

Background: The mechanism of pre-transfer editing by which aaRSs regulate translational fidelity is not well understood.
Results: Yeast mitochondrial ThrRS, MST1, hydrolyzes seryl adenylate at the aminoacylation active site more rapidly than the cognate threonyl adenylate.
Conclusion: MST1 discriminates against serine and reduces mischarging of threonine tRNA by employing pre-transfer editing.
Significance: The mechanism of misactivation and pre-transfer editing of serine by ThrRS is provided.
Keywords: Aminoacyl tRNA Synthetase, Enzyme Mechanisms, Protein Synthesis, Transfer RNA (tRNA), X-ray Crystallography, Proofreading, Translational Quality Control
Abstract
Accurate translation of mRNA into protein is a fundamental biological process critical for maintaining normal cellular functions. To ensure translational fidelity, aminoacyl-tRNA synthetases (aaRSs) employ pre-transfer and post-transfer editing activities to hydrolyze misactivated and mischarged amino acids, respectively. Whereas post-transfer editing, which requires either a specialized domain in aaRS or a trans-protein factor, is well described, the mechanism of pre-transfer editing is less understood. Here, we show that yeast mitochondrial threonyl-tRNA synthetase (MST1), which lacks an editing domain, utilizes pre-transfer editing to discriminate against serine. MST1 misactivates serine and edits seryl adenylate (Ser-AMP) in a tRNA-independent manner. MST1 hydrolyzes 80% of misactivated Ser-AMP at a rate 4-fold higher than that for the cognate threonyl adenylate (Thr-AMP) while releasing 20% of Ser-AMP into the solution. To understand the mechanism of pre-transfer editing, we solved the crystal structure of MST1 complexed with an analog of Ser-AMP. The binding of the Ser-AMP analog to MST1 induces conformational changes in the aminoacylation active site, and it positions a potential hydrolytic water molecule more favorably for nucleophilic attack. In addition, inhibition results reveal that the Ser-AMP analog binds the active site 100-fold less tightly than the Thr-AMP analog. In conclusion, we propose that the plasticity of the aminoacylation site in MST1 allows binding of Ser-AMP and the appropriate positioning of the hydrolytic water molecule.
Introduction
Aminoacyl-tRNA synthetases (aaRSs)3 facilitate decoding of the genetic code by pairing each proteinogenic amino acid with the cognate tRNA. By catalyzing formation of aminoacyl-tRNAs (aa-tRNAs), aaRSs provide reaction substrates for the translating ribosome as it ratchets down the mRNA (1). Each aaRS catalyzes a two-step reaction at the synthetic active site: activation of the amino acid with ATP to form an aminoacyl-adenylate (aa-AMP), and the subsequent transfer of the amino acid moiety to the 3′-end of the cognate tRNA. The structural similarity between amino acids presents a major challenge to the accuracy of aa-tRNA synthesis and hence the fidelity of translation. Although beneficial under certain conditions (2–5), compromised accuracy of protein synthesis and the increased frequency of translational errors have been shown to cause growth defects in bacteria (6–8), mitochondrial dysfunction in yeast (9), and neurodegeneration in mice (10). To overcome the lack of selectivity against structurally similar amino acids, aaRSs commonly utilize pre- and post-transfer editing functions to hydrolyze misactivated amino acids and incorrect aa-tRNAs, respectively (11, 12). Whereas it is well documented that post-transfer editing occurs in a tRNA-dependent manner either at a distinct domain appended to the aaRS or by an autonomous trans-editing factor (7, 13–18), the mechanism of the tRNA-independent pre-transfer editing is less understood.
Pre-transfer editing was first reported by Baldwin and Berg (19). Later studies on the lupin valyl-tRNA synthetase indicated that threonyl adenylate (Thr-AMP) bound to valyl-tRNA synthetase is hydrolyzed more rapidly than valyl-AMP at the aminoacylation active site (20). In contrast, studies on isoleucyl-tRNA synthetase imply that misactivated valyl-AMP first translocates from the aminoacylation site to the editing site in the CP1 domain, where it is subsequently hydrolyzed (21). This model was supported further by the observation that both the pre- and post-transfer analogs of valyl-AMP bind the editing pocket in the CP1 domain of isoleucyl-tRNA synthetase (22). More recent studies on aaRSs lacking a post-transfer editing domain suggested that pre-transfer editing in these enzymes primarily occurs at the aminoacylation active site and that a fraction of misactivated amino acids are expelled into solution for hydrolysis (23–26). It remains elusive how misactivated amino acids are hydrolyzed at the aminoacylation site.
In this work, we show that Saccharomyces cerevisiae mitochondrial threonyl-tRNA synthetase (MST1) misactivates serine (Ser) and hydrolyzes seryl adenylate (Ser-AMP) in the absence of the cognate tRNA. We have further determined the crystal structure of MST1 in complex with a nonhydrolyzable analog of the Ser-AMP conjugate (seryl sulfamoyl adenylate or SAM). SAM and the Thr-AMP analog (threonyl sulfamoyl adenylate or TAM) bind to the aminoacylation site in a slightly different manner and with distinct binding affinities. Our structural and biochemical analyses thus provide insights into the pre-transfer editing mechanism of MST1.
