

Background: The biochemical mechanisms underlying α-smooth muscle actin-mediated vascular disease are unknown.
Results: The R256H mutation in actin alters polymerization kinetics and causes misregulation by the nucleation factor, formin.
Conclusion: Mutation-based changes in conformation affect filament stability and regulation of polymerization.
Significance: The Arg-256 residue stabilizes the actin helix and maintains filament conformation required for formin regulation.
Keywords: Actin, Cytoskeleton, Microfilaments, Smooth Muscle, Yeast, Aneurysm, TAAD, Thoracic Aortic Aneurysm and Dissection
Abstract
More than 30 mutations in ACTA2, which encodes α-smooth muscle actin, have been identified to cause autosomal dominant thoracic aortic aneurysm and dissection. The mutation R256H is of particular interest because it also causes patent ductus arteriosus and moyamoya disease. R256H is one of the more prevalent mutations and, based on its molecular location near the strand-strand interface in the actin filament, may affect F-actin stability. To understand the molecular ramifications of the R256H mutation, we generated Saccharomyces cerevisiae yeast cells expressing only R256H yeast actin as a model system. These cells displayed abnormal cytoskeletal morphology and increased sensitivity to latrunculin A. After cable disassembly induced by transient exposure to latrunculin A, mutant cells were delayed in reestablishing the actin cytoskeleton. In vitro, mutant actin exhibited a higher than normal critical concentration and a delayed nucleation. Consequently, we investigated regulation of mutant actin by formin, a potent facilitator of nucleation and a protein needed for normal vascular smooth muscle cell development. Mutant actin polymerization was inhibited by the FH1-FH2 fragment of the yeast formin, Bni1. This fragment strongly capped the filament rather than facilitating polymerization. Interestingly, phalloidin or the presence of wild type actin reversed the strong capping behavior of Bni1. Together, the data suggest that the R256H actin mutation alters filament conformation resulting in filament instability and misregulation by formin. These biochemical effects may contribute to abnormal histology identified in diseased arterial samples from affected patients.
Introduction
Mutations in α-smooth muscle actin, encoded by ACTA2, are the most common genetic cause of familial thoracic aortic aneurysm and dissection (TAAD)3 (1). More than 30 ACTA2 mutations have been identified to cause this potentially fatal disorder and are now known to differentially cause additional vascular diseases, including coronary artery disease, occlusive stroke, and patent ductus arteriosus (2–7). α-Smooth muscle actin is the most abundant protein in the smooth muscle cells that populate the affected arteries and participates in cell proliferation, migration, and force transduction (8–10). Histopathology of patient samples shows loss and disorganization of vascular smooth muscle cells in resected aortic tissue but hyperplasia and obstruction of coronary arteries. In addition, the vascular smooth muscle cells exhibit decreased α-smooth muscle actin content and shortened or fewer actin stress fibers (1, 2).
Vascular smooth muscle cells switch phenotype during development in response to injury and during atherogenesis (9, 11, 12). α-Smooth muscle actin is expressed early in vascular smooth muscle cell differentiation as well as in the quiescent state. Proliferation and migration of vascular smooth muscle cells depend on tight cytoskeletal regulation. Mutations that interfere with actin dynamics may have an adverse effect on normal smooth muscle cell function. Little is known, however, about the biochemical mechanisms by which TAAD mutations in actin contribute to the cell loss or hyperplasia seen in diseased arteries.
Our recent studies have focused on the effects of the R258H mutation on actin dynamics. R258H is one of the most common TAAD actin mutations and is of particular interest because of the range and severity of associated clinical disorders. In addition to TAAD, this mutation causes the congenital cardiovascular defect, patent ductus arteriosus, as well as moyamoya disease, a distinctive pattern of occlusive stroke (2). The clinical findings support the idea that the mutation affects both developmental arterial function and ongoing maintenance. To better understand the biochemical effects of the mutation R258H, we utilized the budding yeast model. Yeast actin is 86% identical and 94% similar to α-smooth muscle actin, and the residues altered by human ACTA2 mutations are conserved between isoforms. The single actin isoform in yeast and ability to correlate in vivo findings with in vitro analyses have allowed us to successfully use this system to understand the biochemical mechanisms underlying not only TAAD (13) but other actin-mediated diseases (14–16) .
The residue Arg-258 in α-smooth muscle actin corresponds to Arg-256 in yeast actin. (The yeast numbering system will be used for descriptions from here on.) Recent analyses of filamentous actin structure indicate that Arg-256, located in actin subdomain 4, is involved in cross-strand interactions within the filament (Fig. 1). As such, residue Arg-256 may play an important role in filament stabilization (17). Furthermore, structural abnormalities in the filament induced by the mutation potentially could interfere with actin interaction with the proteins that regulate filament dynamics. In this work, we present our investigations of the effects of the R256H mutation on cytoskeletal function and actin polymerization properties. In addition, we describe analyses of formin regulation of mutant actin filament formation.
FIGURE 1.

Model of residue R256H in the actin monomer and filament. A, back view of yeast actin monomer crystal structure (43), modified from Protein Data Bank code 1YAG using the PyMOL Molecular Graphics System (version 1.3 (Schrödinger, LLC)). The position of the R256H mutation studied is color-highlighted in red and labeled. The remaining known TAAD missense mutations are color-highlighted in blue. ATP is depicted in orange, and Mg2+ is shown as a yellow sphere. The numbers denote the actin subdomains, and N and C mark the respective termini. B, model of an actin sextmer based on the filament model by Fujii et al. (17) with the R256H mutation color-highlighted and labeled as described above. The filament model displays the location of the mutation along the interstrand interface. The numbers denote the actin subdomains of the individual monomer. Subunits of one strand are in varied shades of blue, and subunits of the opposing strand are in shades of gray.
EXPERIMENTAL PROCEDURES
Materials
DNase I (grade D) was purchased from Worthington. DE52 DEAE-cellulose was obtained from Whatman. Micro Bio-Spin P-30 Tris columns and Affi-Gel 10-activated resin were purchased from Bio-Rad. ATP was purchased from Sigma. Rhodamine-phalloidin, FM 4-64, and DAPI were purchased from Molecular Probes. The QuikChange[regr] site-directed mutagenesis kit was from Stratagene, and oligodeoxynucleotides were purchased from Integrated DNA Technologies. Yeast cakes for wild type actin preparations were purchased from a local bakery. All other chemicals were reagent-grade quality.
Construction of Mutant Yeast Strains
Mutations were introduced into the centromeric plasmid pRS314 (15) containing the yeast actin coding sequence driven by the ACT1 promoter using the QuikChange[regr] mutagenesis kit according to the manufacturer's instructions. Plasmid containing the desired mutation was introduced into a recipient yeast strain containing a deleted chromosomal ACT1 gene and a plasmid expressing wild type actin (pCENWT) as described previously (18). Plasmid shuffling yielded a viable haploid strain with mutant actin as the sole actin in the cell. The plasmids containing the mutant constructs were re-isolated from these strains and sequenced to confirm the presence of the desired mutation.
