

Background: P5 ATPases are omnipresent putative Ca2+ pumps in eukaryotes, but their substrate specificity remains disputed.
Results: Phosphoenzyme stability of a plant P5A ATPase is influenced by Ca2+ ions independently of transport.
Conclusion: Ca2+ ions might modulate P5A ATPase activity but are not transported ligands.
Significance: The effect of P5A ATPases on Ca2+ homeostasis is indirect, possibly via the vesicular transport machinery.
Keywords: ATPases, Endoplasmic Reticulum (ER), Phosphorylation, Plant, Transport, Yeast, HvP5A1, P5 ATPase, P5A ATPase, Spf1p
Abstract
P5 ATPases constitute the least studied group of P-type ATPases, an essential family of ion pumps in all kingdoms of life. Although P5 ATPases are present in every eukaryotic genome analyzed so far, they have remained orphan pumps, and their biochemical function is obscure. We show that a P5A ATPase from barley, HvP5A1, locates to the endoplasmic reticulum and is able to rescue knock-out mutants of P5A genes in both Arabidopsis thaliana and Saccharomyces cerevisiae. HvP5A1 spontaneously forms a phosphorylated reaction cycle intermediate at the catalytic residue Asp-488, whereas, among all plant nutrients tested, only Ca2+ triggers dephosphorylation. Remarkably, Ca2+-induced dephosphorylation occurs at high apparent [Ca2+] (Ki = 0.25 mm) and is independent of the phosphatase motif of the pump and the putative binding site for transported ligands located in M4. Taken together, our results rule out that Ca2+ is a transported substrate but indicate the presence of a cytosolic low affinity Ca2+-binding site, which is conserved among P-type pumps and could be involved in pump regulation. Our work constitutes the first characterization of a P5 ATPase phosphoenzyme and points to Ca2+ as a modifier of its function.
Introduction
With the rise of genomic sequencing, a group of eukaryotic sequences was discovered clustering together as the fifth subfamily of P-type ATPase pumps (P5 ATPases) (1). P5 ATPases have been identified in every single eukaryotic genome analyzed so far, whereas no homologues can be detected in bacterial genomes (2). Although several members of this family are involved in severe human diseases (3–5), the substrate specificity of any P5 ATPase remains a mystery.
P-type ATPases form a large superfamily of primary active pumps coupling ATP hydrolysis to the transport of a substrate across the membrane (6, 7). The name “P-type” is derived from the fact that they are phosphorylated at a conserved aspartate residue during each catalytic cycle. They are remarkably simple with only a single catalytic subunit and carry out large domain motions during transport. Atomic structure of P-type ATPases in different conformations together with ample mutagenetic evidence have provided detailed insights into the pumping mechanism of these biological nanomachines (8, 9).
P-type ATPases are very divergent with respect to primary structure but can be recognized in genomes by conserved core sequences and signature motifs. Phylogenetically, P-type ATPases are divided into five subfamilies named P1 through P5 (1). These differ with respect to transported ligands and the way they are regulated. P1-P3 ATPases are well characterized ion pumps as follows: P1A pumps are part of the bacterial K+ transport systems; P1Bs are heavy metal pumps; P2A and P2B are Ca2+ pumps; P2C Na+/K+ are pumps of animals; P2Ds are Na+ pumps of fungi and mosses, and P3As are plasma membrane H+ pumps. In contrast to ion transport, P4 pumps participate in lipid flipping across membranes, whereas P5 ATPases have no assigned specificity so far.
P5 ATPases can be identified by a signature sequence, the PPXXP motif, found in transmembrane segment M4, whereas most other P-type ATPases have a PXXX(P/L) motif at the same position that is involved in substrate binding (2, 10). P5 ATPases can be phylogenetically divided into two distinct subgroups (P5A and P5B ATPases) having different subcellular localizations and physiological roles (10). Whereas P5A ATPases are found in all eukaryotic genomes, P5B ATPases have been lost in some lineages of multicellular eukaryotes, including land plants (2).
P5A ATPases appear to be exclusively expressed in the endoplasmic reticulum (ER),6 and loss of the P5A ATPase gene function leads to broad unspecific phenotypes related to an inability to control basic ER functions (11–17). The best characterized P5A ATPase is Spf1p in the yeast Saccharomyces cerevisiae (18). Spf1p was identified in a screen for mutants showing insensitivity to the killer toxin SMKT secreted by the competing yeast Pichia farinosa (hence the name Sensitivity to P.farinosa 1; SPF1) (18). As the Spf1p protein is in the ER and the toxin interacts with the plasma membrane, the effect is most likely indirect (19). Other phenotypes of spf1 cells indicate that Spf1p exerts its function in the ER where loss of its function leads to impairment of ER processes such as protein folding and processing (12, 14, 17–20). In the plant Arabidopsis thaliana, disruption of the single P5A ATPase gene, AtP5A1/MIA/PDR2, compromises male fertility (13) and root meristem patterning in response to phosphate availability (16). In both cases, these effects correlate with sensitizing of a subset of ER quality control responses (13, 16). Many of the observed phenotypes could be related to an effect of P5A ATPases on the ER Ca2+ homeostasis, suggesting that they might be ER Ca2+ pumps.
This study constitutes characterization of a P5A ATPase from barley employing phosphoenzyme formation and degradation as a tool to get insight into the catalytic mechanism. We demonstrate that Ca2+ merely has a modifying effect on protein function but is not likely to be the transported ion. We further show that residues in the phosphorylation and dephosphorylation motifs and the putative substrate-binding site in M4 are important for the function of the P5A ATPase.
EXPERIMENTAL PROCEDURES
Cloning of Full-length HvP5A1 cDNA from Barley
Basic Local Alignment Search Tool (BLAST) at TIGR was used to extract sequence information from barley to generate primers for cloning. TIGR contains 370,546 expressed sequence tags (ESTs) from barley. This information was supplemented with the wheat EST information (580,155 available ESTs). BLAST was performed with the sequence of the rice P5A ATPase obtained using PlantsT. ESTs used for cloning were CA497971 (from ATG), BU980814, and CB877529 (3′ UTR).
Young leaves were isolated from Hordeum vulgare (barley) variety Golden Promise, and total RNA was extracted using FastRNA Pro green kit (Qbiogene). HvP5A1 gene-specific primers were designed from ESTs and used to generate cDNA fragments with SuperScript III first strand synthesis system (Invitrogen). Primers Oli35 5′-ATGGCGCGGTTCGAG-3′ (forward) and Oli36 5′-AGTCCGACAAAATTCTCCTAAACTAC-3′ (reverse) were used to generate HvP5A1.1 and HvP5A1.2, respectively. A BstEII restriction site 1,620 bp within the HvP5A1 sequence was used to ligate the two cDNA clones together to generate a full-length HvP5A1 cDNA clone in the 2.1-TOPO vector (pAM1). The full-length sequence corresponds to GenBankTM accession number BAJ85414. Prediction of transmembrane helices was carried out using the TOPCONS prediction server (21).
Functional Complementation of the Arabidopsis mia Insertion Mutant with HvP5A1
Arabidopsis wild-type (WS-2) seeds and seeds homozygous for the mia tDNA insertion mutant allele (13) were placed in a controlled growth chamber with short day conditions (8 h light, 200 μmol m−2 s−1, 70% humidity, 20 °C). After 3 weeks, the plants were transferred to long day conditions (16 h light, 200 μmol m−2 s−1, 70% humidity, 20 °C).
Vectors for plant transformation were constructed by insertion of HvP5A1 from KpnI/XbaI-digested plasmid pAM1 into the binary plant transformation vector pPZP211 under the control of the 35S promoter (pAM7). Arabidopsis mia seeds were transformed according to the floral dip method (22) with pAM7 and empty plasmid pPZP211. For kanamycin selection, plants were grown on agar plates containing 0.5× Murashige and Skoog (MS) medium (23) supplied with 50 mg/liter kanamycin.
Kanamycin-selected plants were investigated for the presence of HvP5A1 by PCR on genomic DNA using Oli35 5′-ATGGCGCGGTTCGAG-3′ (forward) and Oli36 5′-AGTCCGACAAAATTCTCCTAAACTAC-3′ (reverse). RNA was isolated with the Qiagen RNeasy kit, and quantitative real time PCR was performed to investigate expression levels of HvP5A1. Gene-specific primers for actin (reverse 5′-TCTGTGAACGATTCCTGGAC-3′ and forward 5′-CTTCCCTCAGCACATTCCAG-3′) were employed for normalization.
Subcellular Localization of HvP5A1
HvP5A1 and GFP were PCR-amplified and introduced into the TOPO entry Gateway vector pENTR.SD (Invitrogen). A Gateway LR clonase II (Invitrogen) reaction (24) resulted in vector pAM10 encoding HvP5A1 fused in its C-terminal end to the N-terminal end of green fluorescent protein (GFP). As a control for ER localization, we used GFP tagged with an ER retention signal (25).