EXPERIMENTAL PROCEDURES
Expression and Purification of MST1
MST1 was cloned into the pET28a (Novagen) expression vector with an N-terminal His6 tag. The recombinant protein was overexpressed for 18 h in the Rosetta pLysS (Novagen) Escherichia coli expression strain at 15 °C. The expressed protein was captured from the cell lysate on a Ni2+ affinity column (GE Healthcare) following a standard purification protocol. The affinity column eluate was dialyzed against 4 liters of 100 mm Tris (pH 7.5), 300 mm NaCl, 1 mm dithiothreitol overnight at 4 °C. The dialyzed sample was filtered through 0.22 μm filter, flash-frozen in liquid nitrogen, and stored at −80 °C prior to use.
Pyrophosphate Exchange Assay
The reaction was performed in the presence of 100 mm Na-HEPES (pH 7.2), 30 mm KCl, 10 mm MgCl2, 2 mm potassium fluoride, 2 mm ATP, 2 mm 32PPi (1 cpm/pmol), 0.2 μm MST1, 0.2–5 mm Thr or 10–1000 mm Ser. The resulting [32P]ATP was measured as described in Ref. 27.
Pre-transfer Editing Assays
The pre-transfer editing activity of MST1 was measured at 37 °C in the presence of 100 mm Na-HEPES (pH 7.2), 30 mm KCl, 10 mm MgCl2, 9 μm MST1, 20 mm amino acid, 2 mm cold ATP, 0.1 mCi/ml [γ-32P], or [α-32P]ATP, and 0.01 mg/ml inorganic pyrophosphatase. 2 μl of the reaction mix was added to an equal volume of acetic acid at each time point to stop the reaction. Phosphate (Pi) was separated from [γ-32P]ATP on polyethylenimine (PEI) cellulose plates in 0.1 m potassium phosphate buffer (pH 3.4). AMP, aa-AMP, and [α-32P]ATP were separated on PEI-cellulose plates in 0.1 m ammonium acetate plus 5% acetic acid. The spots were visualized and quantified with phosphorimaging. For the chase experiment, the reaction was performed with 0.1 mm cold ATP and 0.1 mCi/ml [α-32P]ATP for 2 min followed by the addition of 20 mm cold ATP.
Inhibition Assay
Aminoacylation of mitochondrial tRNAThr was performed in the presence of 100 mm Na-HEPES (pH 7.2), 30 mm KCl, 10 mm MgCl2, 40 nm MST1, 20 μm [14C]Thr (44 μCi/ml), 2 mm cold ATP, 50–1,000 nm SAM or TAM. The apparent Ki (Kiapp) was calculated according to the following equation (28):
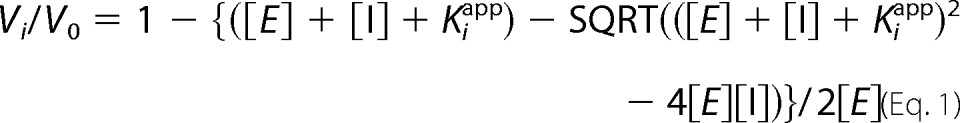 |
where Vi and V0 are the initial velocities in the presence and absence of the inhibitor, and [E] and [I] denote the concentrations of the enzyme and inhibitor, respectively.
Crystallization and Structure Determination of the MST1-SAM Binary Complex
Crystals of MST1 were obtained by sitting drop vapor-diffusion method at 12 °C by mixing equal volumes of the solution containing MST1 and tRNA2Thr and the well buffer (0.1 m Na2HPO4/KH2PO4 (pH 6.2), 0.2 m NaCl, 10% polyethylene glycol (PEG) 8,000). The crystals, which contained only apo-MST1, grew to a maximum size after 2–4 weeks. To obtain the binary complex, the crystals were incubated with 20 mm SAM for 12–18 h at 12 °C and then cryoprotected in the crystallization buffer supplemented with 12% PEG 8,000 and 20% glycerol. Data were collected at liquid nitrogen temperature at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beam line at the Advanced Photon Source, Argonne National Laboratory. The diffraction data were processed in HKL2000 (29). The crystal structure of the binary MST1-SAM complex was determined by molecular replacement in Phaser (30) using the structure of apo-MST1 (Protein Data Bank (PDB) ID code 3UGQ) as a search model. The structure refinement was performed in Phenix (31), and the model building was done in COOT (32, 33). All figures were produced in PyMOL (The PyMOL Molecular Graphics System, Version 1.2, Schrödinger, LLC).
RESULTS
MST1 Misactivates and Edits Serine in the Absence of Threonine tRNA
Previous studies have shown that bacterial threonyl-tRNA synthetases (ThrRSs) misactivate Ser and possess both pre- and post-transfer editing activities against Ser (26, 34). Yeast MST1 is homologous to bacterial ThrRSs but lacks the N-terminal editing domain that hydrolyzes misacylated Ser-tRNAThr (35, 36), prompting us to investigate the fidelity of MST1 for different near-cognate amino acids such as Ser, Val, Ala, and Cys. We first measured the activation rates of Thr and Ser by MST1 using a pyrophosphate exchange assay. The kcat value for Ser is approximately 2-fold lower than that for Thr, whereas the Km is 400-fold higher (Table 1). Collectively, MST1 activates Ser 710-fold less efficiently than Thr, and such a misactivation rate is higher than the commonly accepted rate of amino acid misincorporation (10−4 to 10−3) in proteins (11, 37).