Growth Behavior in Liquid Culture
Cells were grown in complete liquid YPD medium (1% yeast extract, 2% peptone, and 2% dextrose) with 0.01% adenine (YPAD) at 30 °C on a shaking platform. Growth was determined by diluting an overnight culture of each strain into fresh medium at an initial A600 of 0.1 and following growth at 30 °C with agitation. The absorbances of the cultures were recorded as a function of time. The absorbances were back-calculated following the appropriate dilutions to lower the cell density to the linear range of the spectrometer.
Growth under Stress Conditions
Temperature sensitivity of mutant actin was examined by plating four serial 10-fold dilutions of the cultures on YPD plates followed by incubation at 24, 30, or 37 °C. Colony size was assessed as a function of time. To assess mitochondrial function, cells were grown on medium containing glycerol as the sole carbon source. Cultures were plated on YPG medium (YPD medium with the dextrose replaced with 2% glycerol) and incubated at 30 °C. To test for hyperosmolar sensitivity, cells were plated on YPD plus 0.5 m and 0.9 m NaCl agar plates and incubated at 30 °C.
Cytology
Cell structures were imaged with an Olympus IX81 microscope and a Hamamatsu camera (Model C10600-10B-H). Camera control and image enhancement were performed using Slidebook5 (3i Inc., Denver, Co.). Vacuoles were imaged following exposure of the cells to the dye FM4-64 as described previously (19). Nuclear and mitochondrial DNA was stained with DAPI as described previously (20). Mitochondria were visualized in living cells by expressing a fusion protein of green fluorescent protein (GFP) conjugated to the mitochondrial signal sequence of citrate synthase kindly provided by Dr. Liza A. Pon (21). Cells expressing the plasmid were grown to an A600 of 0.3–0.5 in YPAD. An aliquot of cells was resuspended and imaged as described below. The actin cytoskeleton was visualized by fluorescence microscopy after staining fixed cells with rhodamine-phalloidin as described previously (20). Cytoskeletal morphology was assessed in budding cells when the daughter was less than half of the size of the mother cell.
To measure cell size, mounted samples were visualized by differential interference contrast microscopy. The long axis of the cell was measured using ImageJ software. For assaying the morphologies of the actin cytoskeleton and mitochondria, images were taken at 0.2-μm intervals in a Z-series through the cell. Out-of-focus light was removed from each section by deconvolution using Slidebook5. The Z-series of deconvolved images were stacked into a two-dimensional image with ImageJ software. Presentation of cell images was done using Adobe Photoshop. All cellular statistical analyses were based on cell counts of >100 for each sample.
Latrunculin A Sensitivity
Sterile filtered discs (0.5 cm in diameter) were presoaked in 2 μl of dimethyl sulfoxide (control), 0.1, 0.5, or 1 mm of latrunculin A (22). Soaked discs were placed on YPD plates containing 100 μl of evenly spread wild type or mutant cells (A600 = 0.1). The plates were incubated at 30 °C for 48 h, and the area of growth inhibition surrounding the drug eluting disc was measured via ImageJ software.
Cells were grown in liquid YPD medium to an A600 of 0.3–0.5 and treated with 5 μm latrunculin A for 1 min. Cells were washed three times with fresh YPAD medium (YPD with 0.01% adenine) and incubated in 5 ml of medium at room temperature with intermittent shaking. Cells were removed in 250-μl aliquots at designated time points, fixed with formaldehyde, stained with rhodamine-phalloidin, and imaged using an Olympus fluorescence microscope as described previously (20). More than 50 cells were analyzed for each sample, and the percentage of cells with actin cables was quantified.
Protein Purification
Wild type and mutant yeast actin were purified from lysates of frozen yeast cells using a combination of DNase I-agarose affinity chromatography, DEAE-cellulose chromatography, and polymerization/depolymerization cycling as described previously (23). The quality of actin preparations was assessed using SDS-PAGE and Coomassie Blue staining of the gels. The concentration of G-actin was determined from the absorbance at 290 nm using an extinction coefficient of 0.63 m−1 cm−1. Actin was stored as G-actin in G buffer (10 mm Tris-HCl, pH 7.5, 0.2 mm ATP, 0.2 mm CaCl2, and 0.1 mm dithiothreitol). Purified actin was stored at 4 °C and used within 3 days of purification.
The His-tagged Bni1 yeast formin FH1-FH2 C-terminal fragment was expressed in and purified from Saccharomyces cerevisiae strain (BDY502) as described (24, 25). Purified formin fragments were stored in stock buffer (10 mm Tris, pH 7.5, 150 mm KCl, 0.2 mm MgCl2, 1 mm dithiothreitol, and 10% glycerol) at a concentration of 5–10 μm at −80 °C. Protein concentration was determined from absorbance at 280 nm with an extinction coefficient of 14650 m−1 cm−1.
Actin Polymerization
Polymerization of G-actin in a total volume of 120 μl was induced by the addition of MgCl2 and KCl to final concentrations of 2 and 50 mm, respectively. Polymerization was monitored at 25 °C by following the increase in light scattering of the sample using a FluoroMax-3 fluorescence spectrometer outfitted with a computer-controlled thermostatted four position multisample exchanger (HORIBA Jobin Yvon, Inc.). The excitation and emission wavelengths were both set to 360 nm with the slit widths set at 1 nm.
Actin polymerization experiments involving Bni1 used pyrene-labeled actin and followed the same methods as above with exceptions listed below. N-(1-pyrenyl) maleimide (Sigma) was covalently attached to actin at Cys-374 (pyrene-actin) as described previously (26). Wild type and mutant samples each contained 10% pyrene-labeled actin and were preincubated at room temperature for 15 min prior to induction of polymerization. For admixture experiments, wild type and mutant actin were individually labeled and then combined. The excitation and emission wavelengths were set at 365 and 385 nm, respectively. For most experiments, the designated concentration of the Bni1 fragment was added to the reaction mixture prior to induction of polymerization. Where noted, the Bni1 fragment was added to the reaction mixture midway into the elongation phase of polymerization. For polymerization experiments using phalloidin, unlabeled phalloidin (Sigma p2141) was added to the reaction mixture prior to induction of polymerization, as described previously (27). All polymerization experiments were performed at least three times with at least three different actin preparations.
Critical Concentration Determination
The critical concentration (Cc) of each actin was determined from the net change in light scattering of an actin polymerization reaction and measured as a function of increasing actin concentration. The critical concentration of actin was obtained by drawing a line through the points and determining its intersection in the x axis.