Onion (Allium cepa L.) epidermal strips were placed on agar containing MS salt mixture (1 × MS salt (Invitrogen) 2% agar, 3% sucrose, pH 5.8) and bombarded with either of the three vectors pAM10, GFP, and ER-control using a PDS-1000/He biolistic particle delivery system (Bio-Rad). A total of 10 μg of each expression vector was coated onto 1-μm gold particles and transferred into cells using a hepta adaptor and a helium pressure of 1,100 p.s.i. Following bombardment, Petri dishes containing bombarded epidermal strips were sealed with MicroporeTM tape (Neuss, Germany) and placed in darkness at 22 °C for 18 h. Transformed cells expressing the HvP5A1-GFP fusion protein were visualized using a Zeiss LSM Pascal 5 confocal laser scanning microscope equipped with a C-Apochromat 40 × 1.2 water-immersion lens. The excitation wavelength for GFP was 488 nm. A total of 50 cells for each construct was observed, and images were processed using the ImageJ software (National Institutes of Health).
Functional Complementation of the Yeast spf1 Mutant with HvP5A1
A fragment containing the HvP5A1 full-length clone was excised from plasmid pAM1 and inserted into the yeast expression vector YEp351 under control of the constitutive PMA1 promoter (pAM2). The phosphorylation site of HvP5A1 was destroyed by mutation of Asp-488 into Asn hereby generating plasmid pAM3 containing HvP5A1-D488N. A histidine tag (RGS-His6) was introduced in the C terminus of HvP5A1 and HvP5A1-D488N by PCR, generating plasmid pAM5 (HvP5A1-RGS-His6) and pAM6 (HvP5A1-D488N-RGS-His6).
Plasmids YEp351 (empty control), pAM5, and pAM6 were expressed in three yeast strains (Euroscarf) Y00000 (wild type; Mata, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0), Y00272 (spf1; Mat a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, YEL031w::kanMX4), and Y01587 (ypk9; Mat a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, YOR291w::kanMX4). Experiments were performed at 30 °C in synthetic minimal media supplemented with galactose (2%), histidine (30 μg/ml), methionine (30 μg/ml), and uracil (30 μg/ml) with indicated amounts of caffeine. Standard SDS-PAGE and Western blot experiments were performed to investigate expression level of proteins using anti-RGS-His6 antibody for detection.
Protein Expression in Yeast RS72 and Purification of Crude Membranes
Produced vectors were transformed into the yeast expression strain RS72 (MATa, ade1-100, his4-519, leu2-3,112, pPMA1:pGAL) by the lithium acetate method (26), and transformants were selected on minimal media plates containing 0.7% yeast nitrogen base (without amino acids; Difco) supplemented with adenine (40 μg/ml), l-histidine (30 μg/ml) and 2% galactose (SGAH). Positive transformants were used to saturate a 100 ml of SGAH pre-culture grown at 30 °C, 150 rpm. At saturation, the cells were transferred to 1 liter of rich medium containing 2% glucose or galactose as indicated, 1% yeast extract and 2% bactopeptone, and grown for 22 h at 25 °C, 150 rpm.
For purification of crude membranes, RS72 cultures with transformed constructs were harvested after 22 h by centrifugation (3,000 rpm, 5 min). Homogenization buffer used consisted of 29% glycerol, 10 mm EDTA, 75 mm MES, pH 6.5, with N-methyl-d-glucamine, and 1 mm DTT. Between purification steps all samples were kept on ice. For each sample, 1 ml of homogenization buffer/g of yeast cells and 40 μl of 0.1 m PMSF and 1 mg/ml pepstatin were added. Ice-cold glass beads (BioSpec) were added to half the volume, and the samples were vortexed five times for 1 min with a minimum of 1 min between shakes on ice. The samples were centrifuged (3,000 rpm, 15 min, 2 °C) followed by ultracentrifugation of the supernatant (50,000 rpm, 1 h, 2 °C). The membrane pellet was dissolved in homogenization buffer, and total protein concentration was determined by Bradford assay (Bio-Rad). The samples were aliquoted, frozen in liquid nitrogen, and stored at −80 °C.
Measuring Formation and Degradation of the HvP5A1 Phosphoenzyme
The phosphorylation status of HvP5A1 protein expressed in yeast RS72 crude membranes was measured at 0 °C essentially as described (27, 28). Microsomes of yeast expressing HvP5A1-RGS-His6 as either wild type or with the indicated mutations were diluted in homogenization buffer to 10 μg/μl. 40 μg of total protein was added to a final volume of 50 μl of either standard reaction buffer (20 mm MOPS, pH 6.5, with N-methyl-d-glucamine, 1 mm MgCl2, 1 mm DTT) or the standard reaction buffer with additions as indicated in the figure legends. Free concentrations of cations and EGTA were calculated by the extended version of MaxChelator. The assay was started by addition of 2.5 mCi of [γ-32P]ATP, 7 μm ATP and was left on ice for 60 s or as indicated before quenching by addition of 400 μl of stop buffer (20% trichloroacetic acid, 1 mm phosphoric acid). For dephosphorylation experiments, chase solutions were added in 5-μl volumes giving the indicated final concentrations before quenching at the indicated time points. For determination of pH dependence, low (pH 5.2) and high (pH 9.6) pH buffers, similar to the standard reaction buffer but with MES replacing MOPS in the low pH buffer, were mixed to give a pH gradient. Similar concentrations of enzyme as used in the reaction measurements were added to 200 μl of standard reaction buffer followed by a short incubation on ice. Following addition of ATP, the final pH of the assay buffers was measured using a pH electrode adjusted to 0 °C. Phosphorylation with inorganic phosphate was carried out in 1 mm MgCl2, 0.1 mm 32Pi, 20 or 40% DMSO, 20 mm MOPS, pH 6.5, adjusted with N-methyl-d-glucamine, at 25 °C.
After phosphorylation, all samples were incubated on ice for 10 min followed by centrifugation at 16,000 × g for 15 min. The pellet was washed once in 400 ml of stop buffer and dissolved in loading buffer (50 μm NaH2PO4, pH 6.0, 0.01% SDS, 20 mm mercaptoethanol, 0.014% bromphenol blue, and 0.1 g/ml Li-SDS). An equal amount of protein for each sample was loaded onto 8% acid SDS-polyacrylamide gels. After running, radioactivity was visualized in a phosphorimager scanner (Storm 860, GE Healthcare), and individual bands corresponding to HvP5A1 or the mutated proteins were quantified. The gels were subsequently stained by Coomassie Brilliant Blue to ensure an equal loading of protein. Graphs were fitted to Michaelis-Menten or exponential decay kinetics using GraphPad Prism 5.0, and rates constants were estimated from the program. We routinely rehydrated SDS-polyacrylamide gels and stained with Coomassie Brilliant Blue to ensure that an equal amount of protein was loaded for each sample.
RESULTS
Expression of a Barley P5A ATPase in Arabidopsis Rescues the mia Insertion Mutant
The only plant P5A ATPase characterized so far is AtP5A1/MIA/PDR2, the single P5A ATPase in the model plant A. thaliana (13, 16). As biochemical characterization of the MIA gene product in our hands proved difficult, no matter whether the gene was expressed in planta or in fungal expression systems, we pursued cloning of P5A ATPases from other plants, including barley. One barley EST showed high similarity to the rice gene OsP5 (29), and the retrieved sequence information was used for cloning of the full-length barley P5A ATPase, which we named HvP5A1. The resulting cDNA encodes a polypeptide of 1,174 amino acids with a predicted molecular mass of 131 kDa and contains signature motifs present in all known P-type ATPases (Fig. 1) (6). HvP5A1 also contains the P5 ATPase-specific signature sequence PPXXPXX located in the transmembrane helix preceding the large cytoplasmic loop that includes the phosphorylated aspartate residue (6). The presence of two glutamate residues in the motif (PPELPME) and the presence of 12 predicted membrane-spanning regions (underlined and numbered Mb, Ma, and M1–10; Fig. 1) further links HvP5A1 to the P5A subgroup (2, 7, 10).
FIGURE 1.

HvP5A1cDNA encodes a P5A P-type ATPase. A, primary sequence of HvP5A1. Underlined residues are predicted to be transmembrane helices; residues in gray are P-type ATPase-associated motifs, including the P5A-specific motif PPXXP found in M4. P-type ATPase-specific motifs include the phosphorylation motif DKTGTLT, in which the aspartate residue is phosphorylated during the reaction cycle, and the LTGES, KG(A/S)PE, and M(I/V/C)TGD motifs (5). B, representation of the 12 predicted transmembrane helices. P indicates phosphorylation of the conserved aspartate in the phosphorylation site.