TABLE 1.
Pyrophosphate exchange by MST1 in the presence of either Thr or Ser
The results are the average of three measurements with standard deviations indicated.
| kcat | Km | kcat/Km | Selectivity | |
|---|---|---|---|---|
| min−1 | (mm) | mm−1 min−1 | ||
| Thr | 199 ± 41 | 0.30 ± 0.03 | 671 ± 123 | 1 |
| Ser | 110 ± 4 | 120 ± 19 | 0.94 ± 0.20 | 710 |
Next, we measured the editing activity of MST1 using a [γ-32P]ATP hydrolysis assay. Wild-type (WT) MST1 stimulated hydrolysis of ATP in the presence of Thr and Ser, but not in the presence of Val, Ala, or Cys (Fig. 1, A and B), suggesting that MST1 preferentially misactivates and edits Ser among the near-cognate amino acids. WT MST1 hydrolyzed ATP 2.6-fold faster in the presence of Ser (20 mm) than Thr (20 mm), and such rates were not significantly enhanced by the addition of tRNAThr (Fig. 1B and supplemental Table S1). The contribution of aminoacylation to the overall ATP consumption is negligible given the errors and the relatively low tRNA concentration used in the assay. Despite the pre-transfer editing activity against Ser, MST1 still formed Ser-tRNAThr in vitro (Fig. 1C), indicating that post-transfer editing could be essential for aminoacylation fidelity in ThrRS enzymes. The lack of an appended post-transfer editing domain, however, makes MST1 an ideal system to study the mechanism of tRNA-independent pre-transfer editing.
FIGURE 1.

Activation and editing of Ser by WT MST1. A and B, hydrolysis of [γ-32P]ATP by MST1 (9 μm) in the presence of amino acids (20 mm each) with or without yeast mitochondrial tRNAThr (3 μm). The experiment was repeated three times with S.D. (error bars) indicated. C, serylation of two mitochondrial tRNAThr species (3 μm) by MST1 (3 μm).
Ser-AMP Is Selectively Hydrolyzed and Released into Solution by MST1
Pre-transfer editing of misactivated amino acids can be promoted either by a water molecule once the aa-AMP conjugate is released from the aaRS into solution or be catalyzed by the aaRS (Fig. 2). To discern which of the two scenarios occurs in the case of MST1, we monitored the formation of aa-AMP and AMP over time using [α-32P]ATP. The steady-state rates of Thr-AMP and Ser-AMP formation are 0.33 and 0.91 min−1, respectively, with the end concentration exceeding that of MST1 active sites (Fig. 3, A and B). This clearly shows that a fraction of Thr-AMP and Ser-AMP formed is released from the aminoacylation active site. The rates of AMP formation in the presence of Thr and Ser are 1.20 and 4.34 min−1, respectively (Fig. 3, A and C), which are significantly higher than the spontaneous hydrolysis rates (k4 in Fig. 2) of Thr-AMP (0.16 min−1) and Ser-AMP (0.13 min−1) under the reaction condition (supplemental Fig. S1). MST1 thus catalyzes the hydrolysis of both Thr-AMP and Ser-AMP. The apparent rate of AMP formation is the sum of the enzyme-catalyzed (k3 in Fig. 2) and spontaneous ATP hydrolysis rates (k4), which allows calculation of the k3 values for Thr-AMP (1.04 min−1) and Ser-AMP (4.21 min−1). These values suggest that MST1 selectively hydrolyzes Ser-AMP over Thr-AMP. Further, assuming that the level of the enzyme-bound aa-AMP remains constant during the steady-state phase, the rate of aa-AMP formation would also equal k2 − k4, thus yielding the k2 values of 0.49 min−1 and 1.04 min−1 for Thr-AMP and Ser-AMP, respectively. In conclusion, our results show that MST1 preferentially hydrolyzes Ser-AMP over Thr-AMP. Based on the steady-state rates of Ser-AMP and AMP formation (Fig. 3), we estimate that ∼80% of Ser-AMP is hydrolyzed by MST1 with the remaining 20% being released into solution.
FIGURE 2.

Kinetic scheme of pre-transfer editing by MST1. k1 and k−1 are the forward and reverse rates of aa-AMP formation, respectively. k2 represents the dissociation rate of an aa-AMP from MST1, and k3 is the rate of enzyme-catalyzed hydrolysis of aa-AMP.
FIGURE 3.

Formation of Thr-AMP, Ser-AMP, and AMP by WT MST1. A, hydrolysis of [α-32P]ATP by MST1 (9 μm) in the presence of either Thr or Ser (20 mm). B and C, product formation was quantified over time. The final results were the average of three measurements with S.D. (error bars) indicated.