Electron Microscopy
To visualize actin filament morphology, samples of 1.5 μm wild type actin and 2.25 μm mutant actin were deposited onto carbon-coated Formvar grids, negatively stained with 1% uranyl acetate, and imaged with a JOEL 1230 transmission electron microscope (University of Iowa Central Electron Microscopy Facility) equipped with a Gatan UltraScan 1000 CCD 2000/2000-pixel camera. Accelerating voltage of the transmission electron microscope was 100 kV. ImageJ software was used to process the images and measure the individual filament lengths for R256H and wild type actin.
Statistical Analysis
All data are presented as means ± S.D. Results for wild type and mutant actin were compared by using a paired t test with a p value < 0.05 considered significant.
RESULTS
Effect of Mutations on Cytoskeletal Morphology and Cell Growth
Our studies began with analyses of the actin cytoskeleton. The two major components of the cytoskeleton in yeast are actin cables and patches. Cables are organized along the mother-bud axis during polarized cell growth and facilitate movement of organelles to the daughter (28). Endocytic patches are distributed in a cell cycle-dependent manner and localize in the bud early in the cell cycle (29–31). Cytoskeletal morphology of mutant and wild type cells were determined by analysis of >500 cells stained with fluorescent phalloidin by three masked reviewers. In cells expressing R256H actin, patch distribution and staining intensity was normal but cables were less pronounced and difficult to visualize (Fig. 2). Stepwise analyses of Z-series images with contrast enhancement allowed identification of polarized cables in mutant cells, but overall, the cables were faint. These cytoskeletal findings were far more common in R256H mutant cells compared with wild type cells (41% versus 6% respectively, p < 0.01).
FIGURE 2.

Effect of the R256H mutation on cytology. A, fluorescence microscopy of cells expressing wild type or R256H yeast actin. Cells assessed were those in which the bud was between one-half and one-third the size of the mother cell. The cytoskeleton was visualized after staining fixed cells with rhodamine-phalloidin. White arrows identify actin cables. Vacuoles were observed following exposure of the cells to the dye FM4-64. Mitochondria were visualized with GFP. Scale bar, 2 mm. B, quantified results are based on assessment of 100 cells for each sample. The bar height indicates the percentage of the cell population that exhibited abnormal morphology with error bars denoting the S.D. Differences were statistically significant compared with wild type actin (p < 0.05).
Disruption of the cytoskeleton may lead to loss of polarization during budding or growth deficits. Despite the high rate of cytoskeletal abnormalities, we found no differences between wild type and R256H mutant actin on cell size (4.6 ± 0.7 μm versus 4.7 ± 0.7 μm, respectively), doubling time (∼ 2.1 h) or extent of growth in liquid medium. To further evaluate cytoskeletal effects, we monitored growth under stress conditions that require rapid physiologic reorganization of the actin cytoskeleton (32, 33). Growth was monitored in hypothermic (24 °C), hyperthermic (37 °C), and hyperosmolar conditions (medium containing 0.9 m NaCl), and no differences were identified between wild type and R256H strains (data not shown). Thus, despite the morphologic abnormalities, the cytoskeleton conserves sufficient functionality to maintain growth and accommodate environmental stress.
Despite maintenance of cell size and growth, we questioned whether actin function was sufficient to preserve morphology and distribution of organelles. Vacuoles, mitochondria, and nuclei were analyzed to assess morphology and localization in the cell. Vacuoles typically consist of one to four small, evenly sized lobes. Abnormal morphology was infrequently identified in wild type cells (7%). In comparison, nearly half of the R256H cells had abnormal morphology (49%, p < 0.02; Fig. 2). The dominant phenotype was that of a single lobe greater than two-thirds the size of the cell or hypovesiculation, although 12% of abnormal cells were hypervesiculated with more than four lobes of varied size.
Disorganization of the polarized actin cytoskeleton can also lead to abnormal fission/fusion events that affect mitochondrial integrity and function (34). We evaluated the effect of the mutation on both mitochondrial and genomic DNA distribution by staining with DAPI. We found no differences in DNA distribution between wild type and R256H cells (data not shown). Despite the normal mitochondrial DNA distribution, mitochondrial morphology was affected by the actin mutation. Normally, mitochondrial are long, tubular structures aligned along the mother-bud axis, as was identified in 95% of wild type cells. Nearly a third of mutant cells, however, had fragmented and aggregated mitochondrial tubules. Overall, the incidence of abnormal mitochondrial morphology was 5% in wild type cells and 34% in R256H cells, p value < 0.01. Regardless, mutant cells were able to maintain growth on glycerol based medium (YPG), indicative of sufficient mitochondrial function (data not shown).
Impact of R256H Mutation on Actin Cytoskeleton Assembly in Vivo
The morphologic abnormalities identified in the actin cytoskeleton led us to further investigate whether the R256H actin causes a defect in actin assembly. To address this question, we examined the sensitivity of wild type and R256H cells to latrunculin A, a drug that sequesters actin monomers and reversibly promotes rapid depolymerization of actin filaments (22, 35, 36). Plate cultures of wild type and mutant cells were exposed to filter discs saturated with a range of concentrations of latrunculin A. The area of growth inhibition surrounding each disc was quantified. Mutant cells were more sensitive to latrunculin A than wild type cells (Fig. 3). For example, the area of growth inhibition around the 1 mm latrunculin A disc was 4.38 ± 0.08 cm2 for the R256H cells versus 1.18 ± 0.01 cm2 for wild type (p < 0.001).
FIGURE 3.

Effect of the R256H mutation on sensitivity to cytoskeletal disassembly. A, sterile filtered discs (0.5 cm in diameter) were presoaked in either 2 μl of dimethyl sulfoxide (control) or 2 μl of 0.1, 0.5, or 1 mm latrunculin A and placed on YPD plates containing 100 μl of evenly spread wild type or mutant cells (A600 = 0.1). The plates were incubated at 30 °C for 48 h. B, quantification of growth inhibition by latrunculin A. The bar height indicates the area of growth inhibition (cm2) for the four concentrations of latruncilin A (mm) with error bars denoting the S.D. The area of growth inhibition for R256H cells was 1.9-fold higher for the highest latrunculin A concentration. Differences were statistically significant compared with wild type actin for all concentrations (p < 0.05).
Our data thus far suggest that the cytoskeletal abnormalities have minimal growth effects on a steadily growing population of cells. We questioned whether the abnormalities would manifest in a situation where the reassembly of the cytoskeleton was required. To answer this, we again used latrunculin A to disassemble the cytoskeleton and then monitored reestablishment of the cytoskeleton. Analyses revealed that mutant cells had marked delay in recovery to base-line cable morphology. For wild type cells, half had normal cables 10 min after treatment, and nearly all cells were restored by 50 min. In contrast, R256H cells took 5-fold longer, 50 min, for half of the population to have base-line cable morphology. The difference persisted with mutant cells requiring >90 min for the whole population to reestablish actin cables (Fig. 4).