Mutation of Arabidopsis AtP5A1/MIA results in reduced plant stature, reduced sizes of siliques, reduced fertility, and reduced seed yield (13). To test whether MIA and HvP5A1 are functional homologues, the mia mutant was transformed with HvP5A1 under the control of the cauliflower mosaic virus 35S promotor. HvP5A1-transformed mia mutants showed a restoration of silique length and seed yield to that of wild-type levels, whereas transformation with empty plasmid control did not result in rescue of the mia phenotype (Fig. 2). Furthermore, the number of infertile ovules was compared with the number of fertile ovules, and we found that the fertility was restored in the mia HvP5A1 plants (92% compared with 94% in the wild type, n = 30). This demonstrates that HvP5A1 is a functional homologue of MIA.
FIGURE 2.

Expression of HvP5A1 functionally complements the Arabidopsis mia insertion mutant. A, normal siliques of wild-type plant (upper left) resemble those from the mia mutant transformed with HvP5A1 (upper right), and siliques from the mia mutant left untransformed (lower left) or transformed with an empty vector (lower right) have markedly reduced sizes. B, mia mutant has a much reduced seed yield as compared with wild-type plant. Expression of HvP5A1, but not the empty vector control (pPZP211), restores seed yield to wild-type levels. C, quantitative real time PCR demonstrates expression of HvP5A1 in transgenic Arabidopsis. HvP5A1 is expressed to about the same level in mia plants as MIA is expressed in wild-type plants. n.d., not detectable.
HvP5A1 Localizes to the ER
So far, all examined P5A ATPases have been identified in membranes of the secretory pathway (12–15, 20). To test whether this is also true for HvP5A1, we investigated its subcellular localization in planta.
A construct containing an HvP5A1:GFP fusion driven by the cauliflower mosaic virus 35S promoter was generated and transformed into onion epidermal cells by particle bombardment. Epidermal leaf cells expressing this HvP5A1:GFP construct exhibited dense fluorescent nuclei (inset in Fig. 3A) and also a cortical ER-like network (Fig. 3A). The network aligned the cortical cytoplasm and did not extend into trans-vacuolar cytoplasmic strands. A control construct in which GFP was fused to retention signal for the ER had a similar fluorescence pattern (Fig. 3C). Cells expressing GFP alone showed diffuse fluorescence throughout the cytoplasm and nucleus of all transformed cells (Fig. 3E). These findings indicate that HvP5A1 localizes to ER membranes when expressed transiently in a plant host.
FIGURE 3.

HvP5A1 localizes to the ER membrane. HvP5A1-GFP (A and B), GFP-HDEL (C and D), and GFP (E and F) were individually expressed in onion epidermal cells. HvP5A1-GFP and GFP-HDEL show distinct fluorescent cortical networks, whereas GFP alone exhibits characteristic diffuse fluorescence throughout the cytoplasm and nucleus. A, C, and E are stacked images of the upper 10 μm of transformed fluorescent cells. Insets are maximum intensity images of Z-projections of all confocal planes of the same individual cells. B, D, and F are bright field images. Scale bars, 100 μm.
Heterologous Expression of HvP5A1 in Yeast Results in a Functionally Active HvP5A1 Protein
Heterologous yeast systems have previously been used with success to produce a variety of plant P-type ATPases in high yield for functional and biochemical analysis (30–32). We therefore wanted to test whether such a system could be used to produce a catalytically active form of the barley HvP5A1 protein suited for biochemical characterization.
Mutants in the single P5A ATPase gene of S. cerevisiae, SPF1, have previously been associated with growth-deficient phenotypes on compounds affecting ER functions such as tunicamycin (12), DTT (33), lovastatin (11), and caffeine (34). We observed that the caffeine phenotype appears to be specific for P5A ATPases, as the spf1 mutant was sensitive to caffeine (Fig. 4A), whereas a deletion mutant of the single P5B ATPase YPK9, which localizes to vacuolar membranes (35), was not (Fig. 4B).
FIGURE 4.
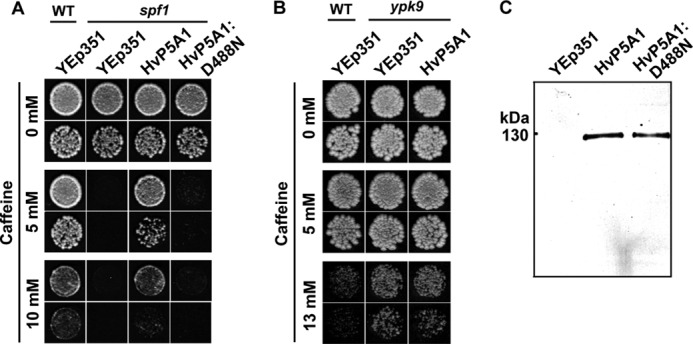
Expression of HvP5A1 functionally complements yeast spf1 knock-out cells. A, wild-type cells transformed with empty vector (YEp351) tolerate growth on caffeine, whereas knock-out of spf1 results in growth deficiency under the same conditions. Transformation of spf1 cells with HvP5A1 restores growth to the level of wild-type, whereas transformation with empty YEp351 has no effect. Mutation of Asp-488 into Asn (HvP5A1-D488N) results in a catalytically dead protein unable to rescue spf1 knock-out cells. Growth was recorded on YPD plates containing increasing amounts of caffeine, and yeast cells were diluted to A600 0.01 (top rows) or 0.001 (bottom rows) before spotting. B, yeast ypk9 knock-out cells do not display a growth-deficient phenotype on caffeine (similar conditions as in A). C, HvP5A1 and HvP5A1-D488N are both expressed to similar levels in the spf1 strain as indicated by Western blotting against the C-terminal RGS-His6 tag. 15 μg of total protein as determined by Bradford assay was loaded in each lane.
We transformed C-terminally RGS-His6-tagged HvP5A1 (HvP5A1-RGS-His6; hereafter referred to as wildtype HvP5A1) into the yeast spf1 strain and recorded growth of transformed cells on medium containing increasing concentrations of caffeine (Fig. 4A). Under these conditions, expression of HvP5A1 functionally complemented spf1. The effect was specific for HvP5A1, as the spf1 strain transformed with empty vector control failed to complement the growth deficiency. This demonstrates that the product of the HvP5A1 gene is functionally equivalent to the yeast Spf1p.
Next, we transformed the spf1 strain with a mutated version of HvP5A1 in which the conserved aspartate residue in the phosphorylation sequence DKTGLT is replaced by Asn (HvP5A1-D488N). This would be anticipated to render the protein catalytically inactive as the phosphorylation site is destroyed. Indeed, in contrast to wild-type HvP5A1, the Asp-488 mutated version of HvP5A1 failed to complement the caffeine phenotype. Western blotting against the RGS-His6 tag located in the C terminus of both proteins revealed that these were expressed to the same levels (Fig. 4C). We hereby conclude that the ability of HvP5A1 to complement spf1 is linked to expression of a catalytically active protein.
HvP5A1 Shows Phosphorylation on the Catalytic Asp-488 Residue When Expressed in Yeast Membranes
An obligatory step in the catalytic cycle of P-type ATPases is the phosphorylation of a conserved aspartate residue by Mg2+-ATP and subsequent dephosphorylation to release Pi (36). ATPase activity can thus be determined as a function of released Pi. We were, however, not able to observe any significant difference in ATPase activity between membranes from spf1 yeast expressing either wild-type HvP5A1 or the catalytically dead HvP5A1-D488N mutant protein. A low expression level, slow turnover rate, or an inability of the gene product to undergo the full catalytic cycle could all explain the absence of detectable ATP hydrolytic activities of HvP5A1.
As an alternative approach to measure HvP5A1 activity, we tried to measure the steady-state phosphorylation levels of HvP5A1 following incorporation of 32P from [γ-32P]ATP. This method is more sensitive than detection of released phosphate by colorimetric methods and furthermore detects enzyme stalled in its catalytic cycle. For this purpose, we expressed HvP5A1 in the RS72 yeast strain in which expression of the predominant P-type ATPase in yeast, the plasma membrane H+-ATPase PMA1, is switched off on glucose medium (37). This approach allowed us to significantly lower the level of Pma1p during expression of HvP5A1 thus removing a likely artifact from of the assay (data not shown).
Membranes of RS72 expressing either wild-type HvP5A1 or, as controls, empty vector or the catalytically dead HvP5A1-D488N mutant protein were incubated with 7 μm [γ-32P]ATP in the presence of Mg2+ and subsequently fractionated by SDS-gel electrophoresis under acidic conditions, which preserve the phosphoenzyme intermediate. Autoradiography showed a band at the expected size of HvP5A1 (131 kDa) (Fig. 5A). The band was found to be sensitive to hydroxylamine, a compound that cleaves aspartyl-phosphate bonds (38), and was stable only during acidic to neutral conditions (pH below 10), which corresponds to hydrolysis of the same aspartyl-phosphate bond at alkaline pH levels (Fig. 5B). Most importantly, however, the band was absent in membranes of yeast expressing the HvP5A1-D488N-mutated protein (Fig. 5A). These findings indicate that the observed phosphoenzyme is the true reaction intermediate of the catalytic Asp-488 residue and does not include phosphorylation of other residues on HvP5A1 caused, for instance, by yeast endogenous protein kinase activity present in the membrane preparations.
