

Background: GnT-Vb(IX) branches the α-mannose to the O-Manβ(1,2)-GlcNac in brain.
Results: GnT-Vb does not synthesize N-linked structures in vivo. GnT-V, however, can compensate for GnT-Vb activity in vivo.
Conclusion: GnT-Vb is predominantly involved in synthesizing branched O-mannosyl glycans in mouse brain.
Significance: It is shown for the first time in vivo that GnT-Vb and -V have different activity in the synthesis of N- and O-linked glycans.
Keywords: Development, Glycobiology, Glycomics, Glycosyltransferases, Neurodevelopment, GnT-IX, GnT-V, GnT-Vb, HNK-1, O-Linked Mannose
Abstract
The severe phenotypic effects of altered glycosylation in the congenital muscular dystrophies, including Walker-Warburg syndrome, muscle-eye-brain disease, Fukuyama congenital muscular dystrophy, and congenital muscular dystrophy 1D, are caused by mutations resulting in altered glycans linked to proteins through O-linked mannose. A glycosyltransferase that branches O-Man, N-acetylglucosaminyltransferase Vb (GnT-Vb), is highly expressed in neural tissues. To understand the expression and function of GnT-Vb, we studied its expression during neuromorphogenesis and generated GnT-Vb null mice. A paralog of GnT-Vb, N-acetylglucosaminyltransferase (GnT-V), is expressed in many tissues and brain, synthesizing N-linked, β1,6-branched glycans, but its ability to synthesize O-mannosyl-branched glycans is unknown; conversely, although GnT-Vb can synthesize N-linked glycans in vitro, its contribution to their synthesis in vivo is unknown. Our results showed that deleting both GnT-V and GnT-Vb results in the total loss of both N-linked and O-Man-linked β1,6-branched glycans. GnT-V null brains lacked N-linked, β1,6-glycans but had normal levels of O-Man β1,6-branched structures, showing that GnT-Vb could not compensate for the loss of GnT-V. By contrast, GnT-Vb null brains contained normal levels of N-linked β1,6-glycans but low levels of some O-Man β1,6-branched glycans. Therefore, GnT-V could partially compensate for GnT-Vb activity in vivo. We found no apparent change in α-dystroglycan binding of glycan-specific antibody IIH6C4 or binding to laminin in GnT-Vb null mice. These results demonstrate that GnT-V is involved in synthesizing branched O-mannosyl glycans in brain, but the function of these branched O-mannosyl structures is unresolved using mice that lack these glycosyltransferases.
Introduction
Correct O-mannosyl glycosylation of proteins is critical for the structure and function of both muscle and brain tissues. Genetic disorders that affect O-mannosyl glycosylation lead to forms of congenital muscular dystrophies that are characterized by muscular dystrophy, type II lissencephaly, and eye abnormalities (1–8). Studies have demonstrated that disruption of the activity of several key enzymes in the O-mannosyl glycosylation pathway likely mediates their pathological effects through altered glycosylation of α-dystroglycan (9–11). Relatively little is known, however, about the effects that can be observed when the enzymes that further modify these glycan structures are disrupted; moreover, reports of additional glycoproteins that display O-mannosyl glycosylation are very limited despite evidence of an abundance of O-mannosyl glycans in brain (12–15). Converging evidence has demonstrated the severe phenotypic effects of altered glycosylation in congenital muscular dystrophies (CMD).3 Walker-Warburg syndrome, muscle-eye-brain disease, Fukuyama congenital muscular dystrophy, and congenital muscular dystrophy 1D (MDC1D) are caused by mutations in genes that encode known or putative glycosyltransferases or glycan-modifying enzymes (16–26). The disrupted genes underlying these diseases appear to encode proteins that contribute to the O-mannosyl glycosylation pathway.
O-Linked mannosyl glycans have in common an α-linked Man attached to serine or threonine. The CMD-causing genes, protein O-mannosyltransferase 1 and 2 (POMT1 and 2) (27, 28), and protein O-mannose N-acetylglucosaminyltransferase 1 (POMGnT1) (25, 29), are known to be involved in the synthesis of O-linked mannosyl glycans, whereas the LARGE is involved in the modification of O-mannosyl glycans (26, 30). POMT1 and -2 function together to transfer mannose to Ser or Thr residues forming an α-linkage. POMGnT1 is involved in transferring the β1,2-linked N-acetylglucosamine (GlcNAc) to O-linked mannose (O-Man) (Scheme 1).
SCHEME 1.

Depiction of products of GnT-Vb and GnT-V after typical elongation by additional glycosyltransferases. The GlcNAc residue transferred by each enzyme is highlighted in red.
A common feature of all these genetic deficiencies is the aberrant or hypo-glycosylation of α-dystroglycan (9–11), a cell surface glycoprotein that binds the transmembrane β-dystroglycan (31–34). α-Dystroglycan binds with high affinity to several extracellular matrix components, laminin, agrin, and neurexin (32, 35–38), and it is heavily glycosylated with O-linked mannosyl glycans (39–43). Mutations in POMT1, POMT2, POMGnT1, and LARGE lead to altered glycosylation of α-dystroglycan with abolished laminin binding activity and are believed to be a major underlying mechanism of muscular dystrophy and type II lissencephaly (10, 44–46). In addition, there is now evidence that glycoproteins other than α-dystroglycan express O-mannosyl glycans, because a mouse model that lacks α-dystroglycan expression shows little alteration in the expression pattern of glycans containing O-Man (47).
Some O-mannosyl glycans in vertebrate brain are clearly modified and extended from the O-Manβ(1,2)-GlcNAc disaccharide; however, the function of these structures and the enzymes that synthesize them are unclear. A recent study identified a unique, extended O-Man glycan located in the non-mucin domain of α-dystroglycan that is essential for binding to laminin (30). Recent work has shown that the glycosyltransferase N-acetylglucosaminyltransferase Vb (GnT-Vb, also referred to as GnT-IX) branches the α-mannose in the O-linked trisaccharide, O-Manβ(1,2)GlcNAc, with a β1,6-linked GlcNAc (Scheme 1) (48–52). GnT-Vb is therefore responsible for the branching of at least some O-mannosyl linked glycans, and these branched structures are clearly found in significant amounts in rabbit brain. Our work has shown that in two cell types GnT-Vb overexpression decreased adhesion to and increased motility on laminin (48, 51, 53). Moreover, in a neuroblastoma cell line, these effects were mediated through the protein receptor tyrosine phosphatase ζ (RPTPζ, also known as RPTPβ), a novel substrate glycoprotein for O-mannosyl glycosylation (48). In addition, we determined that the monoclonal antibody Cat-315 detects the O-mannosyl glycan on RPTPζ synthesized by GnT-Vb.
In this study, we explored the expression and function of GnT-Vb in the nervous system to investigate the role it plays during neural development, and our initial results showed that GnT-Vb is specifically and highly expressed in both developing and adult nervous systems. To determine whether its deletion produces phenotypes similar to those seen after the deletion of other enzymes that function in the O-mannosyl glycosylation pathway, and to assess further its function, we generated an animal in which GnT-Vb was genetically ablated. In the brains of these animals, we document a dramatic reduction in β1,6-branched O-mannosyl structures using newly developed linear ion-trap mass spectrometry methods for analysis of released, permethylated glycans. The enzyme, GnT-V, is known to synthesize N-linked glycans by transferring GlcNAc in β1,6-linkage to Man (Scheme 1), but its ability to branch O-mannosyl glycans in vivo is not known. We also show that in the absence of GnT-Vb, GnT-V can synthesize low levels of some branched O-mannosyl glycans. In the absence of GnT-V, however, GnT-Vb does not branch N-linked glycans.
EXPERIMENTAL PROCEDURES
General Methods and Buffers
Restriction enzyme digests, DNA ligations, PCR, Southern blotting, Northern blotting, and other recombinant DNA procedures were performed using standard protocols. All DNA constructs were verified by DNA sequencing, which was performed by Integrated Biotech Labs, University of Georgia.