FIGURE 4.
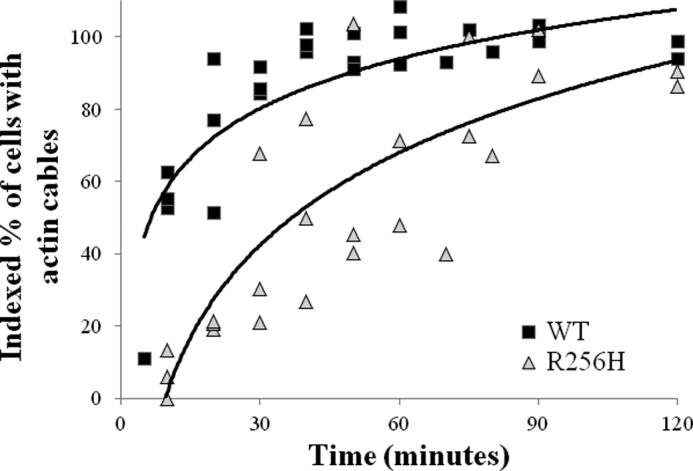
Impact of the R256H mutation on actin cytoskeleton assembly in vivo. A scatter plot of the indexed percentage of cells with actin cables relative to time after treatment with latrunculin A. Cells were transiently treated with latrunculin A to disassemble the cytoskeleton, washed, and incubated. Aliquots of cells were removed at designated time points, fixed, stained with rhodamine-phalloidin, and imaged as described under “Experimental Procedures.” The percentage of cells with actin cables was quantified for each sample and indexed to the pretreatment fraction. At least 50 cells were analyzed per sample, and the experiments were repeated three times. The scatter plot graph displays the indexed percentage of cells with actin cables relative to time for all analyses. Logarithmic trend lines depict the differences in rate of actin cable reassembly between wild type and R256H actin.
Effects of R256H mutation on Actin Polymerization
The in vivo data indicate that the mutant actin cytoskeleton is less stable with problems reestablishing the cable network. These anomalies could derive from mutation-mediated defects in actin polymerization. The location of Arg-256 along the inter-strand interface within the filament supports the idea that a change in that the residue might alter polymerization behavior characterized by changes in nucleation or elongation rates or critical concentration. The effects of the R256H mutation on actin polymerization kinetics were assessed by quantifying the change in light scattering during actin polymerization. Wild type and R256H mutant actin were purified from yeast cells, and polymerization was induced and monitored over time. As suspected, the R256H mutation led to polymerization defects; specifically, an extended nucleation phase and a lower final extent of polymerization (Fig. 5A). The lower final extent of polymerization signifies a higher critical concentration for the mutant actin. Additional polymerization studies established a critical concentration for R256H as 1.36 μm compared with 0.60 μm for wild type actin (p < 0.001; Fig. 5B). In verification, electron micrographs of wild type and R256H actin filaments also demonstrated a difference in filament length (electron micrographs not shown). Mutant actin filaments were shorter measuring 3.10 ± 0.89 μm compared with wild type 3.76 ± 1.29 μm (p < 0.01). Together, these results are consistent with filament destabilization due to the mutation.
FIGURE 5.

Polymerization kinetics of wild type and R256H actin. A, polymerization of 2.25 μm actin was initiated by the addition of magnesium and potassium chloride, and the change in light scattering was monitored as a function of time at 25 °C. Shown are representative plots of experiments performed at least three times with three independent actin preparations. B, critical concentration (Cc) of actin was determined from the net change in light scattering upon polymerization performed as in A as a function of increasing actin concentration. The critical concentration of actin was determined by drawing a line through the points and determining the intersection at the x axis. A.U., arbitrary units.
Nucleation of Actin Polymerization by Bni1
The cytoskeletal morphology in R256H cells and the delay in reestablishment of the cytoskeleton suggest a defect in filament nucleation. Formins are proteins that regulate actin filament nucleation. Formins have FH1 and FH2 domains near the C terminus. The FH2 domains of two formins form a collar around the barbed end of the actin filament and facilitate nucleation (37). This formin collar maintains association with the growing barbed end of the filament in a manner termed processive capping (25). We assessed the effects of formin on actin polymerization using the C-terminal fragment of the yeast formin, Bni1, which contains the FH1 and FH2 domains (38). This Bni1 fragment has been previously shown to cause a concentration dependent increase in the nucleation rate of wild type actin with no observable change in final extent of polymerization (39).
We initially screened 10% pyrene-labeled wild type and R256H actin polymerization with the addition of a range of concentrations of the Bni1 fragment. As a control, we compared the polymerization kinetics of 10% pyrene-labeled actin based on fluorescence to the polymerization kinetics of unlabeled actin based on light scattering. No differences were seen for wild type or mutant actin between the two assays (data not shown). Wild type responded as previously, a concentration-dependent increase in the nucleation rate. R256H actin, however, had the opposite response with a dose-dependent decrease in nucleation and final extent. Given the differences in critical concentration, we determined the concentration at which wild type and R256H actin reached the same final extent of polymerization, namely 1.5 μm of wild type actin and 2.25 μm of R256H actin (data not shown). Our subsequent experiments used these concentrations to account for the difference in critical concentration between wild type and mutant actins. Even with the higher amount of mutant actin, the differences persisted with a concentration-dependent decrease in nucleation and the final extent of polymerization of R256H mutant actin in the presence of the Bni fragment (Fig. 6). For example, 100 nm of the Bni fragment decreased the final extent of R256H actin polymerization to one-third of the light scattering measured for mutant actin polymerization alone. To determine whether the change in formin regulation of mutant actin was due to a capping phenomenon or isolated defect in nucleation, we quantified the effect of formin on oligomers that have progressed past the nucleation phase. To do so, the formin fragment was added to the reaction midway into the elongation phase of polymerization. The result was similar to that observed when the Bni fragment was added prior to induction of polymerization (Fig. 7). These findings support the hypothesis that the R256H mutation in actin alters formin regulation leading to capping rather than normal processive association with the barbed end of the filament.
FIGURE 6.

Actin polymerization kinetics in the presence of the formin, Bni. A, actin polymerization was quantified in the presence of various concentrations of the Bni1 FH1-FH2 fragment. Ten percent of the actin was labeled with pyrene, and the polymerization-dependent increase in fluorescence was quantified over time. To account for the increased critical concentration of R256H mutant actin, actin concentrations with equivalent final extents of polymerization were selected to ensure the same amount of polymerizable actin was used. Accordingly, concentrations for wild type and R256H actin were 1.5 and 2.25 μm, respectively, as described under “Experimental Procedures.” Shown are representative plots of experiments performed at least three times with three independent actin preparations. A.U., arbitrary units.
