-synuclein. Exp. Neurol. 209, 5–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., Riess O. (1998) A30P mutation in the gene encoding α-synuclein in Parkinson disease. Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 4. Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B., Llorens V., Gomez Tortosa E., del Ser T., Muñoz D. G., de Yebenes J. G. (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 [DOI] [PubMed] [Google Scholar]
- 5. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 6. Chartier-Harlin M. C., Kachergus J., Roumier C., Mouroux V., Douay X., Lincoln S., Levecque C., Larvor L., Andrieux J., Hulihan M., Waucquier N., Defebvre L., Amouyel P., Farrer M., Destée A. (2004) α-Synuclein locus duplication as a cause of familial Parkinson disease. Lancet 364, 1167–1169 [DOI] [PubMed] [Google Scholar]
- 7. Singleton A. B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M. R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K. (2003) α-Synuclein locus triplication causes Parkinson disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- 8. Brown D. R. (2007) Interactions between metals and α-synuclein. Function or artifact? FEBS J. 274, 3766–3774 [DOI] [PubMed] [Google Scholar]
- 9. Oueslati A., Fournier M., Lashuel H. A. (2010) Role of post-translational modifications in modulating the structure, function, and toxicity of α-synuclein implications for Parkinson disease pathogenesis and therapies. Prog. Brain Res. 183, 115–145 [DOI] [PubMed] [Google Scholar]
- 10. Hasegawa M., Fujiwara H., Nonaka T., Wakabayashi K., Takahashi H., Lee V. M., Trojanowski J. Q., Mann D., Iwatsubo T. (2002) Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076 [DOI] [PubMed] [Google Scholar]
- 11. Anderson J. P., Walker D. E., Goldstein J. M., de Laat R., Banducci K., Caccavello R. J., Barbour R., Huang J., Kling K., Lee M., Diep L., Keim P. S., Shen X., Chataway T., Schlossmacher M. G., Seubert P., Schenk D., Sinha S., Gai W. P., Chilcote T. J. (2006) Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752 [DOI] [PubMed] [Google Scholar]
- 12. Nonaka T., Iwatsubo T., Hasegawa M. (2005) Ubiquitination of α-synuclein. Biochemistry 44, 361–368 [DOI] [PubMed] [Google Scholar]
- 13. Paleologou K. E., Oueslati A., Shakked G., Rospigliosi C. C., Kim H. Y., Lamberto G. R., Fernandez C. O., Schmid A., Chegini F., Gai W. P., Chiappe D., Moniatte M., Schneider B. L., Aebischer P., Eliezer D., Zweckstetter M., Masliah E., Lashuel H. A. (2010) Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 30, 3184–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li W., West N., Colla E., Pletnikova O., Troncoso J. C., Marsh L., Dawson T. M., Jäkälä P., Hartmann T., Price D. L., Lee M. K. (2005) Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson disease-linked mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 2162–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu C. W., Giasson B. I., Lewis K. A., Lee V. M., Demartino G. N., Thomas P. J. (2005) A precipitating role for truncated α-synuclein and the proteasome in α-synuclein aggregation. Implications for pathogenesis of Parkinson disease. J. Biol. Chem. 280, 22670–22678 [DOI] [PubMed] [Google Scholar]
- 16. Zhou W., Long C., Reaney S. H., Di Monte D. A., Fink A. L., Uversky V. N. (2010) Methionine oxidation stabilizes nontoxic oligomers of α-synuclein through strengthening the autoinhibitory intramolecular long range interactions. Biochim. Biophys. Acta 1802, 322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohrfelt A., Zetterberg H., Andersson K., Persson R., Secic D., Brinkmalm G., Wallin A., Mulugeta E., Francis P. T., Vanmechelen E., Aarsland D., Ballard C., Blennow K., Westman-Brinkmalm A. (2011) Identification of novel α-synuclein isoforms in human brain tissue by using an online nanoLC-ESI-FTICR-MS method. Neurochem. Res. 36, 2029–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown J. L., Roberts W. K. (1976) Evidence that approximately 80% of the soluble proteins from Ehrlich ascites cells are Nα-acetylated. J. Biol. Chem. 251, 1009–1014 [PubMed] [Google Scholar]