Binding of the Ser-AMP Analog Stabilizes the Active-site Lid in a Conformation Similar to yet Distinct from That Observed in the Presence of the Thr-AMP Mimic
To understand the mechanism of pre-transfer editing of MST1 at the structural level, we determined the crystal structure of MST1 in complex with the nonhydrolyzable analog of Ser-AMP to 2.87 Å resolution (supplemental Table S2). The overall structure of the binary complex is similar to the previously reported structure of MST1 complexed with the nonhydrolyzable analog of Thr-AMP (TAM) (36), with the main differences noted in the conformations of the active-site lid and the anticodon-binding domain. The binary complex crystals, which belonged to a monoclinic space group (C21), contained two MST1 homodimers in the asymmetric unit. The MST1-SAM model contained 1,706 amino acids, 8 SAM molecules, 4 Zn2+ ions, and 341 water molecules and was refined to a final Rwork/Rfree of 17.5/22.9% (supplemental Table S2). Each MST1 monomer binds two SAM molecules; one SAM, which is referred to as SAM 1, interacts with the aminoacylation active-site groove as expected, whereas the second SAM molecule, which is referred to as SAM 2, is bound to the anticodon-binding domain (Fig. 4A and supplemental Fig. S2). The binding of SAM 2 to MST1 is likely an artifact under the crystallization condition (see supplemental text).
FIGURE 4.

Ser-AMP analog binds to two sites in MST1 and stabilizes the closed conformation of the aminoacylation domain. A, ribbon diagram of the crystal structure of the MST1-SAM binary complex determined at 2.87 Å resolution. SAM 1 is bound to the aminoacylation site along with a Zn2+ ion (orange sphere), whereas SAM 2 binds to the site in the anticodon-binding domain implicated in the anticodon sequence recognition (see supplemental Figs. S4 and S5 for more details). Helices, strands, and loops are dark red, gray, and olive, respectively. SAM molecules are shown as blue sticks. B, superimpositioning of the apo-MST1 structure (beige; PDB ID code 3UGQ) onto MST1-SAM (dark red) reveals a structural rearrangement of the active-site lid. In particular, loop β5-β6 becomes disordered, and helix α4 breaks into two smaller helices (labeled here as α4 and α4′), which are now positioned at an angle of ∼90°. C, comparison of the crystal structures of MST1-SAM (dark red) and MST1-TAM (gray; PDB ID code 3UH0) reveals that helices α4 and α4′ move closer to the active site when MST1 binds SAM. SAM (blue balls-and-sticks) and Zn2+ (orange sphere) are shown as reference points.
The superimpositioning of the apo-MST1 (PDB ID code 3UGQ) onto the MST1-SAM yields a root mean square deviation value of 0.66 Å (2,870 Cα atoms used in calculation). The main differences between the two structures are in the conformations of the active site and the anticodon-binding domain, and the level of disorder of loop β5-β6 in the N-terminal aminoacylation domain. The binding of the nonhydrolyzable analog of Ser-AMP promotes conformational rearrangements in MST1 reminiscent of those observed on binding of the Thr-AMP analog (Fig. 4, B and C), which suggests that MST1 employs the same general mechanism for activation of the cognate and near-cognate aa-AMPs. Indeed, the superimpositioning of MST1-SAM onto MST1-TAM yields a lower root mean square deviation value of 0.41 Å. As in the case of TAM, the binding of SAM stabilizes an “open” conformation of MST1, in which loop β5-β6 is completely disordered. On the other hand, helix α4, which serves as a lid of the active-site groove, adopts a more “closed” conformation in MST1-SAM compared with MST1-TAM (Fig. 4C). Also, whereas the residues 97–113 form a single helix α4 in the apo-MST1, the same stretch of amino acids forms two α-helices, termed α4 (residues 97–102) and α4′ (residues 105–113), in the MST1-SAM binary complex. These helices, which are linked with a two-residue-long loop, are oriented at an angle of almost 90°. Because the side chains in the α4-α4′ loop and helix α4′ participate directly in binding the aa-AMP conjugates, the more closed conformation could affect the rate by which a given aa-AMP conjugate is hydrolyzed by MST1 (see below).
Differences in the Binding of Ser-AMP and Thr-AMP to the Aminoacylation Site of MST1
Whereas the overall structural similarity between the MST1-SAM and MST1-TAM complexes suggests that MST1 recognizes Thr and Ser by a similar mechanism, important structural differences in and around the aminoacylation site of MST1 in the two complexes provide insights into the mechanism by which MST1 hydrolyzes Ser-AMP more efficiently than Thr-AMP.
A nonhydrolyzable analog of Ser-AMP binds to the active-site crevice of MST1 in a fashion similar to the mimic of Thr-AMP (Fig. 5A and supplemental Fig. S2A). Superimpositioning of the MST1-TAM onto the corresponding atoms in MST1-SAM reveals slight yet important structural differences in the N-terminal domain in general and the aminoacylation site in particular. First, the active-site lid (i.e. helix α4) adopts a more closed conformation in MST1-SAM (Fig. 4C), and consequently, the side chains of Tyr109 and Asp112 are positioned closer to the Ser-AMP analog (Fig. 5A). Second, a potential hydrolytic water molecule, Wat1, is positioned, through direct H-bonding interactions with Lys273 and indirect H-bonding with Tyr109, above the sulfur atom at a distance of 4.5 Å and at an angle that is optimal for nucleophilic attack (Fig. 5B). Moreover, the side chains of Arg162 and Gln287 stabilize the sulfamoyl moiety in a configuration optimal for the nucleophilic attack (Fig. 5B). In contrast, in the MST1-TAM crystal, a similar water molecule, Wat1′, forms a H-bond with a nonbridging oxygen atom of the sulfamoyl group, and its orientation is not optimal for the nucleophilic attack (Fig. 5A). Finally, whereas the adenine ring and ribose of SAM interact with MST1 like the corresponding groups in TAM (see Fig. 2 in Ref. 36), the interactions of the seryl moiety with Zn2+ do not fully resemble that of the threonyl moiety. In particular, the γ-OH group of SAM is positioned 2.4 and 3.1 Å away from the Oδ1 atom of Asp182 and Zn2+, respectively, whereas the same distances in the MST1-TAM binary complex crystal were 2.6 and 2.2 Å (data not shown). Also, α-NH2 of SAM is positioned closer to a nearby water molecule (distance of 2.35 Å), which is held in place by the backbone amide of Asn132 and Oδ2 of Asp182, than to the Zn2+ ion (distance of 2.7 Å). This is in contrast to TAM, whose α-NH2 is 2.2 and 3.0 Å away from Zn2+ and water, respectively (data not shown).