RNA Extraction and RT-PCR Analysis
Total RNA was isolated from the brain tissues of GnT-V(+/+)Vb(+/+), GnT-V(−/+)Vb(−/+), GnT-V(−/−)Vb(+/+), GnT-V(+/+)Vb(−/−), and GnT-V(−/−)Vb(−/−) mice using a TRIzol reagent (Invitrogen) followed by treatment with RNase-free DNase (Promega). Semiquantitative real time one-step reverse transcription (RT)-PCR was performed using the Superscript III first-strand synthesis system (Invitrogen). The Platinum Blue PCR SuperMix (Invitrogen) was employed to amplify a 319-bp GnT-Vb cDNA region containing exon 6 and 7 with primers JKL103 (5′-CCTCTGACCCCTGCTACGCCTTCTTTG-3′) and JKL33(5′-TTGCTTCTGGTCCCTCCGAATGTCCCC-3′). GnT-V expression was also analyzed with primer JKL51 and JKL24. The PCR products were analyzed by agarose gel electrophoresis. Primer sequences used for genotyping and RT-PCR are JKL33 (5′-TTGCTTCTGGTCCCTCCGAATGTCCCC-3′), JKL91 (5′-GAATCCAGTACAGGGAACTTGCTTGG-3′), JKL92 (5′-ATGTGTGCGAGGCCAGAGGCCACTTGT-3′), JKL93 (5′-CTGGGGACCCAATCAGCCCTGAATCAC-3′), JKL97 (5′-GACTGCATGATGTCGGTGCATGAAGAG-3′), and JKL103 (5′-CCTCTGACCCCTGCTACGCCTTCTTTG-3′).
L-PHA Blotting
Brain tissue samples from adult mice were homogenized in extraction buffer (50 mm MES, pH 7.4, 150 mm NaCl, 1% Triton X-100) using a Dounce homogenizer. A protein inhibitor mixture complete Mini (Roche Diagnostics) was added. The concentration of proteins in the homogenate was established by the BCA method. 50 μg of proteins were added to each lane of 7.5% gels (SDS-PAGE). Proteins were blotted to a PVDF-membrane in a tank blotter. The membranes were blocked in TBS, 0.1% Tween 20 containing 5% dry milk and incubated overnight at 4 °C. The membranes were probed with 0.5 mg/ml L-PHA (Vector Laboratories), followed by incubation with a rabbit antibody against L-PHA (1:1,000 dilution) and with HRP-conjugated goat antibody against rabbit IgG (Santa Cruz Biotechnology).
Mouse Brain and Sample Preparation
All mice were used at an average of 8 weeks old, and these mice were killed by CO2 asphyxiation and rapid decapitation. The brain was removed and isolated on ice. All brains for this study were immediately frozen at −80 °C until glycomic analysis. The frozen whole brains were homogenized and delipidated by extraction twice for 3 h at room temperature on the rocker with a mixture of chloroform/methanol/water (4:8:3, v/v/v) as described (54, 55). The emulsion was centrifuged at 2,800 × g for 15 min at 4 °C to remove the supernatant. The pellets were resuspended in the solvent of acetone/water (10:1, v/v) mixture and incubated on ice for 15 min to wash the pellets twice. The protein pellets were collected by centrifugation and dried on the heating module at 45 °C with the mild nitrogen stream (Reacti-ThermTM, Pierce). The dried protein powder was weighed and stored at −20 °C until analyzed.
Preparation of N-Linked Glycans
3 mg of the mouse brain protein powder was resuspended in 200 μl of 40 mm ammonium bicarbonate (NH4HCO3) by sonication and boiled at 100 °C for 5 min. After cooling to room temperature, 25 μl of trypsin (2 mg/ml in 40 mm NH4HCO3, Sigma) and chymotrypsin (2 mg/ml in 40 mm NH4HCO3, Sigma), respectively, was added. The samples were denatured with 250 μl of 2 m urea in 40 mm NH4HCO3 and incubated overnight (18 h) at 37 °C. After digestion, the peptide samples were centrifuged, and 10 μl of supernatant was collected for protein assay. The peptide amounts were measured by means of a micro-BCA (bicinchoninic acid) protein assay kit (Pierce). The samples were boiled at 100 °C for 5 min and acidified by 500 μl of 10% acetic acid (AcOH) to deactivate proteases. The samples were loaded onto the equilibrated C18 extraction column (BakerBondTM, Mallinckrodt Baker), washed three times with 1 ml of 5% AcOH, and eluted stepwise by 1 ml of 20% isopropyl alcohol in 5% AcOH, 40% isopropyl alcohol in 5% AcOH, and 100% isopropyl alcohol, respectively. The resulting glycopeptides were dried down in a SpeedVac, resuspended in 48 μl of 1× reaction buffer of N-glycosidase F (PNGase F) and 2 μl of PNGase F (Glyko®, Prozyme), and then allowed to incubate for 18 h at 37 °C. Following PNGase F digestion, released oligosaccharides were separated by the C18 extraction column. The mixture was reconstituted to 5% AcOH and loaded onto the equilibrated C18 extraction column. The N-linked oligosaccharides were eluted three times with 1 ml of 5% AcOH and collected as purified N-linked glycans and dried down in a SpeedVac for permethylation processes.
Preparation of O-Linked Glycans
O-Linked oligosaccharides were released by reductive β-elimination and were purified by cation exchange resin as follows. 3 mg of delipidated brain powder was weighted and transferred into a clean glass tube. 500 μl of 50 mm sodium hydroxide (NaOH) and 500 μl of alkaline borohydride solution (a mixture of 2 m sodium borohydride (NaBH4, Sigma) in 50 mm NaOH) were added in a sample tube. The mixture was incubated for 16 h at 45 °C on the heating block, and the reaction was stopped by addition of 10% AcOH with vortexing. The acidified mixture was loaded on an equilibrated cation exchange resin cartridge (AG-50W-X8, Bio-Rad) with 5% AcOH. O-Linked glycans were eluted with 6 ml of 5% AcOH and dried down in a SpeedVac. The sample was resuspended in 1 ml of a methanol/glacial acetic acid (9:1, v/v) solution and dried on the heating module at 45 °C with the mild nitrogen stream to remove borates for permethylation. This step was repeated twice.
Permethylation of Glycans
To facilitate analysis of oligosaccharides by mass spectrometry (MS), the released oligosaccharide mixtures were permethylated as described previously (56). Briefly, glycans were suspended in 200 μl of anhydrous dimethyl sulfoxide (DMSO) and 250 μl of the fresh dehydrated NaOH/DMSO reagent (mixture of 50 mg of NaOH in 2 ml of anhydrous DMSO). After sonication and vortexing in nitrogen gases, 100 μl of iodomethane (CH3I, Sigma) was added, and the mixtures were vortexed vigorously for 5 min. 2 ml of distilled water was added, and the excess iodomethane was removed by bubbling with a nitrogen stream, and 2 ml of dichloromethane (CH2Cl2, Sigma) was added. After vigorous mixing and phase separation by centrifugation, the upper aqueous layer was removed and discarded. The nonpolar organic phase was then extracted four times with distilled water. Dichloromethane was evaporated on the heating module at 45 °C with the mild nitrogen stream. The permethylated glycans were dissolved in adjusted volumes (15–30 μl) of 100% methanol from the results of protein assays for mass spectrometric analysis.
Mass Spectrometric Analysis
For mass spectrometry analysis with five different genotypes of mouse brains performed in duplicate, permethylated glycans were dissolved in a total of 50 μl of sample with 15 μl of sample in 100% methanol plus 35 μl of 1 mm NaOH in 50% methanol and infused directly into a linear ion trap mass spectrometer (LTQ XL, Thermo Fisher Scientific Inc.) using a nanospray ion source with a fused-silica emitter (360 × 75 × 30 μm, SilicaTipTM, New Objective) at a 2.0-kV capillary voltage, 200 °C capillary temperature, and a syringe flow rate of 0.4 μl/min. Full ion trap mass spectrometry spectra in positive ion and profile mode were collected at 400–2000 m/z for 30 s with 5 microscans and a 150 maximum injection time (ms). The centroid MS/MS spectra following collision-induced dissociation were obtained from 400 to 2000 m/z at 34 and 28% normalized collision energy for N- and O-linked glycans respectively, 0.25 activation Q, and 30.0-ms activation time by total ion mapping. Parent mass step size and isolation width were set at 2.0 m/z and 2.8 m/z, respectively, for automated MS/MS spectra with total ion mapping scans. MS3 experiments in the LTQ XL were manually carried out in profile mode with the same instrumental parameters as described above for tetraantennary N-linked glycan structure. Glycan precursor ions were isolated for MS3 using an isolation width of 2.5 m/z.