FIGURE 7.
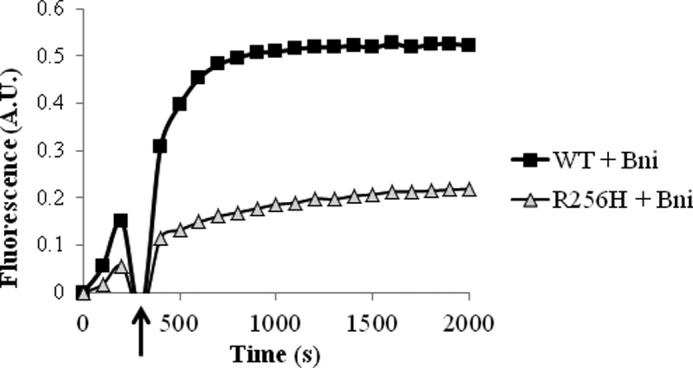
Effect of late addition of Bni1 on actin polymerization kinetics. A, polymerization of 10% pyrene-labeled wild type and R256H actin was fluorescently monitored over time as in Fig. 6 with the exception that 100 nm Bni was added to the reaction midway during elongation. Time of Bni addition is marked by an arrow. Concentrations for wild type and R256H actin were 1.5 and 2.25 μm, respectively. Shown is a representative plot from experiments performed at least three times with three independent actin preparations. A.U., arbitrary units.
We hypothesized that the defect in formin regulation of R256H mutant actin was due to changes in conformation of the filament. To answer this question, we repeated the Bni1 polymerization experiments with the addition of stoichiometric amounts of phalloidin to the reaction mixture. Phalloidin binds actin filaments and reduces flexibility with stabilization intersubunit contacts both along and between the two helical strands. In addition, phalloidin-binding changes the filament conformation causing subdomains 1 and 2 of the actin subunit to rotate outward (40–42). Consistent with our hypothesis, the presence of phalloidin restored polymerization of R256H actin with Bni to wild type kinetics (Fig. 8).
FIGURE 8.

Actin polymerization kinetics in the presence of phalloidin. Polymerization of 10% pyrene-labeled actin was quantified by change in fluorescence over time with and without phalloidin and with and without Bni. Actin concentrations were 2.25 μm as was the concentration of phalloidin when included. 100 nm of the FH1-FH2 fragment of Bni1 was used when included. A, wild type actin. B, R256H mutant actin. Shown are representative plots from experiments performed at least two times with two independent actin preparations. A.U., arbitrary units.
In the clinical disease state, the actin mutations are autosomal dominant with both normal and mutant alleles in the same cell. Incorporation of wild type subunits into the filament may influence conformational changes related to mutant actin. To determine the consequence of wild type actin on R256H actin biochemistry, we examined the polymerization kinetics of admixtures of wild type and mutant actin. Interestingly, wild type actin appeared dominant over the mutant actin. Lesser amounts of wild type actin disproportionately normalized the final extent of polymerization of mutant actin. For example, a 50:50 mixture of wild type and R256H actin had a final extent of polymerization at 80% of wild type actin, nearly twice that of mutant actin alone.
We extended this line of inquiry to the effect of the Bni fragment on polymerization of admixtures of wild type and mutant actin. Consistent with the polymerization experiments, wild type actin had a dominant effect on the interaction of the formin fragment with the mutant actin (Fig. 9). Wild type actin normalized polymerization kinetics of R256H actin with Bni disproportionate to the percentage. Only 25% of wild type actin added to mutant actin led to nearly 50% recovery of the final extent of polymerization.
FIGURE 9.

Polymerization kinetics of admixtures of wild type and mutant actin. A, ratios of wild type and R256H mutant actin were combined with a final concentration of 2.25 μm and induced to polymerize. The polymerization-dependent increase in fluorescence of the 10% pyrene-labeled actin was monitored over time. B, as in A, polymerization kinetics of ratios of wild type and R256H mutant actin was quantified in the presence 100 nm of Bni1. Shown are representative plots of experiments performed at least three times with three independent actin preparations. A.U., arbitrary units.
In summary, the polymerization defects seen with R256H mutant actin are ameliorated by restoration of filament conformation as seen with the addition of phalloidin or wild type actin. These data support that conformation of the barbed end of the actin filament is altered by the R256H mutation leading to flawed filament initiation.
DISCUSSION
Our focus was to determine the biochemical effects of the R256H mutation on actin function in vivo and in vitro. The mutation clearly affected the morphology of actin cables and two organelles whose integrity depends on the cytoskeleton. These alterations, however, did not impact growth of the cells expressing only the mutant actin even under stress conditions. Although mitochondrial patterns are affected, the ability of cells to grow on 2% glycerol indicates that the alterations do not significantly interfere with mitochondrial metabolic capacity. These studies assess growth under conditions where cytoskeletal structure generally propagates off of an existing template. The mutation effects may lie in the ability to assemble the cytoskeletal template. This idea agrees with our latrunculin A results in which mutant cells are less efficient in reestablishing the cytoskeleton than wild type cells.
The R256H actin mutation led to filament instability in vitro demonstrated by the increased critical concentration and a delayed nucleation phase during polymerization. Both of these effects could result from an effect of the mutation on interstrand interactions within the filament leading to altered filament conformation and stability. Models of the actin helix (17, 43) suggest that Arg-256 is involved in interstrand stabilization. Arg-256 and Glu-195 form a triangular unit with the side chain of Lys-113 in the cross-strand monomer (Fig. 10). In this triangle, the guanido group of Arg-256 is 3–4 Å from the side chain of Glu-195 and the Lys-113 side chain amino is within 3 Å of the Glu-195 side chain. Ionic interactions among the three residues would stabilize the unit, and interfering with this complex could lead to filament destabilization. The R256H change could affect this unit via both decreased size and decreased cationic character. Results from another study we are carrying out are in agreement with this hypothesis. The K113E mutation in actin (44) leads to filament destabilization similar to the findings presented here (data not shown).
FIGURE 10.

Model of the actin trimer. A model of the actin trimer based on the filament model of Oda et al. (43). The actin monomer colors are blue, gray, and green. Residue Arg-256 is color-highlighted and labeled (red), residue Glu-195 is color-highlighted and labeled (blue), and residue Lys-113 is color-highlighted and labeled (orange). Models were modified using the PyMOL Molecular Graphics System (version 1.3; Schrödinger, LLC).