FIGURE 5.
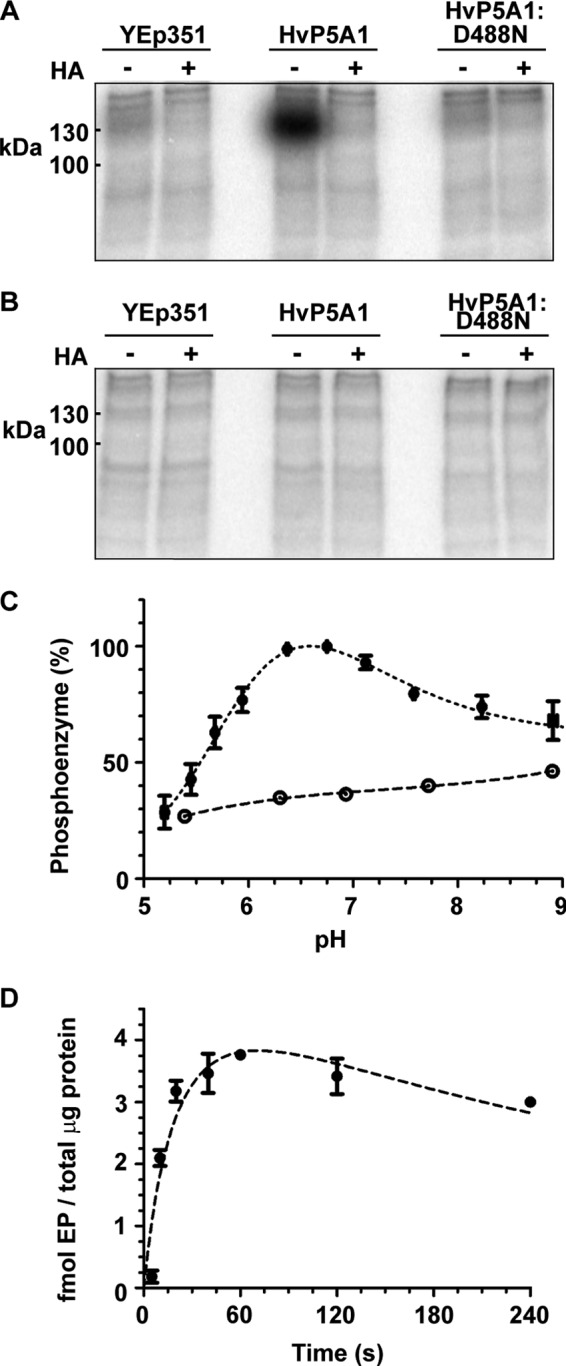
HvP5A1 forms a phosphoenzyme on Asp-488 in yeast crude membranes. A, incubation of crude membranes from RS72 yeast cells expressing HvP5A1-RGS-His6 with radiolabeled [γ-32P]ATP produces a phosphoenzyme at the expected size of HvP5A1. The phosphoenzyme is sensitive to 0.3 m hydroxylamine (HA) and is not detected in membranes of yeast expressing empty YEp351 or the catalytically dead protein (HvP5A1-D488N-RGS-His6). Incubations were performed in 20 mm MOPS adjusted to pH 6.5 with N-methyl-d-glucamine, 1 mm MgCl2, 2 mm DTT and at 0 °C. The reaction was quenched after 60 s with 400 ml 20% TCA, 1 mm phosphoric acid. B, aspartate-phosphate bond is sensitive to alkaline pH. Prior to analysis, the phosphoprotein pellet was washed with 2 m Tris-HCl, pH 10. C, pH dependence of phosphoenzyme formation measured in a MES/MOPS buffer system ranging from pH 5 to pH 9. The phosphoenzyme of RGS-His6-tagged wild-type HvP5A1 (●) shows a maximal presence at the physiological level between pH 6.5 to pH 7.0. Crude membranes from yeast expressing the RGS-His6-tagged and Asp-488 mutated protein (○) establish the background in the same range. D, phosphoenzyme formation reaches a steady-state level after ∼20 s. It is stable for up to 120 s and shows a modest decrease at 240 s.
Phosphorylation of Asp-488 Occurs Spontaneously and at a Slow Rate
In P-type ATPases, hydrolysis of ATP is tightly coupled to binding of the ligands to be transported. This guarantees that ATP is only hydrolyzed when ligand transport can occur (7).
Remarkably, we found that at the conditions used (20 mm MOPS, pH 6.5, d-methyl-N-glucamine, 1 mm MgCl2), HvP5A1 was able to undergo spontaneous catalytic phosphorylation when residing in yeast membranes. Therefore, either a transported substrate is present in the medium employed or in the yeast membranes. Alternatively, the formation of the E1P phosphoenzyme happens independently of ligand binding. As H+ will always be present in the assay, we tested the possibility that protons could influence the E1 to E1P partial reaction step. HvP5A1 showed optimal formation of the phosphoenzyme at pH 6.4 comparable with that of the well characterized plant plasma membrane P-type H+-ATPase (39, 40). Unlike this pump, however, activity was still marked at neutral to alkaline pH (Fig. 5C). Formation of the phosphoenzyme persisted to the same degree when 1 mm EGTA was included in the assay (data not shown).
We proceeded to measure the time dependence of the phosphorylation reaction. Here, we found that a maximal steady-state level was reached after ∼20 s with a half-maximal level observed at ∼10 s (Fig. 5D). The phosphoenzyme was furthermore found to be relatively stable for up to 120 s, although some decay was observed at 240 s. For comparison, the Ca2+-ATPase is under similar conditions (2 μm ATP and 0 °C) fully phosphorylated in less than 1 s (28). The slow phosphorylation kinetics of HvP5A1 is in accordance with a low turnover rate of the phosphoenzyme and readily explains why significant ATPase activity could not be detected.
Overall Low Turnover Rate of HvP5A1 Is Related to a Slow Transition from the E1P to E2P State
To identify whether HvP5A1 undergoes a full reaction cycle (i.e. phosphorylation followed by dephosphorylation) or whether it accumulates as a phosphorylated intermediate, we measured the sensitivity of the phosphoenzyme toward excess amounts of cold ATP (50 mm). If the protein is able to undergo a full catalytic cycle, the phosphoenzyme would under these conditions be expected to exchange the radiolabeled phosphate with unlabeled phosphate. Furthermore, as Mg2+ mediates the transfer of the negatively charged γ-phosphate of ATP to the negatively charged aspartate in the phosphorylation motif of P-type ATPases, we also measured the stability of the phosphoenzyme in the absence of Mg2+ (50 mm EDTA). In both cases, we could observe a decay of the phosphoenzyme to the level of the D488N mutant (Fig. 6A). In contrast, the phosphoenzyme was completely stable in buffer alone (Fig. 6A). This suggests that the protein is able to hydrolyze the 32P-aspartate bond in a catalytic manner.
FIGURE 6.
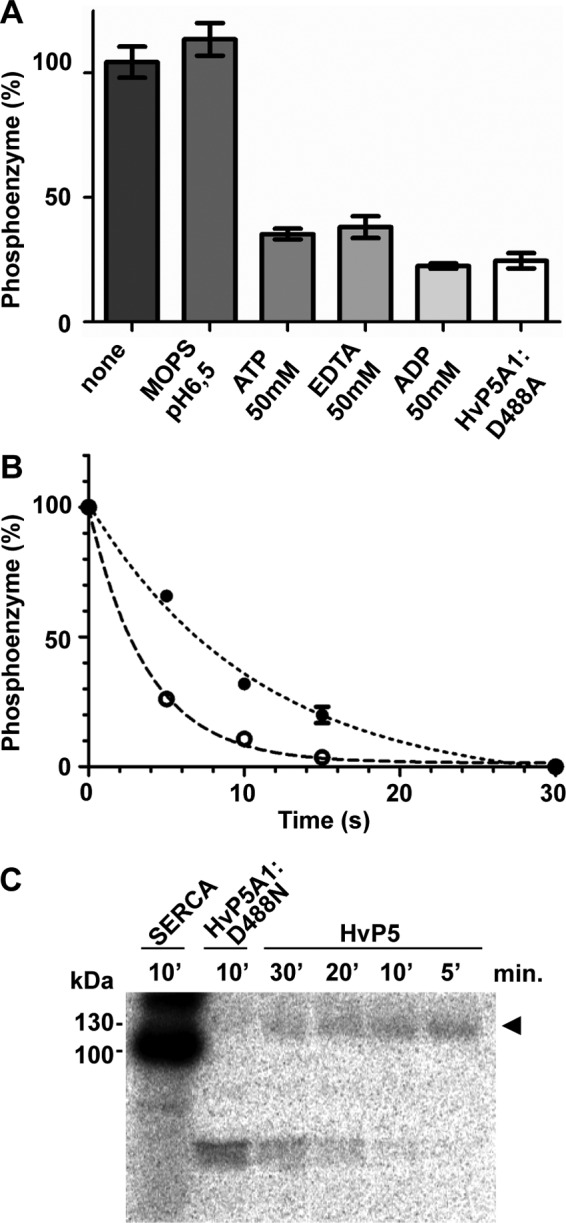
HvP5A1 phosphoenzyme accumulates mainly in the E1P state. A, phosphoenzyme of HvP5A1 is fully stable in MOPS buffer but is sensitive to nonradiolabeled ATP, EDTA, and ADP. Phosphorylation was performed under similar conditions as in Fig. 5A (60 s), and phosphoenzyme stability was measured by adding each of the components at t60 and quenching the reaction after another 30 s by addition of 400 ml of 20% TCA. The lowest levels are comparable with the background observed in membranes from yeast expressing the aspartate mutated HvP5A1-D488N protein. B, time dependence of the dephosphorylation reaction initiated by either 50 mm cold ATP (●) or 50 mm ADP (○). The measurements are adjusted to the lowest level observed after 30 s of dephosphorylation. C, sarcoplasmic reticulum Ca2+-ATPase expressed in crude RS72 yeast membranes readily forms a phosphoenzyme within 10 min after incubation with 0.1 mm 32Pi (in the absence of Ca2+). Wild-type HvP5A1 (arrowhead) hardly forms a phosphoenzyme after 30 min under similar conditions (1 mm MgCl2, 0.1 mm 32Pi, 20% DMSO, 20 mm MOPS, pH 6.5, adjusted with N-methyl-d-glucamine, at 25 °C). Increasing the DMSO content to 40% had no effect on phosphoenzyme formation. The phosphoenzyme observed for wild-type HvP5A1 is absent in the Asp-488 mutated protein (HvP5A1-D488N).