In Situ Hybridization
Frozen brains sectioned coronally or tangentially (15–20 μm thick) were thaw-mounted onto gelatin-coated slides and postfixed in 0.1 m sodium phosphate-buffered 4% paraformaldehyde, pH 7.4. Sections were rinsed in PBS and 2× SSC and acetylated with 0.5% acetic anhydride in 0.1 m triethanolamine, pH 8.0. Sections were rinsed in 2× SSC and PBS, dehydrated in ethanols, and delipidated in chloroform. Sections were prehybridized in 2× SSC and 50% formamide at 55 °C for 1 h. Sections were then hybridized in 0.75 m NaCl, 50% formamide, 1× Denhardt's solution, 10% dextran sulfate, 30 mm dithiothreitol (DTT), 10 mm Tris-HCl, pH 7.5, 1 mm EDTA, 100 μg/ml salmon sperm DNA, 0.5 mg/ml yeast tRNA, and 1.5 × 106 cpm probe per slide for 12–15 h at 55 °C. 33P-Labeled probes were synthesized using an SP6/T7 transcription kit (Hoffmann-La Roche). After hybridization, the sections were washed in 2× SSC, 50% formamide, and 0.1% β-mercaptoethanol at 55 °C for 1 h and treated with 20 μg/ml RNase A in 0.5 m NaCl and 10 mm Tris-HCl, pH 8.0, at 37 °C for 30 min. The sections were then washed in 2× SSC, 50% formamide, and 0.1% β-mercaptoethanol at 60 °C for 30 min and in 0.1× SSC and 0.1% β-mercaptoethanol at 65 °C for 30 min and dehydrated. The slides were exposed to film (Kodak BioMax MR Film) for 2–3 days.
Western Blot Analysis
Brain tissue samples were homogenized in 10 volumes of 25 mm Tris-HCl, pH 7.4, containing protease inhibitor mixture (Complete, EDTA-free, Roche Diagnostics). Aliquots were equilibrated at a final total protein concentration of 1–2 mg/ml in 40 mm Tris-HCl, 40 mm sodium acetate, pH 8.0, containing 5 mm EDTA and treated with 0.25 units/ml of protease-free chondroitinase ABC (Seikagaku, Tokyo, Japan) for 8 h at 37 °C as indicated. Chondroitinase activity was stopped by boiling the samples in the presence of SDS-PAGE gel-loading buffer. Samples (10 μg of total protein) were electrophoresed on reducing 6% SDS-polyacrylamide gels and analyzed by Western blotting. For semiquantitative analysis, immunoblots were developed by chemiluminescence, and the integrated optical density Cat-315 reactive bands were analyzed using the gel analysis package in Image J. Optical density ratios (normalized to brevican expression) were then normalized to expression relative to wild type samples and analyzed by Student's t tests.
Laminin Overlay Assay
Laminin overlay assays were conducted as described by Liu et al. (46). Briefly, to enrich for α-dystroglycan, tissue homogenates were incubated with wheat germ agglutinin-agarose (Ey Laboratories, San Mateo, CA) for 4 h. The samples were then centrifuged and extensively washed, and the protein was eluted with SDS-PAGE loading buffer. Samples were then separated by SDS-PAGE and blotted onto nitrocellulose. IIH6C4 reactivity was detected using our standard Western blotting protocol. For laminin-overlay studies, membranes were incubated with 1.25 μg/ml laminin-1 (Invitrogen) in TBST with 1 mm CaCl2 and 1 mm MgCl2 overnight at 4 °C. Subsequently, the membranes were washed extensively in the same buffer, and bound laminin was detected by standard Western blotting protocols.
Nissl Staining and Stereology
Serial 40- μm frontal sections (20 sections/mouse) spanning the barrel cortex region were subjected to Nissl staining. Sections from animals were matched using histological landmarks (beginning of the corpus callosum), and every 10th section was imaged and analyzed. Stereological analysis was carried out essentially as described (54).
RESULTS
Expression of GnT-Vb Transcripts in Mouse Embryos
Initial quantitative RT-PCR studies showed high levels of GnT-Vb expression in brain and testis, with little or no expression in other human adult tissues. By contrast, GnT-V expression in human adult tissues is relatively ubiquitous, including expression in the nervous system. To initially survey the function of GnT-Vb during development, we first performed in situ hybridizations. GnT-Vb is expressed very early in nervous system development (E7.5, data not shown) and continues to be present with relative specificity in the nervous system throughout adulthood (Fig. 1). Images of in situ hybridizations are shown at different stages of embryonic development from E9 to adult. Early in neural development, GnT-Vb is expressed throughout the growing neuroepithelia. At later stages of development, coincident with neurogenesis, however, GnT-Vb becomes somewhat more restricted in its pattern of expression. Detailed analysis of the developing forebrain shows the expression of GnT-Vb is relatively absent from the ventricular zone but is particularly highly expressed in the subventricular zone into the intermediate zone. In the postnatal brain, GnT-Vb is broadly expressed, but it is highly enriched in certain areas, nuclei and pathways. GnT-Vb is particularly highly expressed in the hippocampus, superficial layers of the cortex, the striatum, nucleus accumbens, a subset of nuclei in the thalamus, inferior colliculus, pontine nucleus in the brain stem, and the rostral migratory stream into the olfactory bulb. GnT-V, by contrast, is also highly expressed in the nervous system, but its expression is not restricted to neural tissues. GnT-V expression in the brain is generally broader and less spatially restricted; for example, it is expressed throughout the cerebral cortex and in the hippocampus. Overall, our data show that GnT-Vb expression is associated with some, but certainly not all, zones of active cell migration in the embryonic forebrain. High levels of GnT-Vb expression are maintained in the mouse nervous system in a distinct, regionally restricted manner.
FIGURE 1.

GnT-Vb is specifically and highly expressed in the developing and adult nervous system. In situ hybridization analyses were conducted on embryos and adult brains to determine the timing and localization of GnT-Vb mRNA. GnT-Vb expression was first detected at embryonic day (E) 7.5 (data not shown), but at all time points it was found almost exclusively within the nervous system. Expression was detected specifically in the nervous system of embryos at E9 (A), E11 (B), and E13 (C). At E15 the expression of GnT-Vb message is shown (D) and the same section was then Nissl-stained (E) to demonstrate the specific localization of GnT-Vb mRNA in neural tissue. F–H, brains from an E15 mouse was removed and sliced horizontally to investigate the spatial distribution of GnT-Vb in the brain. GnT-Vb mRNA (F) and fluorescent Nissl stain (G) of the same section were overlaid (H) to show that GnT-Vb is highly expressed in the subventricular zone (SVZ) and intermediate zone (IZ) and more sparsely in the marginal zone and cortical plate (MZ/CP). It is largely absent from the ventricular zone (VZ). Adult brains were sectioned parasagittally to investigate the spatial distribution of GnT-Vb (I and J) and GnT-V (K). GnT-Vb is highly expressed in the inferior colliculus (IC), nuclei in the thalamus (Thal), the rostral migratory stream (RMS), the dentate gyrus (DG), CA fields of the hippocampus (CA), the striatum and superficial layers of the cortex (I and J). In contrast GnT-V is more broadly expressed throughout the brain although it is largely absent from the striatum (K).
Generation of GnT-Vb Null Mice
To dissect further the function of GnT-Vb, we generated a conditional null mouse using standard cre-lox technology (supplemental Fig. 1). The GnT-Vb conditional mice were crossed to transgenic mouse of ubiquitously expressing Cre and resulted in GnT-Vb null mouse. Crossing GnT-Vb null mice into the GnT-V null background yielded a mouse that was null for both GnT-V and GnT-Vb. When transcripts of GnT-Vb, GnT-V, and control genes were examined in adult brains of animals from these backgrounds (Fig. 2), the transcript expression results for each mouse showed the expected patterns. Lack of expression of either GnT-V or GnT-Vb did not appear to affect the transcript expression of its paralog. Measurements of GnT-V activity using a synthetic trisaccharide acceptor agreed with the transcript results (data not shown).
FIGURE 2.

RT-PCR analysis of different genotypes of GnT-V and GnT-Vb K/O mice. Total RNA was isolated from the brain tissues of GnT-V(+/+)Vb(+/+), GnT-V(−/+)Vb(−/+), GnT-V(−/−) Vb(+/+), GnT-V(+/+)Vb(−/−), and GnT-V(−/−)Vb(−/−) mice. Semi-quantitative, real time, one-step reverse transcription (RT)-PCR was performed to amplify a 319-bp GnT-Vb cDNA region containing exons 6 and 7 as described under “Experimental Procedures.” GnT-V expression was analyzed as described and showed a 119-bp PCR product. −RT, no reverse transcriptase reaction. G6PDH (glutaraldehyde-6-phosphate dehydrogenase) PCRs were used as control.
Binding of L-PHA to Brain Glycoproteins
The lectin, L-phytohemagglutinin (L-PHA), shows binding specificity for the N-linked β1,6-glycan product of GnT-V that terminates with a β1,4-galactose (Scheme 1). Therefore, it was of interest to determine how deletion of GnT-Vb or GnT-V affected the binding of L-PHA to endogenous brain glycans. Fig. 3 shows that deletion of GnT-V results in a significant reduction in biotinylated L-PHA binding to Western blots of mouse brain glycoproteins following SDS-PAGE. By contrast, deletion of GnT-Vb had little, if any, effect on L-PHA binding. These results demonstrate that GnT-V, and not -Vb, synthesizes glycans that are bound by L-PHA.