Interestingly, Lys-113 is part of a helix, Lys-113-Thr-125, that contains two other TAAD mutations we have previously characterized, N115T and R116Q, in addition to Lys-118, the site of two deafness-causing mutations (14, 45). If our hypothesis is correct, the mutations become part of a pathogenic module that, as a unit, can affect F-actin conformation. The nature and site of a particular mutation in this unit would create a potential spectrum of effects on actin filament regulation, which could translate ultimately into the allele-specific symptoms seen in TAAD patients. In this context, the R256H mutation might cause a propagated conformation change to the filament surface leading to an altered interaction with an actin-binding protein such as formin. Two other pieces of evidence correlate with this helix destabilization idea. First, stoichiometric amounts of phalloidin, a filament stabilizing drug, effectively eliminate the nucleation lag and restore the extent of polymerization to near normal values. Second, wild type actin has a dominant effect over the deficits caused by the mutation as evidenced by the admixture experiments. Evidently, dispersion of wild type monomers in the filament can cause propagated changes that restore the overall filament confirmation to a more normal state.
The Bni1 FH2 domain can dimerize and form a collar around the barbed end of the filament. This collar acts as a weak barbed end capping protein. Crystallographic evidence suggests that the FH2 partially occludes the filament barbed end, a hypothesis that is consistent with total internal reflection fluorescence results (46, 47). Models of the actin-formin complex suggest that following nucleation, the formin dimer sidles up the growing filament as it extends (46, 48, 49). The progression alternately exposes a monomer entry site on one strand followed by the entry site on the other strand.
With the R256H actin, however, the FH1-FH2 C-terminal fragment from Bni1 seemed to act as a strong inhibitor of barbed end elongation. Whether this inhibition results from strong capping or slightly leaking capping as described with the vinculin-actin interaction cannot be determined from our data. Polymerization nearly ceased following addition of the fragment at the beginning or midway through the polymerization process. The mutant results suggest that the collar can form but that the altered conformation of the filament prevents formin movement relative to the barbed end. Because the amino acid at position 256 is not part of the FH2 binding site (49), the consequence of the mutation must result from a propagated effect on filament structure that alters the formin binding site (50). Consistent with our hypothesis, the two conditions that restore filament stability, phalloidin or small amounts of wild type actin, overcome this deficiency and restore nucleation facilitated by formin.
The molar ratio of Bni1p to actin in the cell is ∼1 to 500 (51). The concentrations of the Bni fragment used in our experiments are higher than the known cellular concentration of Bni1p. However, Bni1p is highly localized within the cell at the neck or the bud tip so the local concentration relative to actin will be much higher (52). Therefore, the higher concentrations of Bni relative to actin we used for our studies should not present a concern on the grounds of biological significance.
The model of the actin-formin collar proposed by Rosen (49) suggests a molecular explanation for our results. In this model, residue 113 is part of a helix that extends to residue 125 on the actin filament surface, near the formin binding site (Fig. 11). The connection of this helix with residues 256 and Glu-195 of opposing monomer would provide a means for propagating an allosteric signal between the filament exterior and interior. Such an interaction would allow for regulation of filament dynamics by formin binding.
FIGURE 11.

Model of Bni interaction with the actin oligomer. A, model of the actin oligomer based on the filament model of Oda et al. (43) and the Bni1 FH1-FH2 fragment from Rosen et al. (49). The actin monomer colors are blue and gray. Residues are color highlighted: orange, Lys-113; marine blue, Lys-118; yellow, Glu-195; red, Arg-256; and pink, helix containing residues 113–125. The FH1-FH2 domain colors are gold and green. The red rectangle frames this unit of interaction. Models were generated using the PyMOL Molecular Graphics System (version 1.5; Schrödinger, LLC). The two models are a side view and a pointed end view of the structure. B, schematic drawing demonstrating the formin/actin complex colored as above. The post site of the green bridge is free and accessible to recruit an incoming actin subunit.
Our results provide new insight into possible mechanisms by which the R256H mutation leads to TAAD. Histological examination of aortic tissue from patients with this mutation show decreased numbers of actin filaments arranged in a disorganized pattern. This scenario is the end result of a long disease process and leads to the question of how this state arises. Two broad reasons could account for the histological findings. First, the mutation may cause a defect in the initial assembly of the contractile apparatus. Our observations concerning the effects of the R256H mutation on formin function are particularly relevant to this concept. FHOD1, a formin protein, is essential for proper deposition of the actin cytoskeleton. The formin FHOD1 plays an important role in formation and organization of the vascular smooth muscle cell contractile apparatus and a lack of FHOD1 results in fewer and grossly disorganized actin filaments (53). The second potential reason for the arterial pathology is that the mutation may cause defects in maintaining the contractile apparatus in a functional organized state once assembly has occurred. The weakened intermonomer interactions caused by the mutation could result in an inherently less stable structure that is more susceptible to turnover. Alternatively, this weakening could lead to an inability of the actin apparatus to withstand the forces imposed on it by smooth muscle myosin in the generation of contractile force needed for setting and maintaining vascular tone. Similar observations have been made in cases where actin mutations affect striated muscle function (54).
Ultimately, delineating the molecular mechanisms leading to arterial wall weakening will require longitudinally following the process over the course of the disease from birth. Animal models to do this are currently being developed.4 However, understanding the effects of the mutations on actin function per se and its subsequent regulation at the biochemical level with studies as we have reported here will be required to assess how the mutation leads to cellular changes that cause the pathology observed.
Acknowledgment
We thank Rose Lee for participation in the early aspects of this work.
This work was supported by supported by American Heart Association Grant 0830053N (to H. L. B.), National Institutes of Health Grant DC008803 (to P. A. R.), and National Institutes of Health, NCRR Grant UL1RR024979 (to H. L. B. and P. A. R.).
D. Milewicz, personal communication.
- TAAD
- thoracic aortic aneurysm and dissection.