When the decay of the phosphoenzyme was studied as a function of time, it could be fitted with a monoexponential curve with a half-maximal dephosphorylation observed after 8 s (Fig. 6B). The results thus indicate that HvP5A1 is fully active but that its overall turnover rate is very slow. The latter could be related to a slow transition from E1P to E2P. To test the ratio of the phosphoenzyme that is in the E1P or E2P state, we compared the rate of dephosphorylation induced by ATP to that induced by ADP. In other P-type ATPases, only E1P, but not E2P, is capable of ATP synthesis from the ADP and bound phosphate. As a consequence, the E1P state is sensitive to ADP, whereas the E2P state is not (36). For HvP5A1, we observed a small but significant increase in the dephosphorylation rate by addition of ADP, which again could be fitted to a monophasic curve (Fig. 6B).
Our data indicate that the E1 form of HvP5A1 readily undergoes phosphorylation to E1P but is followed by a slow transition from the E1P to E2P state. As a result, the phosphoenzyme mainly populates in the E1P state. In line with this view, only a minor amount of the phosphoenzyme is able to form through the reverse reaction with inorganic phosphate in contrast to what was described for the Ca2+-ATPase (Fig. 6C) (41).
Ca2+ Accelerates Degradation of the HvP5A1 Phosphoenzyme
As many P-type ATPases are stimulated by cations and it remains an open question whether P5A ATPases are directly involved in ion transport, we screened for possible ligands that would stimulate the overall turnover rate of HvP5A1. The Murashige-Skoog plant growth medium (MS), which is commonly used to sustain growth of Arabidopsis, contains all necessary salts and vitamins required for plant growth (23). MS medium therefore seemed as a useful starting point to screen for possible substrate ions related to phosphoenzyme degradation of HvP5A1. We measured the stability of the formed phosphoenzyme after addition of MS to a final concentration of 4.4 mg/liter in a neutral pH buffer (20 mm MOPS, pH 6.5, with N-methyl-d-glucamine, 1 mm MgCl2). We found the phosphoenzyme to be unstable under these conditions (Fig. 7A), suggesting that a transported ligand or cofactor is present in the MS medium. The effect was only apparent with full MS medium and gradually diminished as the medium was diluted (data not shown). By further testing each compound used for formulation of the medium, and in its concentration present in the medium, we found that Ca2+ specifically caused the observed effect (Fig. 7A, upper panel).
FIGURE 7.

HvP5A1 phosphoenzyme is sensitive to Ca2+. A, stability of the HvP5A1 phosphoenzyme was measured after 30 s in buffer alone (MOPS, pH 6.5), in the presence of full plant MS growth medium (MS 4.4 g/liter, pH 6.5), or in the presence of each of the individual components of the MS growth medium as indicated. The experiment was conducted at conditions where the crude membranes were impermeable to ATP (upper panel) and at conditions where the membranes were permeabilized by the addition of 0.4 μg of n-dodecyl β-d-maltoside per μg of total protein (lower panel). Ca2+ is the only ion that produces a major shift in the enzyme to phosphoenzyme equilibrium of HVP5A1 (arrowheads). Phosphorylation was performed for 60 s, and dephosphorylation was performed for 30 s before quenching the reaction (see Fig. 5A). B, formation of phosphoenzyme can be increased by titrating the crude membranes with detergent n-dodecyl β-d-maltoside (DDM) to unmask latent ligand ATP-binding sites. Phosphorylation was performed for 60 s as in Fig. 4A. Measurements are relative to samples without added detergent. C, n-dodecyl β-d-maltoside does not affect phosphoenzyme stability. Formation of the phosphoenzyme was performed for 60 s as in Fig. 4A, and dephosphorylation was performed in the presence of 0.4 μg of n-dodecyl β-d-maltoside per total μg of protein for the indicated amount of time. Measurements are relative to the phosphoenzyme level observed after 60 s, which corresponds to t0 in the figure.
In microsomal membrane fractions, which could be inside-out or right side-out or a mixture of both, only vesicles that expose the cytoplasmic side of HvP5A1 (including the nucleotide-binding site) to the extravesicular medium would be activated by ATP. In case a transported ligand needs to bind from the extracytoplasmic side to induce dephosphorylation, it would have to gain access to the vesicle interior to produce its effect. Thus, to establish conditions where the tested ions had equal access to both sides of the protein, we titrated the microsomal membrane preparation with increasing amounts of the detergent n-dodecyl β-d-maltoside prior to phosphorylation to establish the concentration at which the detergent would solubilize the membranes and allow ATP to enter at latent nucleotide-binding sites (Fig. 7B). Addition of the detergent by itself did not affect the stability of the phosphoenzyme for up to 120 s (Fig. 7C). Under conditions where the crude membrane fraction was permeable for ATP, we again tested the stability of the phosphoenzyme following addition of either full MS medium or each of the single components of the medium. Also, here we found that only Ca2+ affected phosphoenzyme stability under these conditions (Fig. 7A, lower panel). Taken together, these results indicate that Ca2+ exerts its specific effect by acting at the same side of the membrane as ATP, i.e. the cytoplasmic side.
Ca2+ Induces Degradation of the HvP5A1 Phosphoenzyme with Submillimolar Affinity
To determine dephosphorylation kinetics under conditions where a continuous forward E1 to E1P reaction could be disregarded, we established conditions where ATP was depleted specifically during the dephosphorylation reaction. ATP-depleted conditions were obtained by adding hexokinase to the phosphorylation buffer and glucose to the dephosphorylation buffer. This allows hexokinase in a rapid reaction to transfer the γ-phosphate from ATP to glucose when dephosphorylation is initiated. Following addition of hexokinase and glucose to the assay, we thus observed that inhibition of phosphoenzyme formation by hexokinase was glucose-dependent and furthermore that hexokinase and glucose together did not affect the stability of the phosphoenzyme already formed (Fig. 8A).
FIGURE 8.