FIGURE 3.
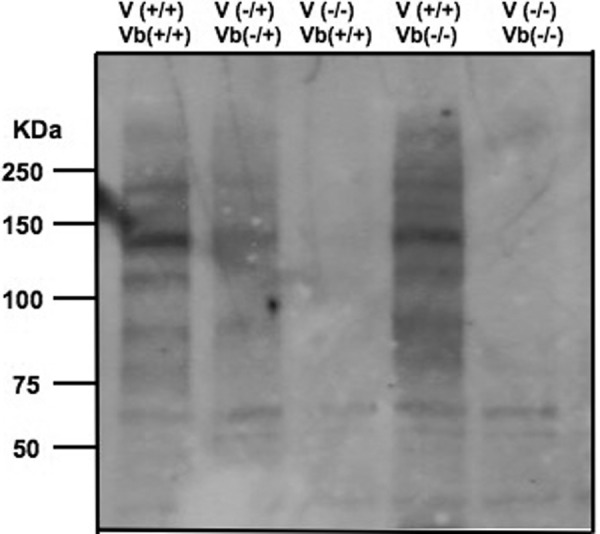
L-PHA-reactive glycoproteins of brain tissue homogenates. Brain tissue samples from adult mice were homogenized in extraction buffer. The proteins were subjected to SDS-PAGE and were blotted to a PVDF membrane. The blot was probed with L-PHA followed by incubation with rabbit IgG against L-PHA and HRP-conjugated goat anti-rabbit IgG.
Relative Expression of N-Linked and O-Linked Glycans
The use of NSI-LTQ-MSn analysis of permethylated glycans allows the identification of glycan structures using fragment ion identification. To determine how deletion of GnT-Vb and GnT-V, separately and together, affected glycan expression, we utilized a recently developed methodology of glycoprotein extraction from adult mouse brain, N-glycan and O-glycan release, followed by permethylation and glycan mass spectrometric analysis (47). The results from the NSI-LTQ-MS/MS analysis of O-linked trisaccharides to heptasaccharides from GnT-V(+/+)Vb(+/+), GnT-V(−/+)Vb(−/+), GnT-V(−/−)Vb(+/+), GnT-V(+/+)Vb(−/−), and GnT-V(−/−)Vb(−/+) mouse brain tissues are shown in supplemental Fig. 2, A–F and are summarized in Table 1. The permethylated O-linked trisaccharide O-Man-(GlcNAc)2-Gal, m/z 779.415, cannot be readily distinguished from the O-linked trisaccharide O-GalNAc-[GlcNAc]-Gal; as expected, a species with this m/z is present in brain tissue from five mouse genotypes. The species with an m/z of 983.515, corresponding to an O-Man tetrasaccharide with two GlcNAc residues and one Gal residue, is present in wild type, heterozygote, and GnT-V null mice, but it is completely absent (<5%, compared with homozygous wild type) from GnT-Vb null and double null brains. This result is consistent with the hypothesis that GnT-Vb encodes the enzymatic activity that transfers the second GlcNAc residue onto the O-Man-GlcNAc disaccharide; as seen in Table 1, a species in which Gal is present on one of the GlcNAc residues to make a tetrasaccharide is identified. (All O-linked structures observed are shown in supplemental Table 1.) A similar absence of structures was observed in the GnT-Vb null and double null brains for the structures corresponding to this tetrasaccharide with a single Fuc residue attached, m/z 1157.604, and a pentasaccharide with two Gal attached, m/z 1187.615. Interestingly, structures corresponding to the next three higher m/z values were observed in the GnTV(+/+)Vb(−/−) brains, with relative amounts ranging from <10 to <60% compared with wild type, but these structures were all absent in the double null brains. The heptasaccharide product (Table 1, number 8) with two sialic acids could be detected by analysis of fragmented ions, but it was too low to allow relative quantification. The trisaccharide was efficiently galactosylated and then was observed with either one sialic acid or fucose. In the absence of GnT-V and GnT-Vb, however, all these O-mannosylated products were not observed. Taken together, these results are consistent with the conclusion that in the absence of GnT-Vb, GnT-V can transfer very low levels of GlcNAc to the O-Man-GlcNAc disaccharide and synthesize branched O-Man structures, which can then be extended by other glycosyltransferases.
TABLE 1.
Specific O-linked oligosaccharides identified by MS/MS mass spectrometry in brain tissues of various genotypes
The relative amounts compared with wild type were calculated as follows: x, <0.05.

a Data are not unique.
b The signals were too low for accurate relative quantification. sc means charged. Red triangle indicates Fuc; green circle indicates Man; yellow circle indicates Gal; blue square indicates GlcNAc; yellow square indicates GalNAc, and red diamond indicates NeuAc.
Next, signature m/z ions corresponding to various tetraantennary N-linked glycans were examined to obtain information on the relative abundance of structures that require the activity of GnT-V for synthesis. The supplemental Fig. 3, A–C shows representative spectra obtained from permethylated PNGase F-released glycans from mouse brains prepared from mice with the five genotypes mentioned above, and Table 2 summarizes the results from these data on N-linked glycan expression. Four signature ions were identified as being diagnostic for tetraantennary glycans that would require the activity of GnT-V or GnT-Vb, and the results show that in the absence of GnT-V, none of the four tetraantennary signature ions could be detected (Table 2). (All N-linked structures observed are shown in supplemental Table 2.) In the absence of GnT-Vb, by contrast, all these tetraantennary structures were observed. These results are consistent with the conclusion that when GnT-V is not expressed, GnT-Vb cannot compensate for the lack of GnT-V activity and synthesize N-linked glycans.
TABLE 2.
Specific N-linked oligosaccharides identified by MS/MS mass spectrometry in brain tissue of various genotypes
O indicates observed; X indicates below level of detection (<0.05 relative to wild type); dc indicates doubly charged; tc indicates triply charged; ( ), denotes triply charged species.
| No. | N-Linked oligosaccharide composition | m/z (mono), [M + Na]* | V(+/+) Vb(+/+), dc(tc) | V(−/+) Vb(−/+), dc(tc) | V(−/−) Vb(+/+),dc(tc) | V(+/+) Vb(−/−), dc(tc) | V(−/−) Vb(−/−), dc(tc) |
|---|---|---|---|---|---|---|---|
| 1 | (Gal)1(GlcNAc)5(Man)3(GlcNAc)2(Fuc)1 | 2775.405 | O | O | X | O | X |
| 2 | (NeuAc)3(Gal)4(GlcNAc)4(Man)3(GlcNAc)2(Fuc)1 | 4226.099 | (O) | (O) | (X) | (O) | (X) |
| 3 | (NeuAc)4(Gal)4(GlcNAc)4(Man)3(GlcNAc)2 | 4413.183 | (O) | (O) | (X) | (O) | (X) |
| 4 | (NeuAc)4(Gal)4(GlcNAc)4(Man)3(GlcNAc)2(Fuc)1 | 4587.273 | (O) | (O) | (X) | (O) | (X) |
Expression of Cat-315 in GnT-Vb Knock-outs and GnT-Vb and -V Double Knock-outs
Our previous work demonstrated that the monoclonal antibody Cat-315 detects a carbohydrate epitope on RPTPζ early in development (48). Cat-315-reactive punctae pre-patterned the surface of neurons prior to synaptogenesis and seemed to define nonsynaptic or perisynaptic sites on the surfaces of the cells. Several pieces of circumstantial evidence led us to postulate that Cat-315 detects an O-mannosyl-linked glycan on RPTPζ that is positive for the HNK-1 epitope, including results which showed that overexpression of GnT-Vb dramatically increased Cat-315 reactivity with RPTPζ after immunoblotting. Therefore, in this study, we hypothesized that elimination of GnT-Vb would specifically reduce Cat-315 reactivity, reflecting altered glycosylation of RPTPζ. To test this hypothesis, we performed immunoblot analysis of extracts from brains of GnT-Vb, GnT-V, and GnT-Vb and -V double knock-out animals. For these studies, whole brains from wild type and knock-out animals were analyzed for reactivities with a set of known anti-RPTPζ antibodies. These experiments showed that when RPTPζ was analyzed in GnT-Vb knock-out brains, the Cat-315-reactive O-mannosyl glycans on RPTPζ are significantly reduced; however, they are not completely eliminated (Fig. 4). We next investigated the source of the remaining Cat-315 reactivity on RPTPζ in the GnT-Vb null brains. We envisioned two possible explanations. First, the remaining reactivity could be due to compensation by GnT-V, which could elaborate some O-mannosyl glycans in the absence of GnT-Vb. Second, it is possible that the Cat-315-reactive glycan could be present to some extent on both the β1,2- and β1,6-branches of the elaborated O-mannosyl glycan. To distinguish between these two possibilities, we examined the reduction in Cat-315 reactivity in GnT-V/GnT-Vb double knock-outs. Interestingly, we found no obvious further reduction in Cat-315 reactivity. These data indicate that GnT-V is not contributing to synthesis of the Cat-315 epitope in the absence of GnT-Vb. The Cat-315 epitope on O-mannosyl glycans is likely present on the β-1,2 branch, at least when the β-1,6 branch is absent.