REFERENCES
- 1. Guo D. C., Pannu H., Tran-Fadulu V., Papke C. L., Yu R. K., Avidan N., Bourgeois S., Estrera A. L., Safi H. J., Sparks E., Amor D., Ades L., McConnell V., Willoughby C. E., Abuelo D., Willing M., Lewis R. A., Kim D. H., Scherer S., Tung P. P., Ahn C., Buja L. M., Raman C. S., Shete S. S., Milewicz D. M. (2007) Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 39, 1488–1493 [DOI] [PubMed] [Google Scholar]
- 2. Guo D. C., Papke C. L., Tran-Fadulu V., Regalado E. S., Avidan N., Johnson R. J., Kim D. H., Pannu H., Willing M. C., Sparks E., Pyeritz R. E., Singh M. N., Dalman R. L., Grotta J. C., Marian A. J., Boerwinkle E. A., Frazier L. Q., LeMaire S. A., Coselli J. S., Estrera A. L., Safi H. J., Veeraraghavan S., Muzny D. M., Wheeler D. A., Willerson J. T., Yu R. K., Shete S. S., Scherer S. E., Raman C. S., Buja L. M., Milewicz D. M. (2009) Mutations in smooth muscle α-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 84, 617–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoffjan S., Waldmüller S., Blankenfeldt W., Kötting J., Gehle P., Binner P., Epplen J. T., Scheffold T. (2011) Three novel mutations in the ACTA2 gene in German patients with thoracic aortic aneurysms and dissections. Eur. J. Hum. Genet. 19, 520–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morisaki H., Akutsu K., Ogino H., Kondo N., Yamanaka I., Tsutsumi Y., Yoshimuta T., Okajima T., Matsuda H., Minatoya K., Sasaki H., Tanaka H., Ishibashi-Ueda H., Morisaki T. (2009) Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum. Mutat. 30, 1406–1411 [DOI] [PubMed] [Google Scholar]
- 5. Yoo E. H., Choi S. H., Jang S. Y., Suh Y. L., Lee I., Song J. K., Choe Y. H., Kim J. W., Ki C. S., Kim D. K. (2010) Clinical, pathological, and genetic analysis of a Korean family with thoracic aortic aneurysms and dissections carrying a novel D26Y mutation. Ann. Clin. Lab. Sci. 40, 278–284 [PubMed] [Google Scholar]
- 6. Disabella E., Grasso M., Gambarin F. I., Narula N., Dore R., Favalli V., Serio A., Antoniazzi E., Mosconi M., Pasotti M., Odero A., Arbustini E. (2011) Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle α-actin 2 (ACTA2). Heart 97, 321–326 [DOI] [PubMed] [Google Scholar]
- 7. Renard M., Callewaert B., Baetens M., Campens L., Macdermot K., Fryns J. P., Bonduelle M., Dietz H. C., Gaspar I. M., Cavaco D., Stattin E. L., Schrander-Stumpel C., Coucke P., Loeys B., De Paepe A., De Backer J. (2011) Int. J. Cardiol. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fatigati V., Murphy R. A. (1984) Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. J. Biol. Chem. 259, 14383–14388 [PubMed] [Google Scholar]
- 9. Ailawadi G., Moehle C. W., Pei H., Walton S. P., Yang Z., Kron I. L., Lau C. L., Owens G. K. (2009) Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J. Thorac. Cardiovasc. Surg. 138, 1392–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alexander M. R., Owens G. K. (2012) Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 74, 13–40 [DOI] [PubMed] [Google Scholar]
- 11. Owens G. K., Kumar M. S., Wamhoff B. R. (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 [DOI] [PubMed] [Google Scholar]
- 12. Tananari Y., Maeno Y., Takagishi T., Sasaguri Y., Morimatsu M., Kato H. (2000) Role of apoptosis in the closure of neonatal ductus arteriosus. Jpn. Circ. J. 64, 684–688 [DOI] [PubMed] [Google Scholar]
- 13. Bergeron S. E., Zhu M., Thiem S. M., Friderici K. H., Rubenstein P. A. (2010) Ion-dependent polymerization differences between mammalian β- and γ-nonmuscle actin isoforms. J. Biol. Chem. 285, 16087–16095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bryan K. E., Rubenstein P. A. (2009) Allele-specific effects of human deafness γ-actin mutations (DFNA20/26) on the actin/cofilin interaction. J. Biol. Chem. 284, 18260–18269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bryan K. E., Wen K. K., Zhu M., Rendtorff N. D., Feldkamp M., Tranebjaerg L., Friderici K. H., Rubenstein P. A. (2006) Effects of human deafness γ-actin mutations (DFNA20/26) on actin function. J. Biol. Chem. 281, 20129–20139 [DOI] [PubMed] [Google Scholar]
- 16. Morín M., Bryan K. E., Mayo-Merino F., Goodyear R., Mencía A., Modamio-Høybjør S., del Castillo I., Cabalka J. M., Richardson G., Moreno F., Rubenstein P. A., Moreno-Pelayo M. A. (2009) In vivo and in vitro effects of two novel γ-actin (ACTG1) mutations that cause DFNA20/26 hearing impairment. Hum. Mol. Genet. 18, 3075–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujii T., Iwane A. H., Yanagida T., Namba K. (2010) Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature 467, 724–728 [DOI] [PubMed] [Google Scholar]
- 18. Cook R. K., Sheff D. R., Rubenstein P. A. (1991) Unusual metabolism of the yeast actin amino terminus. J. Biol. Chem. 266, 16825–16833 [PubMed] [Google Scholar]
- 19. Vida T. A., Emr S. D. (1995) A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 128, 779–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKane M., Wen K. K., Boldogh I. R., Ramcharan S., Pon L. A., Rubenstein P. A. (2005) A mammalian actin substitution in yeast actin (H372R) causes a suppressible mitochondria/vacuole phenotype. J. Biol. Chem. 280, 36494–36501 [DOI] [PubMed] [Google Scholar]
- 21. Fehrenbacher K. L., Yang H. C., Gay A. C., Huckaba T. M., Pon L. A. (2004) Live cell imaging of mitochondrial movement along actin cables in budding yeast. Curr. Biol. 14, 1996–2004 [DOI] [PubMed] [Google Scholar]
- 22. Ayscough K. R., Stryker J., Pokala N., Sanders M., Crews P., Drubin D. G. (1997) High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin A. J. Cell Biol. 137, 399–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cook R. K., Blake W. T., Rubenstein P. A. (1992) Removal of the amino-terminal acidic residues of yeast actin. Studies in vitro and in vivo. J. Biol. Chem. 267, 9430–9436 [PubMed] [Google Scholar]
- 24. Wen K. K., Rubenstein P. A. (2009) Differential regulation of actin polymerization and structure by yeast formin isoforms. J. Biol. Chem. 284, 16776–16783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moseley J. B., Sagot I., Manning A. L., Xu Y., Eck M. J., Pellman D., Goode B. L. (2004) A conserved mechanism for Bni1- and mDia1-induced actin assembly and dual regulation of Bni1 by Bud6 and profilin. Mol. Biol. Cell 15, 896–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng L., Kim E., Lee W. L., Miller C. J., Kuang B., Reisler E., Rubenstein P. A. (1997) Fluorescence probing of yeast actin subdomain 3/4 hydrophobic loop 262–274. Actin-actin and actin-myosin interactions in actin filaments. J. Biol. Chem. 272, 16829–16837 [DOI] [PubMed] [Google Scholar]