- 19. Brown J. L. (1979) A comparison of the turnover of α-N-acetylated and nonacetylated mouse L-cell proteins. J. Biol. Chem. 254, 1447–1449 [PubMed] [Google Scholar]
- 20. Caesar R., Warringer J., Blomberg A. (2006) Physiological importance and identification of novel targets for the N-terminal acetyltransferase NatB. Eukaryot. Cell 5, 368–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Persson B., Flinta C., von Heijne G., Jörnvall H. (1985) Structures of N-terminally acetylated proteins. Eur. J. Biochem. 152, 523–527 [DOI] [PubMed] [Google Scholar]
- 22. Polevoda B., Sherman F. (2001) NatC Nα-terminal acetyltransferase of yeast contains three subunits, Mak3p, Mak10p, and Mak31p. J. Biol. Chem. 276, 20154–20159 [DOI] [PubMed] [Google Scholar]
- 23. Arnesen T., Van Damme P., Polevoda B., Helsens K., Evjenth R., Colaert N., Varhaug J. E., Vandekerckhove J., Lillehaug J. R., Sherman F., Gevaert K. (2009) Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. U.S.A. 106, 8157–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Starheim K. K., Gevaert K., Arnesen T. (2012) Protein N-terminal acetyltransferases. When the start matters. Trends Biochem. Sci. 37, 152–161 [DOI] [PubMed] [Google Scholar]
- 25. Polevoda B., Cardillo T. S., Doyle T. C., Bedi G. S., Sherman F. (2003) Nat3p and Mdm20p are required for function of yeast NatB Nα-terminal acetyltransferase and of actin and tropomyosin. J. Biol. Chem. 278, 30686–30697 [DOI] [PubMed] [Google Scholar]
- 26. Arnesen T., Anderson D., Baldersheim C., Lanotte M., Varhaug J. E., Lillehaug J. R. (2005) Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem. J. 386, 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Starheim K. K., Arnesen T., Gromyko D., Ryningen A., Varhaug J. E., Lillehaug J. R. (2008) Identification of the human Nα-acetyltransferase complex B (hNatB). A complex important for cell-cycle progression. Biochem. J. 415, 325–331 [DOI] [PubMed] [Google Scholar]
- 28. Starheim K. K., Gromyko D., Evjenth R., Ryningen A., Varhaug J. E., Lillehaug J. R., Arnesen T. (2009) Knockdown of human Nα-terminal acetyltransferase complex C leads to p53-dependent apoptosis and aberrant human Arl8b localization. Mol. Cell. Biol. 29, 3569–3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zabrocki P., Bastiaens I., Delay C., Bammens T., Ghillebert R., Pellens K., De Virgilio C., Van Leuven F., Winderickx J. (2008) Phosphorylation, lipid raft interaction, and traffic of α-synuclein in a yeast model for Parkinson. Biochim. Biophys. Acta 1783, 1767–1780 [DOI] [PubMed] [Google Scholar]
- 30. Arnesen T. (2011) Toward a functional understanding of protein N-terminal acetylation. PLoS Biol. 9, e1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soppa J. (2010) Protein acetylation in archaea, bacteria, and eukaryotes. Archaea 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hejjaoui M., Haj-Yahya M., Kumar K. S., Brik A., Lashuel H. A. (2011) Toward elucidation of the role of ubiquitination in the pathogenesis of Parkinson disease with semisynthetic ubiquitinated α-synuclein. Angew. Chem. Int. Ed. Engl. 50, 405–409 [DOI] [PubMed] [Google Scholar]
- 33. Hejjaoui M., Butterfield S. M., Fauvet B., Vercruysse F., Cui J., Dikiy I., Prudent M., Olschewski D., Zhang Y., Eliezer D., Lashuel H. A. (2012) Elucidating the role of C-terminal post-translational modifications using protein semisynthesis strategies. α-Synuclein phosphorylation at tyrosine 125. J. Am. Chem. Soc. 134, 5196–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blanco-Canosa J. B., Dawson P. E. (2008) An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew. Chem. Int. Ed. Engl. 47, 6851–6855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang H., Rabenstein D. L. (1999) A cleavage cocktail for methionine-containing peptides. J. Pept. Res. 53, 548–553 [DOI] [PubMed] [Google Scholar]
- 36. Johnson E. C., Kent S. B. (2006) Insights into the mechanism and catalysis of the native chemical ligation reaction. J. Am. Chem. Soc. 128, 6640–6646 [DOI] [PubMed] [Google Scholar]