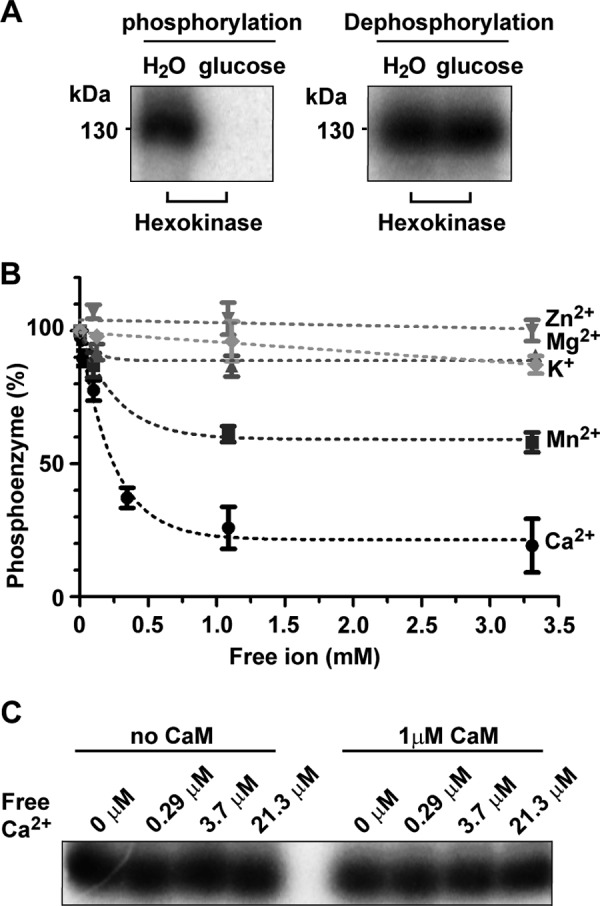
Ca2+ induces degradation of the HvP5A1 phosphoenzyme with submillimolar affinity. A, hexokinase and glucose can be used to inhibit the spontaneous E1 to E1P reaction through depletion of ATP. When phosphorylation was performed for 60 s in the presence of hexokinase and glucose, but not in the presence of hexokinase and H2O, [32P]ATP could be depleted resulting in complete inhibition of the phosphorylation reaction (left panel). Addition of glucose or H2O to the phosphoenzyme formed for 60 s in the presence of hexokinase shows that this depletion does not affect phosphoenzyme stability (right panel). B, stability of the phosphoenzyme was determined in increasing amounts of either CaCl2 (●), MnCl2 (■), MgCl2 (▴), ZnCl2 (▾), or KCl (♦) and at conditions where spontaneous phosphorylation was inhibited. Phosphorylation was performed for 60 s with 50 units of hexokinase present in the reaction buffer. Dephosphorylation was initiated by adding 5 μl of reaction mix producing the indicated final concentrations of cations and 2 mm glucose at t60. After another 30 s, the reaction was quenched with 400 μl of 20% TCA. C, inclusion of exogenous calmodulin in the assay did not affect the stability of the phosphoenzyme at low concentrations of Ca2+.
Under ATP-depleted conditions, we next determined the level of dephosphorylation at different Ca2+ concentrations. When the phosphoenzyme of HvP5A1 was titrated with Ca2+ in the presence of 50 μm EGTA, 2 mm glucose, and 50 units of hexokinase, we observed that the phosphoenzyme degradation had a low apparent K0.5 value for Ca2+ (∼0.2 mm) indicating that Ca2+ exerts its effect with low affinity (Fig. 8B). The related cation Mn2+ only partly catalyzed degradation of the phosphoenzyme, whereas Zn2+, Mg2+, and K+ showed no effect on its stability (Fig. 8B). The regulatory protein calmodulin often increases the Ca2+ affinity of Ca2+-dependent enzymes, but inclusion of exogenous calmodulin in the assay did not increase the apparent affinity for Ca2+ (Fig. 8C).
Mutations in the Putative Substrate-binding Site in M4 Hampers in Vivo Function of HvP5A1 but Does Not Affect Phosphoenzyme Formation or Ca2+-dependent Phosphoenzyme Degradation
P5A ATPases are distinguished by the presence of two highly conserved glutamate residues at the putative substrate- binding site in M4 (2). In the corresponding transmembrane helix of the Ca2+-ATPase, the side chain group of a conserved glutamate interacts directly with Ca2+ in the transport binding pocket (42).
To analyze whether residues in M4 are important for HvP5A1 function, we performed alanine substitutions of highly conserved residues, including the conserved glutamates (Glu-447 and Glu-451, see Fig. 9A). The mutated versions of HvP5A1 were subsequently tested for their ability to rescue spf1 cells during caffeine pressure. None of the single-point mutations introduced in M4 rendered HvP5A1 unable to complement spf1 (Fig. 9B). However, when both of the glutamates were mutated simultaneously (HvP5A1-E447A/E451A), we observed a loss-of-function effect on HvP5A1 (Fig. 9B).
FIGURE 9.

Mutations in the putative substrate-binding site in M4 hampers in vivo function of HvP5A1 but does not affect phosphorylation or Ca2+-dependent degradation of the phosphoenzyme. A, alignment of transmembrane helix M4 from P5A ATPases of barley (HvP5A), Arabidopsis (MIA/PDR2), yeast (Spf1p), and human (ATP13A1). Highly conserved residues from Ref. 10 are shown in boldface. The sarcoplasmic reticulum Ca2+-ATPase sequence is noted in italic below where residues involved in Ca2+ binding are noted in bold italic and residues conserved between P5A ATPases and SERCA are indicated by asterisks. Mutations introduced in M4 of HvP5A1 are indicated by the arrowheads. B, alanine mutations of individual residues in M4 do not affect the ability of HvP5A1 to rescue the caffeine-induced phenotype of spf1 cells. Only the double E447A/E451A mutation affects the ability of HvP5A1 to rescue spf1 in vivo. A protein blot against the C-terminal RGS-His6 tag of all proteins is shown below confirming the presence of protein. Fifteen μg of total protein as determined by Bradford assay was loaded in each lane and growth was observed on YPD plates containing increasing amounts of caffeine. Yeast cells were spotted for each measuring point diluted to A600 0.01 (top rows) or 0.001 (bottom rows). C, kinetics of phosphoenzyme formation is comparable between wild-type HvP5A1 (●) and HvP5A1-E447A/E451A (○) mutated protein when expressed in yeast RS72 cells. D, Ca2+ induced degradation of the phosphoenzyme is independent of the glutamates in M4. Phosphorylation was performed for 60 s as in Fig. 7B, and dephosphorylation was measured in the presence (dark gray bars) or absence (white bars) of 1 mm Ca2+ before quenching the reaction after 30 s. The levels observed after 30 s are comparable with the background observed in Figs. 5A and 6A.
To analyze further the double glutamate mutated version of HvP5A1, we expressed the protein in RS72 cells and analyzed the kinetics of phosphoenzyme formation and degradation. The introduced mutations did not abolish the ability to phosphorylate on Asp-488 and apparently did not alter phosphorylation kinetics of the enzyme (Fig. 9C). Furthermore, mutation of both glutamates had no effect on the Ca2+-dependent phosphoenzyme degradation (Fig. 9D).
The complementation data indicated that M4 and more specifically the conserved glutamates in M4 are important for the function of HvP5A1. However, Glu-447 and Glu-451 are not required for the spontaneous phosphorylation of HvP5A1 or for the Ca2+-dependent phosphoenzyme degradation. We can conclude that the effect of Ca2+ on HvP5A1 is not mediated by any of the negatively charged residues on M4.
Ca2+ Induces Degradation of the Phosphoenzyme Independently of the Conserved Phosphatase Motif
To establish whether phosphatase function is required for the effect of Ca2+ on phosphoenzyme degradation, we measured the stability of phosphoenzymes of wild-type HvP5A1 and of a mutant in which the phosphatase sequence is altered. In the rabbit Ca2+-ATPase SERCA1, Glu-183, which locates to the highly conserved TGES motif in the A domain (Fig. 10A), is essential for the phosphatase action related to catalytic dephosphorylation of the EP state (43). We therefore mutated the corresponding residue in the conserved TGES stretch of HvP5A1, Glu-313, to analyze if Ca2+-induced degradation of the phosphoenzyme was dependent on catalytic dephosphorylation.
FIGURE 10.
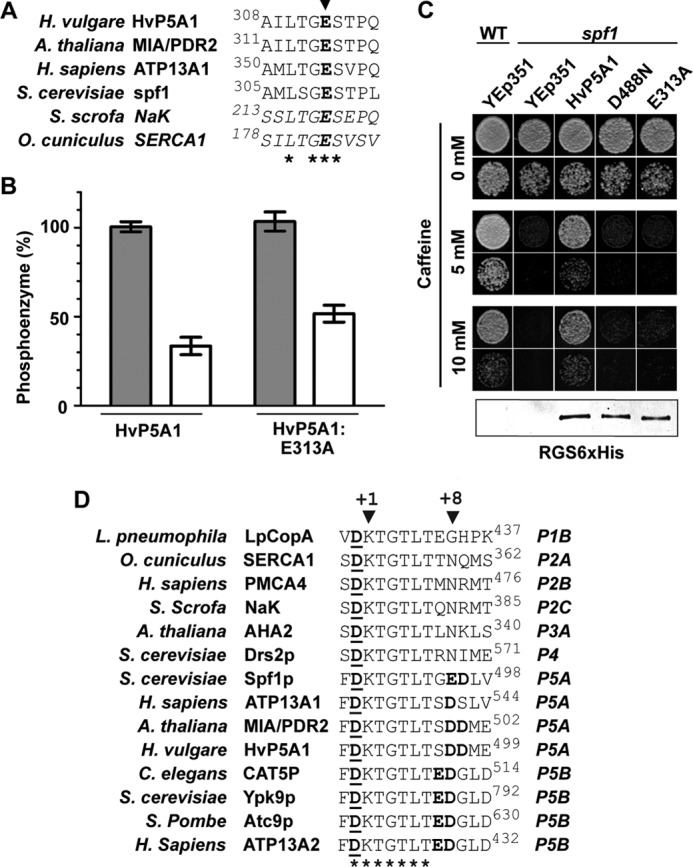
Ca2+-dependent degradation of the phosphoenzyme is independent of an intact phosphatase motif. A, TGES phosphatase motif is highly conserved in the A domain of both the sarcoplasmic reticulum Ca2+-ATPase, Na+/K+-ATPase, and the P5A ATPases. Asterisks indicate conserved residues, and Glu-313 in HvP5A1 is indicated by the arrowhead. B, Ca2+-induced degradation of the phosphoenzyme is independent of an intact phosphatase motif. Phosphorylation was performed for 60 s as in Fig. 7B, and dephosphorylation was measured in the presence (dark gray bars) or absence (white bars) of 1 mm Ca2+ before quenching the reaction after 30 s. The levels observed after 30 s are comparable with the background observed in Figs. 4A and 5A. C, mutation of Glu-313 into Ala results in a protein unable to rescue the caffeine-induced phenotype of spf1 cells. The level of growth is comparable with that observed from yeast transformed with the Asp-488 mutated HvP5A1. A protein blot against the C-terminal RGS-His6 tag of all proteins is shown below. Fifteen μg of total protein as determined by Bradford assay was loaded in each lane. Growth was observed on YPD plates containing increasing amounts of caffeine, and yeast cells were spotted for each measuring point diluted to A600 0.01 (top rows) or 0.001 (bottom rows). D, P5A and P5B ATPases have negatively charges conserved at positions +7, +8, and/or +9 (boldface) of the phosphorylated aspartate residue (underlined). Asterisks indicate residues conserved between P1 to P5 type ATPases.