FIGURE 4.

Western blot analysis reveals that genetic disruption of GnT-Vb reduces the expression of the Cat-315 epitope on RPTPζ. A, postnatal day 0 (P0) brains were analyzed by Western blot analysis with known antibodies against RPTPζ, including 3F8, H300, and Cat-315. H300 detects the RPTPζ protein core, whereas 3F8 and Cat-315 detect specific carbohydrate epitopes on RPTPζ at this age. Brevican (Bcan) was detected to control for protein loading. B, semiquantitative analysis of Cat-315 reactivity from wild types (Wt) and knock-outs (KO). The Cat-315 reactivity relative to brevican has been analyzed as described under “Experimental Procedures.” The combined group of single or double knock-outs were used for KO because they did not differ from each other. n = 4 for wild types and 6 for knock-outs (2 of each genotype).
Analysis of α-Dystroglycan Glycosylation and Laminin Binding in Wild Type and GnT-Vb and -V Double Knock-outs
Previous work showed that in animal models in which O-mannosyl glycosylation is altered, such as POMGnT1 knock-out animals, that staining of α-dystroglycan with glycan-specific antibody IIH6C4 is lost as is the ability of α-dystroglycan to bind laminin (46). To test if a similar alteration were found in the GnT-V and -Vb double knock-out, we performed Western blot analysis with the IIH6C4 antibody and laminin overly assays (Fig. 5). These studies showed that there is no alteration in the reactivity of α-dystroglycan with IIH6C4 or any change in its affinity for laminin. Therefore, it is likely that glycosylation of α-dystroglycan is not functionally altered in the GnT-V and -Vb double knock-outs.
FIGURE 5.

Analysis of α-dystroglycan glycosylation and laminin overlay assay. Western blot and overlay analysis indicates that double deletion of GnT-V and GnT-Vb does not alter the reactivity of α-dystroglycan to the 116C4 carbohydrate-specific epitope or its ability to bind laminin. Wheat germ agglutinin-enriched samples from wild type (+/+, +/+) or GnT-V and -Vb double knock-out (−/−, −/−) brains were analyzed by Western blot and laminin overlay assays. Each two mice brain tissues were applied. The IIH6C4 antibody detects a specific carbohydrate epitope that is lost from α-dystroglycan in many forms of CMDs and animal models in which O-mannosylation is disrupted. It is, however, unaffected by the absence of GnT-Vb and -Va. The ability of α-dystroglycan to bind laminin seems similarly unaffected.
Neural Development and Function Are Not Obviously Perturbed in GnT-Vb and -V Double Knock-outs
We next examined if there were any morphological differences between the brains of knock-out animals. Surprisingly, we found no quantifiable anatomic changes in the brains of GnT-Vb and -V and double knock-out mice compared with wild type controls. Nissl-stained coronal sections from representative wild type, heterozygote, and knock-out animals showed that there were no reproducible, obvious differences in brain morphology, cell number, or lamination (Fig. 6).
FIGURE 6.

Nissl-stained brain sections from GnT-Vb and -V double knock-outs show no obvious malformations or changes in cell number or architecture. Thin (3 μm) sections from wild type (Wt) and GnT-Vb and -V double knock-outs were analyzed histologically and using stereological cell counts to determine whether loss of these two enzymes alters brain structure. We found no obvious changes in the morphology of the brain and therefore no changes in the cell number or cellular organization of the brain. These data argue that GnT-Vb and GnT-V are relatively dispensable for normal brain development. Modification of O-mannosyl glycans by these enzymes does not contribute any phenotypes similar to those found in CMDs.
DISCUSSION
Expression of GnT-Vb Transcripts in Mouse Embryos
Our in situ hybridization analyses showed that early in development GnT-Vb is expressed relatively specifically and highly in the neuroepithelia. This is an area of active cell proliferation, differentiation, and motility in the embryonic brain and suggestive that GnT-Vb may play a role in one or more of these processes. A recent study has also demonstrated that the brain-specific expression of GnT-Vb (IX) can be epigenetically regulated and that two regulatory proteins, NeuroD1 and CTCF, activate the promoter of this enzyme (57). This seminal study documented for the first time that the expression of a glycosyltransferase is epigenetically regulated, underscoring the complex and precise regulation of GnT-Vb expression.
Work from several laboratories has confirmed that brain abnormalities in some congenital muscular dystrophies are often the result of disruption of the normal developmental process of migration due to aberrant O-mannosyl glycosylation. In particular, the hypoglycosylation of α-dystroglycan leads to fragility of the pial basement membrane and over-migration of developing neurons past this normally limiting membrane (11, 58–69). The timing and localization of GnT-Vb raise the possibility that it could also contribute to these disorders. Consistent with this hypothesis, we demonstrated previously that overexpression of GnT-Vb in a neuroblastoma cell line decreased adhesion to and increased motility on laminin (51). However, this study found no obvious changes in the migration or positioning of neurons in either GnT-Vb null or GnT-V/GnT-Vb double null brains. These data suggest that these enzymes are dispensable for normal neuronal migration and brain development and that these enzymes likely do not contribute to the pathogenesis of CMDs.
In Vivo Function of GnT-Vb in Synthesis of β1,6-Branched Brain Glycans
The discovery of a paralog of GnT-V immediately suggested that it could be involved in the synthesis of O-mannosylated β1,6-branched glycans whose presence had been documented in vertebrate brain. Although ubiquitous in yeast, fungi, and other nonvertebrates, until only the last 10 years have the contributions of O-Man-containing glycans in vertebrate tissues been investigated. Structural studies have shown that O-Man-containing glycans constitute 30% of O-linked glycans in rabbit brain, and the results from our laboratories using mouse brain confirm this proportion. In both rabbit and mouse brain, a substantial fraction of the O-mannosylated glycans contain a GlcNAc-β1,6-Man. Taken together with the results of this study, which demonstrates that GnT-Vb is likely responsible for nearly all of the brain O-linked β1,6-branched mannose, it is clear that GnT-Vb in vivo modifies substantial amounts of the Man-β1,2-GlcNAc glycans throughout the brain. Evidence also suggests some of these products of GnT-Vb are further extended with poly-N-acetyllactosamine repeats, as well as the HNK-1 epitope.
Although GnT-V and GnT-Vb show 45% identity and 53% similarity at the amino acid level, our data clearly illustrate that GnT-Vb cannot serve the role of GnT-V in N-linked glycan synthesis and that GnT-V can partially, but not completely, serve the role of GnT-Vb in O-Man branching in vivo in the absence of GnT-V activity. The structure/function relationships of these two enzymes that underlie these findings are not understood and are an avenue of future exploration.
A recent study used mass spectrometry to study O-Man glycans in mice that were α-dystroglycan null. The conclusion from these experiments was that there was little overall change in the total amount of O-Man glycans when α-dystroglycan was not expressed, suggesting that the majority of this class of glycans is actually found expressed on glycoproteins other than α-dystroglycan. Our previous in vitro experiments identified RPTPζ as a potential target of the O-mannosylation pathway (48). Here, we provide the first in vivo evidence that RPTPζ is indeed a physiological substrate of the O-mannosylation pathway. We show that Cat-315 reactivity detects the elaboration of O-mannosyl glycans on RPTPζ and that this reactivity is reduced in knock-out animals. The in vitro studies showed that overexpressing GnT-Vb in neuroblastoma cells resulted in altered association of RPTPζ on the cell surface, which inhibited its phosphatase activity, resulting in destabilized E-cadherin and inhibition of cell-cell adhesion, measured by either aggregation or migration assays (48). It will be interesting to determine whether other members of the RPTP family express O-Man glycans and if their functions are altered when specific O-Man glycans are deleted or overexpressed.
Our results concerning expression of the Cat-315 epitope showed in the GnT-Vb knock-outs or double knock-outs that reactivity is reduced but not lost. This result suggests that the β1,2-branch likely also expresses distal glycans that makes the O-Man glycan reactive for Cat-315. In the absence of GnT-Vb activity in the knock-out animals, it is clearly possible that the more distal sugars, such as the HNK-1 epitope and poly-N-acetyllactosamine, may then show increased expression on the β1,2-GlcNAc. In this case, the functions of RPTPζ could be maintained by this O-mannosyl branch, resulting in the lack of altered function. Future experiments will test this hypothesis.