- 37. Trexler A. J., Rhoades E. (2012) N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 21, 601–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paleologou K. E., Schmid A. W., Rospigliosi C. C., Kim H. Y., Lamberto G. R., Fredenburg R. A., Lansbury P. T., Jr., Fernandez C. O., Eliezer D., Zweckstetter M., Lashuel H. A. (2008) Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of α-synuclein. J. Biol. Chem. 283, 16895–16905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mbefo M. K., Paleologou K. E., Boucharaba A., Oueslati A., Schell H., Fournier M., Olschewski D., Yin G., Zweckstetter M., Masliah E., Kahle P. J., Hirling H., Lashuel H. A. (2010) Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 285, 2807–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marley J., Lu M., Bracken C. (2001) A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75 [DOI] [PubMed] [Google Scholar]
- 41. Eliezer D., Kutluay E., Bussell R., Jr., Browne G. (2001) Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073 [DOI] [PubMed] [Google Scholar]
- 42. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 43. Johnson B. A., Blevins R. A. (1994) NMRView, A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 44. Wislet-Gendebien S., Visanji N. P., Whitehead S. N., Marsilio D., Hou W., Figeys D., Fraser P. E., Bennett S. A., Tandon A. (2008) Differential regulation of wild-type and mutant α-synuclein binding to synaptic membranes by cytosolic factors. BMC Neurosci. 9, 92. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Johnson M., Coulton A. T., Geeves M. A., Mulvihill D. P. (2010) Targeted amino-terminal acetylation of recombinant proteins in E. coli. PLoS One 5, e15801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weinreb P. H., Zhen W., Poon A. W., Conway K. A., Lansbury P. T., Jr. (1996) NACP, a protein implicated in Alzheimer disease and learning, is natively unfolded. Biochemistry 35, 13709–13715 [DOI] [PubMed] [Google Scholar]
- 47. Fauvet B., Mbefo M. K., Fares M. B., Desobry C., Michael S., Ardah M. T., Tsika E., Coune P., Prudent M., Lion N., Eliezer D., Moore D. J., Schneider B., Aebischer P., El-Agnaf O. M., Masliah E., Lashuel H. A. (2012) α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li C., Charlton L. M., Lakkavaram A., Seagle C., Wang G., Young G. B., Macdonald J. M., Pielak G. J. (2008) Differential dynamical effects of macromolecular crowding on an intrinsically disordered protein and a globular protein. Implications for in-cell NMR spectroscopy. J. Am. Chem. Soc. 130, 6310–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maldonado A. Y., Burz D. S., Shekhtman A. (2011) In-cell NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 59, 197–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ito Y., Selenko P. (2010) Cellular structural biology. Curr. Opin. Struct. Biol. 20, 640–648 [DOI] [PubMed] [Google Scholar]
- 51. Dedmon M. M., Lindorff-Larsen K., Christodoulou J., Vendruscolo M., Dobson C. M. (2005) Mapping long range interactions in α-synuclein using spin label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 127, 476–477 [DOI] [PubMed] [Google Scholar]
- 52. Bertoncini C. W., Jung Y. S., Fernandez C. O., Hoyer W., Griesinger C., Jovin T. M., Zweckstetter M. (2005) Release of long range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 1430–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sung Y. H., Eliezer D. (2007) Residual structure, backbone dynamics, and interactions within the synuclein family. J. Mol. Biol. 372, 689–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kang L., Moriarty G. M., Woods L. A., Ashcroft A. E., Radford S. E., Baum J. (2012) N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 21, 911–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Uversky V. N., Eliezer D. (2009) Biophysics of Parkinson disease. Structure and aggregation of α-synuclein. Curr. Protein Pept. Sci. 10, 483–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vamvaca K., Volles M. J., Lansbury P. T., Jr. (2009) The first N-terminal amino acids of α-synuclein are essential for α-helical structure formation in vitro and membrane binding in yeast. J. Mol. Biol. 389, 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Choi W., Zibaee S., Jakes R., Serpell L. C., Davletov B., Crowther R. A., Goedert M. (2004) Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 576, 363–368 [DOI] [PubMed] [Google Scholar]