The stability of the phosphoenzymes from wild-type and E313A-mutated protein (HvP5A1-E313A) remained comparable in the presence or absence of Ca2+ (Fig. 10B). This shows that Ca2+ could produce its effect independently of Glu-313, i.e. in a mutant that is expected to be catalytically inactive. Indeed, the residue is required for the in vivo function of HvP5A1 as the E313A-mutated protein fails to rescue spf1 cells during caffeine pressure (Fig. 10C). Therefore, our data show that Ca2+-induced degradation of the phosphoenzyme is unrelated to catalytic turnover of HvP5A1. In contrast, our data are in line with a stimulating effect of Ca2+ on the spontaneous dephosphorylation of the EP.
DISCUSSION
P5A-type ATPases are found in all eukaryotes, pointing to an essential but so far unidentified function in cell biology. It has been proposed that P5A-ATPases transport Ca2+ into the ER (11, 44). In this work, we have carried out an extensive biochemical analysis of the barley HvP5A1 P5A P-type ATPase and describe for the first time the formation and degradation of a P5A-type phosphoenzyme intermediate. We show that Ca2+ has a modulatory effect on phosphoenzyme stability, but it is not likely to be a transported ligand.
P5A-ATPases Are Tightly Linked to Caffeine Sensitivity and Altered Ca2+ Homeostasis in Yeast
Heterologous expression in yeast of HvP5A1 fully complemented the caffeine-sensitive phenotype described for a null mutation of the native yeast P5A ATPase (34). As caffeine is known to affect intracellular Ca2+ homeostasis in yeast (45), it is a tempting hypothesis that the phenotype of the SPF1 null mutant relates to a change in Ca2+ homeostasis of affected cells (34). The spf1 mutant shows an activated unfolded protein response and a defect in protein-linked oligosaccharide trimming (14), both of which are processes regulated by Ca2+. In addition, spf1 knock-out cells show increased cellular Ca2+ levels, but only in cooperation with the Golgi located Pmr1p Ca2+/Mn2+ pump (12). Moreover, in fission yeast Cta4p/Spf1p modulates the Ca2+ filling of the ER (44). These results indicate that P5A ATPases play a role in controlling cellular Ca2+ homeostasis. This effect could be direct, for example as a result of P5A ATPases being Ca2+ pumps located in the ER, or indirect via other mechanisms.
Ca2+ Plays a Modulatory Role on the Biochemical Activity of P5A-type ATPases but Is Not Likely to be a Transported Substrate
Although the phenotype of yeast Spf1 deletion mutants is tightly linked to Ca2+, our results demonstrate that neither HvP5A1 nor Spf1p are likely to transport Ca2+. This conclusion is based on five observations. First, we observed spontaneous phosphorylation of HvP5A1 in the absence of Ca2+ and in the presence of EGTA. Under these conditions HvP5A1 undergoes a full, albeit slow, catalytic cycle. Accordingly, Ca2+-dependent ATPase activity could not be demonstrated for the yeast Spf1p (12). Because ATP hydrolysis in all P-type ATPases is tightly coupled to substrate transport (7), it seems unlikely that HvP5A1 and/or Spf1p directly transport Ca2+.
Second, we determined an effect of Ca2+ on the dephosphorylation kinetics of HvP5A1 that, however, was independent of catalytic dephosphorylation of the pump. We identified this effect of Ca2+ by screening all inorganic factors required for plant growth and show that the effect occurs independently of the phosphatase motif of the pump.
Third, modulation by Ca2+ occurred independently of conserved negatively charged residues in M4 that in the SERCA Ca2+ pump are essential for coordination of Ca2+ (42), and in other P-type ATPases they contribute to binding of transported ligands (7).
Fourth, the Ca2+ effect was evident in sealed vesicles in which the ATP-binding site faced the extravesicular medium. This suggest that Ca2+ exerts its effect from the cytoplasmic side of the membrane and rules out the possibility that Ca2+ triggers dephosphorylation by binding as a transported counterion from the extracytoplasmic side of the membrane.
Fifth, the fact that ER Ca2+ pumps like animal SERCA or plant ACA3 does not complement the loss of Spf1 with respect to Hmg2p degradation (12) indicates that Spf1p is not merely an ER Ca2+ pump, and it has roles other than ER Ca2+ filling.
Our results suggests that HvP5A1 does not transport Ca2+ but is modulated by Ca2+ via a cytosolic Ca2+-binding site that communicates with the aspartyl-phosphate group of the protein. The modulatory effect might be a common feature of all P5A-ATPases, because above 100 μm, Ca2+ also reduced the ATPase activity of purified Spf1p (12). With an apparent K0.5 for dephosphorylation of about 0.2 mm Ca2+, the effect is to be characterized as low affinity and in a range above normal in vivo cytosolic Ca2+ concentrations. The P-type plasma membrane H+-ATPase AHA2 from Arabidopsis has a low affinity regulatory Ca2+-binding site in the nucleotide binding domain (46), which in other species might have high (submicromolar) affinity for Ca2+ (47). We therefore cannot rule out that other P5A ATPases may have higher Ca2+ affinities or that the Ca2+ affinity of HvP5A1 under certain circumstances can increase, e.g. as a result of post-translational modification or binding of regulatory proteins. However, we show that at least calmodulin has no effect on the apparent Ca2+ affinity of HvP5A1.
If Ca2+ is not the transported ligand, how is P5A function then related to Ca2+ homeostasis? In the yeast ER, where no SERCA Ca2+ pump is found, Ca2+ filling critically depends on vesicle transport (48). Pmr1p is a P-type Ca2+-ATPase responsible for loading of the Golgi apparatus with Ca2+ (49). Retrograde transport from the Golgi to the ER is the most important mechanism to load the ER with Ca2+, which explains the dominant effect of Pmr1p on the ER Ca2+ concentration (48). Mutations in vesicular transport proteins are known that indirectly disrupt Ca2+ homeostasis in yeast (50). Accordingly, P5A ATPases might be important for vesicular trafficking required to load the ER with Ca2+ (12, 44).
Mechanism of the Ca2+ Effect
How Ca2+ stimulates the noncatalytic dephosphorylation of HvP5A1 remains elusive. The Ca2+ effect on phosphoenzyme degradation was still observed in ATP-depleted conditions in which the Mg2+-ATP-dependent E1 to E1P reaction is prevented. This rules out that the Ca2+-destabilizing effect is due to a displacement of Mg2+ by Ca2+ from the phosphorylation site. Many P-type ATPases bind nontransported cations, which modulate pump function (46, 51, 52). It is possible that Ca2+ stimulates the E1P to E1 reverse reaction or facilitates the attack by H2O in the E2P state, independently from the TGES motif. It is interesting to note that in contrast to other P-type ATPases, only P5A and P5B ATPases contain conserved negatively charged residues at positions +7, +8, and/or +9 of the phosphorylated aspartate (Fig. 10D). These could influence the stability of the phospho-intermediate or might mediate cation binding directly at the site of phosphorylation. Interestingly, the modulatory Ca2+-binding site of the plasma membrane proton ATPase locates to the nucleotide-binding site (46) in close proximity of the conserved negative residues in P5A and P5B ATPases.