FIGURE 5.

Structural rearrangements in the active site of MST1 promoted by SAM binding. A, structural comparison between MST1-TAM (gray; PDB ID code 3UH0) and MST1-SAM (dark red) reveals that a number of side chains and water molecules adopt a different orientation when SAM (blue balls-and-sticks) binds to the active site of MST1. The side chains of Tyr109 and Asp112 (gold sticks) from helix α4′ and water molecules, Wat1 and Wat2 (red spheres), are positioned closer to SAM than to TAM (gray balls-and-sticks). Wat1, the putative hydrolytic water, is positioned differently in MST1-TAM; the water in that complex is designated as Wat1′ and is shown as a gray sphere. B, hydrogen-bonding network in the active site of MST1 complexed with SAM positioning the putative hydrolytic water (Wat1) at a distance and an angle proper for nucleophilic attack onto the mimic of the phosphorus atom. All hydrogen bonds are shown as dashed lines.
The structural differences in the mode of recognition of SAM and TAM have been further verified by biochemical assays. In particular, we determined the binding affinities of SAM and TAM for MST1 using an aminoacylation inhibition assay. The apparent Ki values for SAM and TAM are 450 and 4.5 nm, respectively (Fig. 6). The lower binding affinity of SAM is presumably caused by the weaker interaction between the seryl moiety and the active-site Zn2+ ion. Also, the observations that SAM binds to the aminoacylation site 100-fold less tightly than TAM and causes further conformational changes suggest that the recognition of aa-AMP by MST1 is plastic. The plasticity of the active site thus could explain why MST1 promotes hydrolysis of Ser-AMP more efficiently than that of Thr-AMP (see “Discussion”).
FIGURE 6.
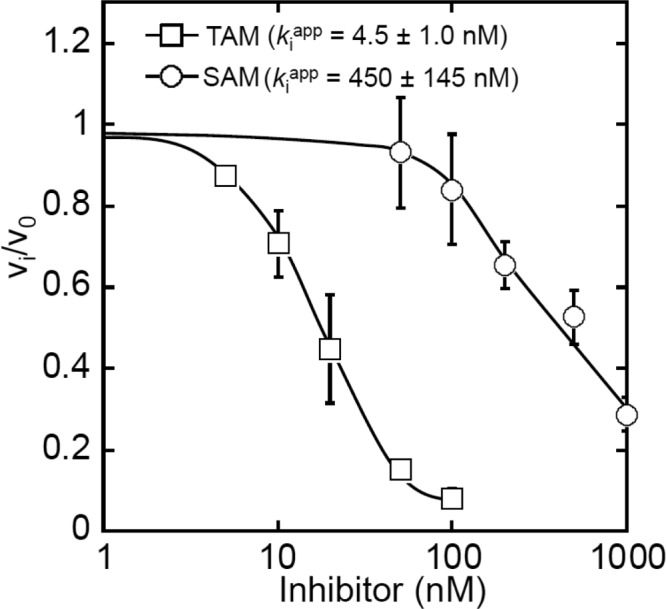
Inhibition of MST1 aminoacylation by SAM and TAM. The aminoacylation was performed in the presence of 20 μm [14C]Thr. Vi and V0 are the initial velocities of aminoacylation in the presence and absence of the inhibitor, respectively. The results were the average of three measurements with S.D. (error bars) indicated.