- 58. Jo E., Fuller N., Rand R. P., St George-Hyslop P., Fraser P. E. (2002) Defective membrane interactions of familial Parkinson disease mutant A30P α-synuclein. J. Mol. Biol. 315, 799–807 [DOI] [PubMed] [Google Scholar]
- 59. Bussell R., Jr., Eliezer D. (2004) Effects of Parkinson disease-linked mutations on the structure of lipid-associated α-synuclein. Biochemistry 43, 4810–4818 [DOI] [PubMed] [Google Scholar]
- 60. Rustom A., Saffrich R., Markovic I., Walther P., Gerdes H. H. (2004) Nanotubular highways for intercellular organelle transport. Science 303, 1007–1010 [DOI] [PubMed] [Google Scholar]
- 61. McNeil P. L., Steinhardt R. A. (2003) Plasma membrane disruption. Repair, prevention, adaptation. Annu. Rev. Cell Dev. Biol. 19, 697–731 [DOI] [PubMed] [Google Scholar]
- 62. Goetze S., Qeli E., Mosimann C., Staes A., Gerrits B., Roschitzki B., Mohanty S., Niederer E. M., Laczko E., Timmerman E., Lange V., Hafen E., Aebersold R., Vandekerckhove J., Basler K., Ahrens C. H., Gevaert K., Brunner E. (2009) Identification and functional characterization of N-terminally acetylated proteins in Drosophila melanogaster. PLoS Biol. 7, e1000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Urbancikova M., Hitchcock-DeGregori S. E. (1994) Requirement of amino-terminal modification for striated muscle α-tropomyosin function. J. Biol. Chem. 269, 24310–24315 [PubMed] [Google Scholar]
- 64. Caesar R., Blomberg A. (2004) The stress-induced Tfs1p requires NatB-mediated acetylation to inhibit carboxypeptidase Y and to regulate the protein kinase A pathway. J. Biol. Chem. 279, 38532–38543 [DOI] [PubMed] [Google Scholar]
- 65. Tercero J. C., Wickner R. B. (1992) MAK3 encodes an N-acetyltransferase whose modification of the L-A gag NH2 terminus is necessary for virus particle assembly. J. Biol. Chem. 267, 20277–20281 [PubMed] [Google Scholar]
- 66. Manning L. R., Manning J. M. (2001) The acetylation state of human fetal hemoglobin modulates the strength of its subunit interactions. Long range effects and implications for histone interactions in the nucleosome. Biochemistry 40, 1635–1639 [DOI] [PubMed] [Google Scholar]
- 67. Lee F. J., Lin L. W., Smith J. A. (1989) N[α]-Acetyltransferase deficiency alters protein synthesis in Saccharomyces cerevisiae. FEBS Lett. 256, 139–142 [DOI] [PubMed] [Google Scholar]
- 68. Kikuchi J., Iwafune Y., Akiyama T., Okayama A., Nakamura H., Arakawa N., Kimura Y., Hirano H. (2010) Co- and post-translational modifications of the 26 S proteasome in yeast. Proteomics 10, 2769–2779 [DOI] [PubMed] [Google Scholar]
- 69. Jakes R., Spillantini M. G., Goedert M. (1994) Identification of two distinct synucleins from human brain. FEBS Lett. 345, 27–32 [DOI] [PubMed] [Google Scholar]
- 70. Polevoda B., Sherman F. (2002) The diversity of acetylated proteins. Genome Biol. 3, reviews0006.0001–0006.0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hol W. G. (1985) The role of the α-helix dipole in protein function and structure. Prog. Biophys. Mol. Biol. 45, 149–195 [DOI] [PubMed] [Google Scholar]
- 72. Cotman C., Blank M. L., Moehl A., Snyder F. (1969) Lipid composition of synaptic plasma membranes isolated from rat brain by zonal centrifugation. Biochemistry 8, 4606–4612 [DOI] [PubMed] [Google Scholar]
- 73. Martínez M., Mougan I. (1998) Fatty acid composition of human brain phospholipids during normal development. J. Neurochem. 71, 2528–2533 [DOI] [PubMed] [Google Scholar]
- 74. Wang W., Perovic I., Chittuluru J., Kaganovich A., Nguyen L. T., Liao J., Auclair J. R., Johnson D., Landeru A., Simorellis A. K., Ju S., Cookson M. R., Asturias F. J., Agar J. N., Webb B. N., Kang C., Ringe D., Petsko G. A., Pochapsky T. C., Hoang Q. Q. (2011) A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U.S.A. 108, 17797–17802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bartels T., Choi J. G., Selkoe D. J. (2011) α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.