Focusing specifically on α-dystroglycan function, the unique phosphorylated O-mannosyl glycan described by Yoshida-Moriguchi et al. (30) that is expressed on α-dystroglycan is essential for its binding to laminin. This glycan contains a Ser/Thr-O-Man with a phosphate group on its 6′-OH, as well as a GlcNAc-linked β1,2. The phosphate group is normally in a diester linkage with a moiety that is the product of the LARGE and whose chemical structure is, as yet, not fully elucidated. Because GnT-Vb transfers GlcNAc to the 6-OH of this O-linked Man, it is possible that GnT-Vb action can inhibit the activity or activities that result in the transfer of the phosphodiester LARGE product, thereby perturbing, and possibly regulating, the adhesion between α-dystroglycan and laminin. Understanding the biosynthesis and regulation of the unique glycan on α-dystroglycan that is responsible for this adhesion is clearly at the forefront of research on the group of congenital muscular dystrophies that are caused by altered glycan expression.
GnT-V knock-out mice have been reported to exhibit several phenotypic differences in immune function and embryonic fibroblast adhesion and migration behaviors, but there are no reports of altered neural function in these animals, despite extensive expression of GnT-V throughout the brain and neural tissues (70–73). Glycan analysis demonstrated that GnT-Vb is primarily responsible for adding the β1,6-branch to O-mannosyl glycans but that GnT-Vb and GnT-V have varying degrees of overlapping or compensatory functions. However, in the absence of both enzymes, the β1,6-branch is eliminated from both O-mannosyl glycans and N-linked glycans, demonstrating that these two enzymes together are responsible for the synthesis of GlcNAc-β1,6-Man linkages. Unexpectedly, however, either elimination of almost all of the branched O-mannosyl glycans in the GnT-Vb knock-out or all branched structures in the double knock-out had no obvious effect on brain morphology, cellular numbers, or architecture of the brain. The specific functions of these GlcNAc-β1,6-Man-branched structures in neural tissue, therefore, remain elusive. Despite not identifying an obvious neural phenotype in these animals, future studies likely will identify neural abnormalities. Future studies will therefore be aimed at determining if the ability to recover from neural insults or damage is impaired in the knock-out mice. Consistent with a role that altered glycosylation of RPTPζ could play in such processes, studies have demonstrated that RPTPζ knock-out animal have impaired recovery after experimental autoimmune encephalitis (74). We cannot presently rule out the possibility that the GnT-Vb animals would be similarly impaired after these types of insults.
Supplementary Material
Acknowledgments
We thank Dr. Hua-Bei Guo and Gail Kelly for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants P41GM103490, RO1CA064462, and RO1NS069660.

This article contains supplemental Experimental Procedures, Figs. 1–3, and Tables 1 and 2.
- CMD
- congenital muscular dystrophy
- GnT-V
- N-acetylglucosaminyltransferase V
- GnT-Vb
- N-acetylglucosaminyltransferase Vb
- RPTPζ
- protein receptor tyrosine phosphatase ζ
- PNGase F
- N-glycosidase F.
REFERENCES
- 1. Dobyns W. B., Kirkpatrick J. B., Hittner H. M., Roberts R. M., Kretzer F. L. (1985) Syndromes with lissencephaly. II. Walker-Warburg and cerebro-oculo-muscular syndromes and a new syndrome with type II lissencephaly. Am. J. Med. Genet. 22, 157–195 [DOI] [PubMed] [Google Scholar]
- 2. Golden J. A. (2001) Cell migration and cerebral cortical development. Neuropathol. Appl. Neurobiol. 27, 22–28 [DOI] [PubMed] [Google Scholar]
- 3. Haltia M., Leivo I., Somer H., Pihko H., Paetau A., Kivelä T., Tarkkanen A., Tom F., Engvall E., Santavuori P. (1997) Muscle-eye-brain disease. A neuropathological study. Ann. Neurol. 41, 173–180 [DOI] [PubMed] [Google Scholar]
- 4. Jimenez-Mallebrera C., Brown S. C., Sewry C. A., Muntoni F. (2005) Congenital muscular dystrophy. Molecular and cellular aspects. Cell. Mol. Life Sci. 62, 809–823 [DOI] [PubMed] [Google Scholar]
- 5. Lian G., Sheen V. (2006) Cerebral developmental disorders. Curr. Opin. Pediatr. 18, 614–620 [DOI] [PubMed] [Google Scholar]
- 6. Parano E., Pavone L., Fiumara A., Falsaperla R., Trifiletti R. R., Dobyns W. B. (1995) Congenital muscular dystrophies. Clinical review and proposed classification. Pediatr. Neurol. 13, 97–103 [DOI] [PubMed] [Google Scholar]
- 7. Ross M. E., Walsh C. A. (2001) Human brain malformations and their lessons for neuronal migration. Annu. Rev. Neurosci. 24, 1041–1070 [DOI] [PubMed] [Google Scholar]
- 8. van der Knaap M. S., Smit L. M., Barth P. G., Catsman-Berrevoets C. E., Brouwer O. F., Begeer J. H., de Coo I. F., Valk J. (1997) Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann. Neurol. 42, 50–59 [DOI] [PubMed] [Google Scholar]
- 9. Grewal P. K., Holzfeind P. J., Bittner R. E., Hewitt J. E. (2001) Mutant glycosyltransferase and altered glycosylation of α-dystroglycan in the myodystrophy mouse. Nat. Genet. 28, 151–154 [DOI] [PubMed] [Google Scholar]
- 10. Michele D. E., Barresi R., Kanagawa M., Saito F., Cohn R. D., Satz J. S., Dollar J., Nishino I., Kelley R. I., Somer H., Straub V., Mathews K. D., Moore S. A., Campbell K. P. (2002) Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 418, 417–422 [DOI] [PubMed] [Google Scholar]
- 11. Takeda S., Kondo M., Sasaki J., Kurahashi H., Kano H., Arai K., Misaki K., Fukui T., Kobayashi K., Tachikawa M., Imamura M., Nakamura Y., Shimizu T., Murakami T., Sunada Y., Fujikado T., Matsumura K., Terashima T., Toda T. (2003) Fukutin is required for maintenance of muscle integrity, cortical histogenesis, and normal eye development. Hum. Mol. Genet. 12, 1449–1459 [DOI] [PubMed] [Google Scholar]
- 12. Chai W., Yuen C. T., Kogelberg H., Carruthers R. A., Margolis R. U., Feizi T., Lawson A. M. (1999) High prevalence of 2-mono- and 2,6-di-substituted manol-terminating sequences among O-glycans released from brain glycopeptides by reductive alkaline hydrolysis. Eur. J. Biochem. 263, 879–888 [DOI] [PubMed] [Google Scholar]
- 13. Finne J., Krusius T., Margolis R. K., Margolis R. U. (1979) Novel mannitol-containing oligosaccharides obtained by mild alkaline borohydride treatment of a chondroitin sulfate proteoglycan from brain. J. Biol. Chem. 254, 10295–10300 [PubMed] [Google Scholar]
- 14. Kogelberg H., Chai W., Feizi T., Lawson A. M. (2001) NMR studies of mannitol-terminating oligosaccharides derived by reductive alkaline hydrolysis from brain glycoproteins. Carbohydr. Res. 331, 393–401 [DOI] [PubMed] [Google Scholar]
- 15. Krusius T., Finne J., Margolis R. K., Margolis R. U. (1986) Identification of an O-glycosidic mannose-linked sialylated tetrasaccharide and keratan sulfate oligosaccharides in the chondroitin sulfate proteoglycan of brain. J. Biol. Chem. 261, 8237–8242 [PubMed] [Google Scholar]
- 16. Beltrán-Valero de Bernabé D., Currier S., Steinbrecher A., Celli J., van Beusekom E., van der Zwaag B., Kayserili H., Merlini L., Chitayat D., Dobyns W. B., Cormand B., Lehesjoki A. E., Cruces J., Voit T., Walsh C. A., van Bokhoven H., Brunner H. G. (2002) Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am. J. Hum. Genet. 71, 1033–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beltran-Valero de Bernabé D., Voit T., Longman C., Steinbrecher A., Straub V., Yuva Y., Herrmann R., Sperner J., Korenke C., Diesen C., Dobyns W. B., Brunner H. G., van Bokhoven H., Brockington M., Muntoni F. (2004) Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet. 41, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brockington M., Blake D. J., Prandini P., Brown S. C., Torelli S., Benson M. A., Ponting C. P., Estournet B., Romero N. B., Mercuri E., Voit T., Sewry C. A., Guicheney P., Muntoni F. (2001) Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am. J. Hum. Genet. 69, 1198–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brockington M., Yuva Y., Prandini P., Brown S. C., Torelli S., Benson M. A., Herrmann R., Anderson L. V., Bashir R., Burgunder J. M., Fallet S., Romero N., Fardeau M., Straub V., Storey G., Pollitt C., Richard I., Sewry C. A., Bushby K., Voit T., Blake D. J., Muntoni F. (2001) Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 10, 2851–2859 [DOI] [PubMed] [Google Scholar]