- 27. Kuang B., Rubenstein P. A. (1997) Beryllium fluoride and phalloidin restore polymerizability of a mutant yeast actin (V266G,L267G) with severely decreased hydrophobicity in a subdomain 3/4 loop. J. Biol. Chem. 272, 1237–1247 [DOI] [PubMed] [Google Scholar]
- 28. Yang H. C., Pon L. A. (2002) Actin cable dynamics in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 99, 751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amberg D. C. (1998) Three-dimensional imaging of the yeast actin cytoskeleton through the budding cell cycle. Mol. Biol. Cell 9, 3259–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Engqvist-Goldstein A. E., Drubin D. G. (2003) Actin assembly and endocytosis: from yeast to mammals. Annu. Rev. Cell Dev. Biol. 19, 287–332 [DOI] [PubMed] [Google Scholar]
- 31. Smith M. G., Swamy S. R., Pon L. A. (2001) The life cycle of actin patches in mating yeast. J. Cell Sci. 114, 1505–1513 [DOI] [PubMed] [Google Scholar]
- 32. Chowdhury S., Smith K. W., Gustin M. C. (1992) Osmotic stress and the yeast cytoskeleton: Phenotype-specific suppression of an actin mutation. J. Cell Biol. 118, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller M. J., Xuong N. H., Geiduschek E. P. (1979) A response of protein synthesis to temperature shift in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 76, 5222–5225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Simon V. R., Karmon S. L., Pon L. A. (1997) Mitochondrial inheritance: Cell cycle and actin cable dependence of polarized mitochondrial movements in Saccharomyces cerevisiae. Cell Motil. Cytoskeleton 37, 199–210 [DOI] [PubMed] [Google Scholar]
- 35. Morton W. M., Ayscough K. R., McLaughlin P. J. (2000) Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2, 376–378 [DOI] [PubMed] [Google Scholar]
- 36. Moseley J. B., Goode B. L. (2006) The yeast actin cytoskeleton: From cellular function to biochemical mechanism. Microbiol. Mol. Biol. Rev. 70, 605–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li F., Higgs H. N. (2005) Dissecting requirements for auto-inhibition of actin nucleation by the formin, mDia1. J. Biol. Chem. 280, 6986–6992 [DOI] [PubMed] [Google Scholar]
- 38. Moseley J. B., Goode B. L. (2005) Differential activities and regulation of Saccharomyces cerevisiae formin proteins Bni1 and Bnr1 by Bud6. J. Biol. Chem. 280, 28023–28033 [DOI] [PubMed] [Google Scholar]
- 39. Wen K. K., McKane M., Stokasimov E., Rubenstein P. A. (2011) Mutant profilin suppresses mutant actin-dependent mitochondrial phenotype in Saccharomyces cerevisiae. J. Biol. Chem. 286, 41745–41757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Steinmetz M. O., Goldie K. N., Aebi U. (1997) A correlative analysis of actin filament assembly, structure, and dynamics. J. Cell Biol. 138, 559–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steinmetz M. O., Stoffler D., Müller S. A., Jahn W., Wolpensinger B., Goldie K. N., Engel A., Faulstich H., Aebi U. (1998) Evaluating atomic models of F-actin with an undecagold-tagged phalloidin derivative. J. Mol. Biol. 276, 1–6 [DOI] [PubMed] [Google Scholar]
- 42. Nyitrai M., Hild G., Lukács A., Bódis E., Somogyi B. (2000) Conformational distributions and proximity relationships in the rigor complex of actin and myosin subfragment-1. J. Biol. Chem. 275, 2404–2409 [DOI] [PubMed] [Google Scholar]
- 43. Oda T., Iwasa M., Aihara T., Maéda Y., Narita A. (2009) The nature of the globular to fibrous actin transition. Nature 457, 441–445 [DOI] [PubMed] [Google Scholar]
- 44. Laing N. G., Dye D. E., Wallgren-Pettersson C., Richard G., Monnier N., Lillis S., Winder T. L., Lochmüller H., Graziano C., Mitrani-Rosenbaum S., Twomey D., Sparrow J. C., Beggs A. H., Nowak K. J. (2009) Mutations and polymorphisms of the skeletal muscle α-actin gene (ACTA1). Hum. Mutat. 30, 1267–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bergeron S. E., Wedemeyer E. W., Lee R., Wen K. K., McKane M., Pierick A. R., Berger A. P., Rubenstein P. A., Bartlett H. L. (2011) Allele-specific effects of thoracic aortic aneurysm and dissection α-smooth muscle actin mutations on actin function. J. Biol. Chem. 286, 11356–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu Y., Moseley J. B., Sagot I., Poy F., Pellman D., Goode B. L., Eck M. J. (2004) Crystal structures of a formin homology-2 domain reveal a tethered dimer architecture. Cell 116, 711–723 [DOI] [PubMed] [Google Scholar]
- 47. Kovar D. R., Pollard T. D. (2004) Insertional assembly of actin filament barbed ends in association with formins produces piconewton forces. Proc. Natl. Acad. Sci. U.S.A. 101, 14725–14730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shemesh T., Otomo T., Rosen M. K., Bershadsky A. D., Kozlov M. M. (2005) A novel mechanism of actin filament processive capping by formin: Solution of the rotation paradox. J. Cell Biol. 170, 889–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Otomo T., Tomchick D. R., Otomo C., Panchal S. C., Machius M., Rosen M. K. (2005) Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature 433, 488–494 [DOI] [PubMed] [Google Scholar]
- 50. Le Clainche C., Dwivedi S. P., Didry D., Carlier M. F. (2010) Vinculin is a dually regulated actin filament barbed end-capping and side-binding protein. J. Biol. Chem. 285, 23420–23432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weng S., Dong Q., Balakrishnan R., Christie K., Costanzo M., Dolinski K., Dwight S. S., Engel S., Fisk D. G., Hong E., Issel-Tarver L., Sethuraman A., Theesfeld C., Andrada R., Binkley G., Lane C., Schroeder M., Botstein D., Michael Cherry J. (2003) Saccharomyces Genome Database (SGD) provides biochemical and structural information for budding yeast proteins. Nucleic Acids Res. 31, 216–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buttery S. M., Yoshida S., Pellman D. (2007) Yeast formins Bni1 and Bnr1 utilize different modes of cortical interaction during the assembly of actin cables. Mol. Biol. Cell 18, 1826–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Staus D. P., Blaker A. L., Medlin M. D., Taylor J. M., Mack C. P. (2011) Formin homology domain-containing protein 1 regulates smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 31, 360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Costa C. F., Rommelaere H., Waterschoot D., Sethi K. K., Nowak K. J., Laing N. G., Ampe C., Machesky L. M. (2004) Myopathy mutations in α-skeletal-muscle actin cause a range of molecular defects. J. Cell Sci. 117, 3367–3377 [DOI] [PubMed] [Google Scholar]