DISCUSSION
Fidelity of Protein Synthesis
Maintenance of translational fidelity has been a major selective pressure during the evolution of life (38), and decreased translational accuracy is associated with severe defects from bacteria to humans (5, 9, 39). Approximately half of the aaRSs use editing mechanisms to ensure the accuracy of aminoacylation (11, 40). The choice of pre- or post-transfer editing pathways could be affected by either the rate of aminoacylation or the property of the misactivated amino acid (26, 41). It has been reported that human mitochondrial leucyl-tRNA synthetase and yeast mitochondrial phenylalanyl-tRNA synthetase lack a functional editing site present in their bacterial and cytosolic counterparts, yet their active sites are more selective against near-cognate amino acids (5, 27, 42). Compromising the high selectivity of yeast mitochondrial phenylalanyl-tRNA synthetase for amino acids leads to a complete loss of mitochondrial respiration (5). The post-transfer editing domain of ThrRS appears to be lost or dysfunctional in yeast mitochondria and mycoplasma (4, 35), raising the question as to whether this results in promiscuous translation. We show here that MST1 poorly discriminates against Ser and employs a pre-transfer editing mechanism to remove Ser-AMP (Table 1 and Fig. 1). The overall editing rate of MST1 is 3-fold faster than the steady-state aminoacylation rate (35), suggesting that the pre-transfer editing activity is important and physiologically relevant in reducing the amount of formed Ser-tRNAThr. It is also plausible that an unidentified trans-editing factor hydrolyzes Ser-tRNAThr or that Ser is simply misincorporated at Thr codons at a high frequency in yeast mitochondria, thus decreasing the overall translational fidelity. It has recently been shown that mistranslation could not only be tolerated, but also preferred in a number of organisms and under certain stress conditions (4). Future studies to determine which of these scenarios plays a major role in regulating both the aminoacylation of tRNAThr and co-translational incorporation of Thr in yeast mitochondria are warranted.
The Mechanism of Pre-transfer Editing by MST1
Pre-transfer editing of aaRSs, discovered >40 years ago, prevents the coupling of the misactivated amino acid to tRNA, and thus is critical for the accuracy of gene translation. Recent biochemical and structural studies have provided evidence for three mechanisms responsible for hydrolysis of the misactivated amino acids. First, in some aaRSs, the misactivated amino acid translocates from the aminoacylation site to an editing site where it gets hydrolyzed (21, 43). In this case, the editing site is capable of binding only the activated near-cognate amino acid(s) and not the cognate one. Second, the near-cognate aa-AMP conjugate is released from the aminoacylation site into solution, and its hydrolysis is then promoted by the solvent (44). Finally, the aminoacylation site of some aaRSs is capable of hydrolyzing the near-cognate aa-AMP (23–26, 45).
Our results suggest that the aminoacylation active site of MST1 is responsible for hydrolyzing the majority (∼80%) of the Ser-AMP formed, whereas the remainder is released into solution and hydrolyzed independently of the enzyme. This is supported by our structural data, which show that Ser-AMP binds into the active-site pocket in a manner resembling Thr-AMP (Fig. 5). However, our binding assays also show that MST1 binds the nonhydrolyzable analog of Ser-AMP with 100-fold less affinity compared with the mimic of Thr-AMP and that it releases Ser-AMP at a faster rate than Thr-AMP (Fig. 3). This is likely a consequence of the differences in the way the particular aminoacyl groups interact with the Zn2+ ion in the active site. Most importantly, whereas the γ-OH of the threonyl moiety interacts primarily with Zn2+, the corresponding group in the seryl moiety interacts more closely with the surrounding water molecules and the side chains in the active site (Fig. 5). This is perhaps the main reason why Ser-AMP binds to the active site with lesser affinity and why it is released into solution at a much faster rate compared with Thr-AMP.
However, because the differences in the binding affinities and dissociation rates between the cognate Thr-AMP and the near-cognate Ser-AMP are not sufficient to prevent the misincorporation of Ser, we postulated that the aminoacylation site of MST1 might be employed for hydrolysis of the misactivated Ser-AMP. Indeed, our data show that MST1 hydrolyzes Ser-AMP at a significantly faster rate than Thr-AMP (Fig. 3). The detailed comparison of the crystal structure of the MST1-SAM binary complex with that of MST1-TAM provides an explanation as to why MST1 hydrolyzes Ser-AMP more rapidly. In the crystal containing the MST1-TAM binary complex, a water molecule is bound in the active site near the Thr-AMP analog but is not positioned properly for a nucleophilic attack. In contrast, in the MST1-SAM crystal, the same water molecule (Wat1) is positioned at an optimal angle (Fig. 5), albeit not at the optimal distance (4.5 Å), for the attack onto the mimic of the phosphoryl group. A structural comparison shows that the binding of SAM induces a conformational change in the active site that promotes the repositioning of Wat1. In particular, the active-site lid adopts a more closed conformation in MST1-SAM compared with MST1-TAM (Fig. 4). The lid residues move toward the active-site groove, and this movement brings Tyr109 and Lys273 closer to Wat1. Interestingly, Tyr109 and Lys273 are highly conserved among ThrRSs (Fig. 5 and supplemental Fig. S3) and, therefore, could be essential catalytic residues. Thus, we propose that SAM binding promotes rearrangements of the putative catalytic residues, Tyr109 and Lys273, which in turn, orient the putative hydrolytic water molecule for the nucleophilic attack onto the phosphoester linkage between the aminoacyl group and AMP. This conformation is stabilized in the presence of Ser-AMP and not in the presence of Thr-AMP, thus providing an explanation as to why MST1 preferentially hydrolyzes the misactivated near-cognate Ser-AMP conjugate. In conclusion, our study reveals how an aaRS that lacks the editing domain is capable of preventing the misacylation events and the subsequent mistranslational errors to occur.
Supplementary Material
Acknowledgment
We thank Dr. Franz Lang for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant GM22854 through the NIGMS (to D. S.). This work was also supported by the American Cancer Society, Illinois Division, Inc. and by National Institutes of Health through the NIGMS (to M. S.). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences Contract W-31-109-Eng-38.