- 20. Currier S. C., Lee C. K., Chang B. S., Bodell A. L., Pai G. S., Job L., Lagae L. G., Al-Gazali L. I., Eyaid W. M., Enns G., Dobyns W. B., Walsh C. A. (2005) Mutations in POMT1 are found in a minority of patients with Walker-Warburg syndrome. Am. J. Med. Genet. A 133, 53–57 [DOI] [PubMed] [Google Scholar]
- 21. de Bernabé D. B., van Bokhoven H., van Beusekom E., Van den Akker W., Kant S., Dobyns W. B., Cormand B., Currier S., Hamel B., Talim B., Topaloglu H., Brunner H. G. (2003) A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J. Med. Genet. 40, 845–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kobayashi K., Nakahori Y., Miyake M., Matsumura K., Kondo-Iida E., Nomura Y., Segawa M., Yoshioka M., Saito K., Osawa M., Hamano K., Sakakihara Y., Nonaka I., Nakagome Y., Kanazawa I., Nakamura Y., Tokunaga K., Toda T. (1998) An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 394, 388–392 [DOI] [PubMed] [Google Scholar]
- 23. Longman C., Brockington M., Torelli S., Jimenez-Mallebrera C., Kennedy C., Khalil N., Feng L., Saran R. K., Voit T., Merlini L., Sewry C. A., Brown S. C., Muntoni F. (2003) Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of α-dystroglycan. Hum. Mol. Genet. 12, 2853–2861 [DOI] [PubMed] [Google Scholar]
- 24. van Reeuwijk J., Brunner H. G., van Bokhoven H. (2005) Glyc-O-genetics of Walker-Warburg syndrome. Clin. Genet. 67, 281–289 [DOI] [PubMed] [Google Scholar]
- 25. Yoshida A., Kobayashi K., Manya H., Taniguchi K., Kano H., Mizuno M., Inazu T., Mitsuhashi H., Takahashi S., Takeuchi M., Herrmann R., Straub V., Talim B., Voit T., Topaloglu H., Toda T., Endo T. (2001) Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell 1, 717–724 [DOI] [PubMed] [Google Scholar]
- 26. Inamori K., Yoshida-Moriguchi T., Hara Y., Anderson M. E., Yu L., Campbell K. P. (2012) Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akasaka-Manya K., Manya H., Nakajima A., Kawakita M., Endo T. (2006) Physical and functional association of human protein O-mannosyltransferases 1 and 2. J. Biol. Chem. 281, 19339–19345 [DOI] [PubMed] [Google Scholar]
- 28. Manya H., Endo T. (2004) Defective O-mannosyl glycosylation causes congenital muscular dystrophies. Tanpakushitsu Kakusan Koso. 49, 2451–2456 [PubMed] [Google Scholar]
- 29. Zhang W., Betel D., Schachter H. (2002) Cloning and expression of a novel UDP-GlcNAc:α-d-mannoside β1,2-N-acetylglucosaminyltransferase homologous to UDP-GlcNAc:α-3-d-mannoside β1,2-N-acetylglucosaminyltransferase I. Biochem. J. 361, 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoshida-Moriguchi T., Yu L., Stalnaker S. H., Davis S., Kunz S., Madson M., Oldstone M. B., Schachter H., Wells L., Campbell K. P. (2010) O-Mannosyl phosphorylation of α-dystroglycan is required for laminin binding. Science 327, 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ervasti J. M., Campbell K. P. (1991) Membrane organization of the dystrophin-glycoprotein complex. Cell 66, 1121–1131 [DOI] [PubMed] [Google Scholar]
- 32. Ervasti J. M., Campbell K. P. (1993) A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122, 809–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ibraghimov-Beskrovnaya O., Ervasti J. M., Leveille C. J., Slaughter C. A., Sernett S. W., Campbell K. P. (1992) Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355, 696–702 [DOI] [PubMed] [Google Scholar]
- 34. Winder S. J. (2001) The complexities of dystroglycan. Trends Biochem. Sci. 26, 118–124 [DOI] [PubMed] [Google Scholar]
- 35. Gee S. H., Blacher R. W., Douville P. J., Provost P. R., Yurchenco P. D., Carbonetto S. (1993) Laminin-binding protein 120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan, and binds with high affinity to the major heparin binding domain of laminin. J. Biol. Chem. 268, 14972–14980 [PubMed] [Google Scholar]
- 36. Montanaro F., Lindenbaum M., Carbonetto S. (1999) α-Dystroglycan is a laminin receptor involved in extracellular matrix assembly on myotubes and muscle cell viability. J. Cell Biol. 145, 1325–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smalheiser N. R., Kim E. (1995) Purification of cranin, a laminin-binding membrane protein. Identity with dystroglycan and reassessment of its carbohydrate moieties. J. Biol. Chem. 270, 15425–15433 [DOI] [PubMed] [Google Scholar]
- 38. Yamada H., Shimizu T., Tanaka T., Campbell K. P., Matsumura K. (1994) Dystroglycan is a binding protein of laminin and merosin in peripheral nerve. FEBS Lett. 352, 49–53 [DOI] [PubMed] [Google Scholar]
- 39. Chiba A., Matsumura K., Yamada H., Inazu T., Shimizu T., Kusunoki S., Kanazawa I., Kobata A., Endo T. (1997) Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve α-dystroglycan. The role of a novel O-mannosyl-type oligosaccharide in the binding of α-dystroglycan with laminin. J. Biol. Chem. 272, 2156–2162 [DOI] [PubMed] [Google Scholar]
- 40. Sasaki T., Yamada H., Matsumura K., Shimizu T., Kobata A., Endo T. (1998) Detection of O-mannosyl glycans in rabbit skeletal muscle α-dystroglycan, Biochim. Biophys. Acta 1425, 599–606 [DOI] [PubMed] [Google Scholar]
- 41. Smalheiser N. R., Haslam S. M., Sutton-Smith M., Morris H. R., Dell A. (1998) Structural analysis of sequences O-linked to mannose reveals a novel Lewis X structure in cranin (dystroglycan) purified from sheep brain. J. Biol. Chem. 273, 23698–23703 [DOI] [PubMed] [Google Scholar]
- 42. Stalnaker S. H., Hashmi S., Lim J. M., Aoki K., Porterfield M., Gutierrez-Sanchez G., Wheeler J., Ervasti J. M., Bergmann C., Tiemeyer M., Wells L. (2010) Site mapping and characterization of O-glycan structures on α-dystroglycan isolated from rabbit skeletal muscle. J. Biol. Chem. 285, 24882–24891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nilsson J., Nilsson J., Larson G., Grahn A. (2010) Characterization of site-specific O-glycan structures within the mucin-like domain of α-dystroglycan from human skeletal muscle. Glycobiology 20, 1160–1169 [DOI] [PubMed] [Google Scholar]
- 44. Kano H., Kobayashi K., Herrmann R., Tachikawa M., Manya H., Nishino I., Nonaka I., Straub V., Talim B., Voit T., Topaloglu H., Endo T., Yoshikawa H., Toda T. (2002) Deficiency of α-dystroglycan in muscle-eye-brain disease. Biochem. Biophys. Res. Commun. 291, 1283–1286 [DOI] [PubMed] [Google Scholar]
- 45. Kim D. S., Hayashi Y. K., Matsumoto H., Ogawa M., Noguchi S., Murakami N., Sakuta R., Mochizuki M., Michele D. E., Campbell K. P., Nonaka I., Nishino I. (2004) POMT1 mutation results in defective glycosylation and loss of laminin-binding activity in α-DG. Neurology 62, 1009–1011 [DOI] [PubMed] [Google Scholar]
- 46. Liu J., Ball S. L., Yang Y., Mei P., Zhang L., Shi H., Kaminski H. J., Lemmon V. P., Hu H. (2006) A genetic model for muscle-eye-brain disease in mice lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1). Mech. Dev. 123, 228–240 [DOI] [PubMed] [Google Scholar]
- 47. Stalnaker S. H., Aoki K., Lim J. M., Porterfield M., Liu M., Satz J. S., Buskirk S., Xiong Y., Zhang P., Campbell K. P., Hu H., Live D., Tiemeyer M., Wells L. (2011) Glycomic analyses of mouse models of congenital muscular dystrophy. J. Biol. Chem. 286, 21180–21190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abbott K. L., Matthews R. T., Pierce M. (2008) Receptor tyrosine phosphatase beta (RPTPβ) activity and signaling are attenuated by glycosylation and subsequent cell surface galectin-1 binding. J. Biol. Chem. 283, 33026–33035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Inamori K., Mita S., Gu J., Mizuno-Horikawa Y., Miyoshi E., Dennis J. W., Taniguchi N. (2006) Demonstration of the expression and the enzymatic activity of N-acetylglucosaminyltransferase IX in the mouse brain. Biochim. Biophys. Acta 1760, 678–684 [DOI] [PubMed] [Google Scholar]
- 50. Kaneko M., Alvarez-Manilla G., Kamar M., Lee I., Lee J. K., Troupe K., Zhang W., Osawa M., Pierce M. (2003) A novel β(1,6)-N-acetylglucosaminyltransferase V (GnT-VB). FEBS Lett. 554, 515–519 [DOI] [PubMed] [Google Scholar]
- 51. Abbott K. L., Troupe K., Lee I., Pierce M. (2006) Integrin-dependent neuroblastoma cell adhesion and migration on laminin is regulated by expression levels of two enzymes in the O-mannosyl-linked glycosylation pathway, PomGnT1 and GnT-Vb. Exp. Cell Res. 312, 2837–2850 [DOI] [PubMed] [Google Scholar]
- 52. Alvarez-Manilla G., Troupe K., Fleming M., Martinez-Uribe E., Pierce M. (2010) Comparison of the substrate specificities and catalytic properties of the sister N-acetylglucosaminyltransferases, GnT-V and GnT-Vb (IX). Glycobiology 20, 166–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee I., Guo H. B., Kamar M., Abbott K., Troupe K., Lee J. K., Alvarez-Manilla G., Pierce M. (2006) N-Acetylglucosaminyltransferase VB expression enhances β1 integrin-dependent PC12 neurite outgrowth on laminin and collagen. J. Neurochem. 97, 947–956 [DOI] [PubMed] [Google Scholar]
- 54. Aoki K., Perlman M., Lim J. M., Cantu R., Wells L., Tiemeyer M. (2007) Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J. Biol. Chem. 282, 9127–9142 [DOI] [PubMed] [Google Scholar]
- 55. Abbott K. L., Aoki K., Lim J. M., Porterfield M., Johnson R., O'Regan R. M., Wells L., Tiemeyer M., Pierce M. (2008) Targeted glycoproteomic identification of biomarkers for human breast carcinoma. J. Proteome Res. 7, 1470–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ciucanu I. (2006) Per-O-methylation reaction for structural analysis of carbohydrates by mass spectrometry. Anal. Chim. Acta 576, 147–155 [DOI] [PubMed] [Google Scholar]
- 57. Kizuka Y., Kitazume S., Yoshida M., Taniguchi N. (2011) Brain-specific expression of N-acetylglucosaminyltransferase IX (GnT-IX) is regulated by epigenetic histone modifications. J. Biol. Chem. 286, 31875–31884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li X., Zhang P., Yang Y., Xiong Y., Qi Y., Hu H. (2008) Differentiation and developmental origin of cerebellar granule neuron ectopia in protein O-mannose UDP-N-acetylglucosaminyltransferase 1 knock-out mice. Neuroscience 152, 391–406 [DOI] [PubMed] [Google Scholar]
- 59. Yang Y., Zhang P., Xiong Y., Li X., Qi Y., Hu H. (2007) Ectopia of meningeal fibroblasts and reactive gliosis in the cerebral cortex of the mouse model of muscle-eye-brain disease. J. Comp. Neurol. 505, 459–477 [DOI] [PubMed] [Google Scholar]
- 60. Hu H., Yang Y., Eade A., Xiong Y., Qi Y. (2007) Breaches of the pial basement membrane and disappearance of the glia limitans during development underlie the cortical lamination defect in the mouse model of muscle-eye-brain disease. J. Comp. Neurol. 501, 168–183 [DOI] [PubMed] [Google Scholar]
- 61. Hu H., Li J., Gagen C. S., Gray N. W., Zhang Z., Qi Y., Zhang P. (2011) Conditional knock-out of protein O-mannosyltransferase 2 reveals tissue-specific roles of O-mannosyl glycosylation in brain development. J. Comp. Neurol. 519, 1320–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Williamson R. A., Henry M. D., Daniels K. J., Hrstka R. F., Lee J. C., Sunada Y., Ibraghimov-Beskrovnaya O., Campbell K. P. (1997) Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1-null mice. Hum. Mol. Genet. 6, 831–841 [DOI] [PubMed] [Google Scholar]
- 63. Moore S. A., Saito F., Chen J., Michele D. E., Henry M. D., Messing A., Cohn R. D., Ross-Barta S. E., Westra S., Williamson R. A., Hoshi T., Campbell K. P. (2002) Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418, 422–425 [DOI] [PubMed] [Google Scholar]
- 64. Satz J. S., Barresi R., Durbeej M., Willer T., Turner A., Moore S. A., Campbell K. P. (2008) Brain and eye malformations resembling Walker-Warburg syndrome are recapitulated in mice by dystroglycan deletion in the epiblast. J. Neurosci. 28, 10567–10575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Han R., Kanagawa M., Yoshida-Moriguchi T., Rader E. P., Ng R. A., Michele D. E., Muirhead D. E., Kunz S., Moore S. A., Iannaccone S. T., Miyake K., McNeil P. L., Mayer U., Oldstone M. B., Faulkner J. A., Campbell K. P. (2009) Basal lamina strengthens cell membrane integrity via the laminin G domain-binding motif of α-dystroglycan. Proc. Natl. Acad. Sci. U.S.A. 106, 12573–12579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hu H., Candiello J., Zhang P., Ball S. L., Cameron D. A., Halfter W. (2010) Retinal ectopias and mechanically weakened basement membrane in a mouse model of muscle-eye-brain (MEB) disease congenital muscular dystrophy. Mol. Vis. 16, 1415–1428 [PMC free article] [PubMed] [Google Scholar]
- 67. Chan Y. M., Keramaris-Vrantsis E., Lidov H. G., Norton J. H., Zinchenko N., Gruber H. E., Thresher R., Blake D. J., Ashar J., Rosenfeld J., Lu Q. L. (2010) Fukutin-related protein is essential for mouse muscle, brain, and eye development, and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum. Mol. Genet. 19, 3995–4006 [DOI] [PubMed] [Google Scholar]
- 68. Willer T., Prados B., Falcón-Pérez J. M., Renner-Müller I., Przemeck G. K., Lommel M., Coloma A., Valero M. C., de Angelis M. H., Tanner W., Wolf E., Strahl S., Cruces J. (2004) Targeted disruption of the Walker-Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc. Natl. Acad. Sci. U.S.A. 101, 14126–14131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kurahashi H., Taniguchi M., Meno C., Taniguchi Y., Takeda S., Horie M., Otani H., Toda T. (2005) Basement membrane fragility underlies embryonic lethality in fukutin-null mice. Neurobiol. Dis. 19, 208–217 [DOI] [PubMed] [Google Scholar]
- 70. Dennis J. W., Laferté S. (1989) Oncodevelopmental expression of –GlcNAcβ1–6Manα1–6Manβ1-branched asparagine-linked oligosaccharides in murine tissues and human breast carcinomas. Cancer Res. 49, 945–950 [PubMed] [Google Scholar]
- 71. Demetriou M., Granovsky M., Quaggin S., Dennis J. W. (2001) Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 409, 733–739 [DOI] [PubMed] [Google Scholar]
- 72. Guo H. B., Lee I., Bryan B. T., Pierce M. (2005) Deletion of Mouse Embryo Fibroblast N-acetylglucosaminyltransferase V stimulates α5β1 integrin expression mediated by the protein kinase C signaling pathway. J. Biol. Chem. 280, 8332–8342 [DOI] [PubMed] [Google Scholar]
- 73. Guo H. B., Lee I., Kamar M., Pierce M. (2003) N-Acetylglucosaminyltransferase V expression levels regulate cadherin-associated homotypic cell-cell adhesion and intracellular signaling pathways. J. Biol. Chem. 278, 52412–52424 [DOI] [PubMed] [Google Scholar]
- 74. Harroch S., Furtado G. C., Brueck W., Rosenbluth J., Lafaille J., Chao M., Buxbaum J. D., Schlessinger J. (2002) A critical role for the protein-tyrosine phosphatase receptor type Z in functional recovery from demyelinating lesions. Nat. Genet. 32, 411–414 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.