This article contains supplemental Figs. S1–S5, Tables S1 and S2, and text.
The atomic coordinates and structure factors (code 4EO4) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- aaRS
- aminoacyl-tRNA synthetase
- aa-AMP
- aminoacyl adenylate
- aa-tRNA
- aminoacyl-tRNA
- PDB
- Protein Data Bank
- SAM
- seryl sulfamoyl adenylate
- Ser-AMP
- seryl adenylate
- TAM
- threonyl sulfamoyl adenylate
- Thr-AMP
- threonyl adenylate
- ThrRS
- threonyl-tRNA synthetase.
REFERENCES
- 1. Ibba M., Söll D. (2000) Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 69, 617–650 [DOI] [PubMed] [Google Scholar]
- 2. Ruan B., Palioura S., Sabina J., Marvin-Guy L., Kochhar S., Larossa R. A., Söll D. (2008) Quality control despite mistranslation caused by an ambiguous genetic code. Proc. Natl. Acad. Sci. U.S.A. 105, 16502–16507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Netzer N., Goodenbour J. M., David A., Dittmar K. A., Jones R. B., Schneider J. R., Boone D., Eves E. M., Rosner M. R., Gibbs J. S., Embry A., Dolan B., Das S., Hickman H. D., Berglund P., Bennink J. R., Yewdell J. W., Pan T. (2009) Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 462, 522–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li L., Boniecki M. T., Jaffe J. D., Imai B. S., Yau P. M., Luthey-Schulten Z. A., Martinis S. A. (2011) Naturally occurring aminoacyl-tRNA synthetases editing-domain mutations that cause mistranslation in Mycoplasma parasites. Proc. Natl. Acad. Sci. U.S.A. 108, 9378–9383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reynolds N. M., Lazazzera B. A., Ibba M. (2010) Cellular mechanisms that control mistranslation. Nat. Rev. Microbiol. 8, 849–856 [DOI] [PubMed] [Google Scholar]
- 6. Bacher J. M., de Crécy-Lagard V., Schimmel P. R. (2005) Inhibited cell growth and protein functional changes from an editing-defective tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 102, 1697–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roy H., Ling J., Irnov M., Ibba M. (2004) Post-transfer editing in vitro and in vivo by the β-subunit of phenylalanyl-tRNA synthetase. EMBO J. 23, 4639–4648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karkhanis V. A., Mascarenhas A. P., Martinis S. A. (2007) Amino acid toxicities of Escherichia coli that are prevented by leucyl-tRNA synthetase amino acid editing. J. Bacteriol. 189, 8765–8768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reynolds N. M., Ling J., Roy H., Banerjee R., Repasky S. E., Hamel P., Ibba M. (2010) Cell-specific differences in the requirements for translation quality control. Proc. Natl. Acad. Sci. U.S.A. 107, 4063–4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee J. W., Beebe K., Nangle L. A., Jang J., Longo-Guess C. M., Cook S. A., Davisson M. T., Sundberg J. P., Schimmel P., Ackerman S. L. (2006) Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 443, 50–55 [DOI] [PubMed] [Google Scholar]
- 11. Ling J., Reynolds N., Ibba M. (2009) Aminoacyl-tRNA synthesis and translational quality control. Annu. Rev. Microbiol. 63, 61–78 [DOI] [PubMed] [Google Scholar]
- 12. Mascarenhas A. P., An S., Rosen A. E., Martinis S. A., Musier-Forsyth K. (2008) in Protein Engineering (RajBhandary U. L., Köhrer C., eds) pp. 153–200, Springer-Verlag, New York [Google Scholar]
- 13. Schmidt E., Schimmel P. (1994) Mutational isolation of a sieve for editing in a transfer RNA synthetase. Science 264, 265–267 [DOI] [PubMed] [Google Scholar]
- 14. Ahel I., Korencic D., Ibba M., Söll D. (2003) Trans-editing of mischarged tRNAs. Proc. Natl. Acad. Sci. U.S.A. 100, 15422–15427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lincecum T. L., Jr., Tukalo M., Yaremchuk A., Mursinna R. S., Williams A. M., Sproat B. S., Van Den Eynde W., Link A., Van Calenbergh S., Grøtli M., Martinis S. A., Cusack S. (2003) Structural and mechanistic basis of pre- and post-transfer editing by leucyl-tRNA synthetase. Mol. Cell 11, 951–963 [DOI] [PubMed] [Google Scholar]
- 16. An S., Musier-Forsyth K. (2004) Trans-editing of Cys-tRNAPro by Haemophilus influenzae YbaK protein. J. Biol. Chem. 279, 42359–42362 [DOI] [PubMed] [Google Scholar]
- 17. Guo M., Chong Y. E., Shapiro R., Beebe K., Yang X. L., Schimmel P. (2009) Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature 462, 808–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dock-Bregeon A. C., Rees B., Torres-Larios A., Bey G., Caillet J., Moras D. (2004) Achieving error-free translation: the mechanism of proofreading of threonyl-tRNA synthetase at atomic resolution. Mol. Cell 16, 375–386 [DOI] [PubMed] [Google Scholar]
- 19. Baldwin A. N., Berg P. (1966) Transfer ribonucleic acid-induced hydrolysis of valyladenylate bound to isoleucyl ribonucleic acid synthetase. J. Biol. Chem. 241, 839–845 [PubMed] [Google Scholar]
- 20. Jakubowski H. (1978) Valyl-tRNA synthetase from yellow lupin seeds: instability of enzyme-bound noncognate adenylates versus cognate adenylate. FEBS Lett. 95, 235–238 [DOI] [PubMed] [Google Scholar]
- 21. Nomanbhoy T. K., Hendrickson T. L., Schimmel P. (1999) Transfer RNA-dependent translocation of misactivated amino acids to prevent errors in protein synthesis. Mol. Cell 4, 519–528 [DOI] [PubMed] [Google Scholar]
- 22. Fukunaga R., Yokoyama S. (2006) Structural basis for substrate recognition by the editing domain of isoleucyl-tRNA synthetase. J. Mol. Biol. 359, 901–912 [DOI] [PubMed] [Google Scholar]
- 23. Gruic-Sovulj I., Rokov-Plavec J., Weygand-Durasevic I. (2007) Hydrolysis of non-cognate aminoacyl-adenylates by a class II aminoacyl-tRNA synthetase lacking an editing domain. FEBS Lett. 581, 5110–5114 [DOI] [PubMed] [Google Scholar]
- 24. Splan K. E., Ignatov M. E., Musier-Forsyth K. (2008) Transfer RNA modulates the editing mechanism used by class II prolyl-tRNA synthetase. J. Biol. Chem. 283, 7128–7134 [DOI] [PubMed] [Google Scholar]
- 25. Boniecki M. T., Vu M. T., Betha A. K., Martinis S. A. (2008) CP1-dependent partitioning of pre-transfer and post-transfer editing in leucyl-tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 105, 19223–19228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Minajigi A., Francklyn C. S. (2010) Aminoacyl transfer rate dictates choice of editing pathway in threonyl-tRNA synthetase. J. Biol. Chem. 285, 23810–23817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roy H., Ling J., Alfonzo J., Ibba M. (2005) Loss of editing activity during the evolution of mitochondrial phenylalanyl-tRNA synthetase. J. Biol. Chem. 280, 38186–38192 [DOI] [PubMed] [Google Scholar]
- 28. Copeland R. A. (2000) in Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis (Copeland R. A., ed) 2 Ed., pp. 305–317, John Wiley and Sons, New York [Google Scholar]
- 29. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 30. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Cowtan K. (2004) COOT: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 33. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of COOT. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dock-Bregeon A., Sankaranarayanan R., Romby P., Caillet J., Springer M., Rees B., Francklyn C. S., Ehresmann C., Moras D. (2000) Transfer RNA-mediated editing in threonyl-tRNA synthetase: the class II solution to the double discrimination problem. Cell 103, 877–884 [DOI] [PubMed] [Google Scholar]
- 35. Su D., Lieberman A., Lang B. F., Simonović M., Söll D., Ling J. (2011) An unusual tRNAThr derived from tRNAHis reassigns in yeast mitochondria the CUN codons to threonine. Nucleic Acids Res. 39, 4866–4874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ling J., Peterson K. M., Simonović I., Cho C., Söll D., Simonović M. (2012) Yeast mitochondrial threonyl-tRNA synthetase recognizes tRNA isoacceptors by distinct mechanisms and promotes CUN codon reassignment. Proc. Natl. Acad. Sci. U.S.A. 109, 3281–3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zaher H. S., Green R. (2009) Fidelity at the molecular level: lessons from protein synthesis. Cell 136, 746–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Drummond D. A., Wilke C. O. (2008) Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Drummond D. A., Wilke C. O. (2009) The evolutionary consequences of erroneous protein synthesis. Nat. Rev. Genet. 10, 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo M., Schimmel P. (2012) Structural analyses clarify the complex control of mistranslation by tRNA synthetases. Curr. Opin. Struct. Biol. 22, 119–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sarkar J., Martinis S. A. (2011) Amino acid-dependent shift in tRNA synthetase editing mechanisms. J. Am. Chem. Soc. 133, 18510–18513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lue S. W., Kelley S. O. (2005) An aminoacyl-tRNA synthetase with a defunct editing site. Biochemistry 44, 3010–3016 [DOI] [PubMed] [Google Scholar]
- 43. Hendrickson T. L., Nomanbhoy T. K., de Crécy-Lagard V., Fukai S., Nureki O., Yokoyama S., Schimmel P. (2002) Mutational separation of two pathways for editing by a class I tRNA synthetase. Mol. Cell 9, 353–362 [DOI] [PubMed] [Google Scholar]
- 44. Hati S., Ziervogel B., Sternjohn J., Wong F. C., Nagan M. C., Rosen A. E., Siliciano P. G., Chihade J. W., Musier-Forsyth K. (2006) Pre-transfer editing by class II prolyl-tRNA synthetase: role of aminoacylation active site in “selective release” of noncognate amino acids. J. Biol. Chem. 281, 27862–27872 [DOI] [PubMed] [Google Scholar]
- 45. Zhu B., Yao P., Tan M., Eriani G., Wang E. D. (2009) tRNA-independent pretransfer editing by class I leucyl-tRNA synthetase. J. Biol. Chem. 284, 3418–3424 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.