Roles of H+ and Mg2+
The only ions present in the basal assay medium in which HvP5A1 carried out a full reaction cycle were H+ and Mg2+. We therefore have to consider the possibility that HvP5A1 transports either H+ or Mg2+. The pH optimum of HvP5A1 was found at pH 6.4, comparable with that of H+-transporting P-type ATPases (39, 40). However, as any enzyme is expected to show an optimal pH condition in the physiological range, the pH optimum needs not be directly related to binding of H+ as a transported ligand. Stabilization of the protein structure by protonation or deprotonation of key residues could be equally important. In fact, it would be rather unusual to have a H+-pump in the ER membrane that is known as being leaky for protons (53).
Mg2+ was required for the Mg2+-ATPase reaction and thus could not be omitted from the assay. Addition of further Mg2+ did not influence phosphoenzyme formation or degradation. Previously, an Mg2+-dependent ATPase activity was observed for purified Spf1p, but the effect was modest, more in line with a modulatory effect of Mg2+ on overall activity (12).
Other Possible Ligands of HvP5A1
The fact that HvP5A1 complements an spf1 mutant suggest that, at least in intact transformed yeast cells, all components required for proper functioning of HvP5A1 (e.g. ligands, lipids, and possible subunits) are present in sufficient amounts. In vitro, we measured HvP5A1 activity in a yeast membrane environment that in vivo is able to support the complementation of the caffeine phenotype in the Spf1 knock-out strain. Furthermore, our studies of the HvP5A1 phosphoenzyme were carried out in the absence of detergent and with no addition of lipids that could destroy enzyme function.
Remarkably, we observed that in vitro HvP5A1 spontaneously undergoes a full catalytic cycle, apparently independent of any ion in the reaction medium. Because all P-type ATPases studied so far tightly couple ATP hydrolysis to the binding of a specific substrate (7), this would suggest that either ATP hydrolytic activity of HvP5A1 is uncoupled from transport or that the transported substrate is not a soluble ligand but is a compound present in the yeast membrane preparation. Because of the complementation of SPF1 by HvP5A1, the substrate must be conserved between plant and yeast. The slow turnover of HvP5A1 is in accordance with a low rate of phosphoenzyme turnover and readily explains why significant ATPase activity of the enzymes could not be detected. Similarly slow phosphoenzyme kinetics have also been observed in other P-type ATPase subfamilies (54, 55). The slow turnover might also indicate that the amount of the endogenous substrate is limiting as compared with the overexpression level of HvP5A1.
A substrate ligand of HvP5A1 in the yeast membrane preparations could be any organic compound. Because the P5 ATPases are most closely related to P4 ATPases than to any other P-type ATPase subgroup (1), it is tempting to speculate that P5 ATPases have a function related to that of P4 ATPases, which are phospholipid flippases involved in initial vesicle budding from endomembranes and the plasma membrane (56). Like P4 ATPases, P5 pumps are restricted to eukaryotes. Interestingly, no P4 flippases so far are known to function in the ER, although continuous vesicle formation and fusion occurs in this compartment. It is therefore a possibility that P5A ATPases might be involved in ER vesicle budding or fusion, e.g. by acting as ER-localized lipid flippases (56).
So far, no direct evidence is presented that any P5A ATPase would mediate vesicle transport. However, in line with a role in vesicular transport, it has been shown that Spf1p is important for the subcellular localization of Sec12p (20), a protein required for the initiation of COPII vesicle formation in ER to Golgi transport. Spf1p is also required for proper transport of CPY to the vacuole (14). In addition, there is a synthetic lethal effect of Spf1 deletion in combination with deletion of GET1/MDM39 or RIC1, the gene products of which are involved in retrieval of HDEL proteins from Golgi to ER and in retrograde transport to the cis-Golgi, respectively (34). A model where Spf1p is important for vesicular transport that plays a role in ER Ca2+ loading is compatible with the pleiotropic phenotype of Spf1p deletion on glycosylation (14), protein folding (14), sorting (14, 20), and resistance to the SMKT killer toxin (18). It would also explain why a stronger Ca2+-dependent phenotype is observed in the Spf1/Pmr1 double knock-out than in the single knock-outs (12). Further support for a role of P5 ATPases in vesicular transport comes from a recent yeast two-hybrid protein screen demonstrating a direct interaction between human P5B ATPase ATP13A2 and proteins involved in ER translocation (SEC61B and FKBP8), ER to Golgi trafficking (FKB8 and YIF1A), and vesicular transport and fusion (VAMP2, SYT11, HSPA8, and AAK1) (57).
Finally, our data do not exclude a receptor role for HvP5A1, e.g. as is the case for the Na+,K+-ATPase (58). Whatever the function of HvP5A1 is, our data show that typical P-type ATPase motifs are required for complementation, such as the phosphorylation site, the TGES dephosphorylation motif, and the putative substrate-binding site in M4.
It has previously been demonstrated that the phenotype of a deletion mutant of the yeast P5A ATPase Spf1p mutant is Ca2+-dependent, whereas a detergent-solubilized and purified form of Spf1p hydrolyzes ATP independently of Ca2+ (12). This lack of correlation was unexpected; no molecular explanation was provided for the ability of Spf1p to influence Ca2+ homeostasis, and it could not be ruled out that detergent solubilization had changed the properties of the enzyme (12). Accordingly, the genetic evidence from knock-out mutants is still used in the literature to suggest that P5A pumps could be Ca2+ pumps (44). In our work, we use an alternative biochemical method, i.e. determination of phosphoenzyme kinetics, and with a P5A ATPase in a undisturbed membrane environment, to demonstrate that Ca2+ influences the stability of the phosphoenzyme of a P5A ATPase, and we show that the effect is unrelated to a Ca2+ pump function for the pump. Furthermore, we provide an alternative testable model for how P5A ATPases regulate Ca2+ homeostasis by regulating vesicle trafficking from and to the endoplasmic reticulum.
Conclusion
We present strong evidence that Ca2+ is not the substrate of the barley P5A ATPase HvP5A1 but that Ca2+ merely has a modulatory effect on phosphoenzyme stability. The link between P5A ATPases and cellular Ca2+ homeostasis could be related to this effect, but alternative models are possible. We are still left with the question as to what ligand(s) are transported by P5A ATPases. As we observe spontaneous phosphoenzyme formation and full enzymatic turnover of HvP5A1 in yeast membranes, the transported substrate, if any, is likely to be conserved between plants and yeast and is present in yeast microsomal membranes.
Footnotes
- ER
- endoplasmic reticulum
- EST
- expressed sequence tag
- SERCA
- sarco/endoplasmic reticulum Ca2+-ATPase.
REFERENCES
- 1. Axelsen K. B., Palmgren M. G. (1998) Evolution of substrate specificities in the P-type ATPase superfamily. J. Mol. Evol. 46, 84–101 [DOI] [PubMed] [Google Scholar]
- 2. Møller A. B., Asp T., Holm P. B., Palmgren M. G. (2008) Phylogenetic analysis of P5 P-type ATPases, a eukaryotic lineage of secretory pathway pumps. Mol. Phylogenet. Evol. 46, 619–634 [DOI] [PubMed] [Google Scholar]
- 3. Ramirez A., Heimbach A., Gründemann J., Stiller B., Hampshire D., Cid L. P., Goebel I., Mubaidin A. F., Wriekat A. L., Roeper J., Al-Din A., Hillmer A. M., Karsak M., Liss B., Woods C. G., Behrens M. I., Kubisch C. (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191 [DOI] [PubMed] [Google Scholar]
- 4. Kwasnicka-Crawford D. A., Carson A. R., Roberts W., Summers A. M., Rehnström K., Järvelä I., Scherer S. W. (2005) Characterization of a novel cation transporter ATPase gene (ATP13A4) interrupted by 3q25-q29 inversion in an individual with language delay. Genomics 86, 182–194 [DOI] [PubMed] [Google Scholar]
- 5. Habtemichael N., Kovacs G. (2002) Cloning the AFURS1 gene, which is up-regulated in senescent human parenchymal kidney cells. Gene 283, 271–275 [DOI] [PubMed] [Google Scholar]
- 6. Kühlbrandt W. (2004) Biology, structure, and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 5, 282–295 [DOI] [PubMed] [Google Scholar]
- 7. Palmgren M. G., Nissen P. (2011) P-type ATPases. Annu. Rev. Biophys. 40, 243–266 [DOI] [PubMed] [Google Scholar]
- 8. Morth J. P., Pedersen B. P., Buch-Pedersen M. J., Andersen J. P., Vilsen B., Palmgren M. G., Nissen P. (2011) A structural overview of the plasma membrane Na+,K+-ATPase and H+-ATPase ion pumps. Nat. Rev. Mol. Cell Biol. 12, 60–70 [DOI] [PubMed] [Google Scholar]
- 9. Møller J. V., Olesen C., Winther A. M., Nissen P. (2010) The sarcoplasmic Ca2+-ATPase. Design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 43, 501–566 [DOI] [PubMed] [Google Scholar]
- 10. Sørensen D. M., Buch-Pedersen M. J., Palmgren M. G. (2010) Structural divergence between the two subgroups of P5 ATPases. Biochim. Biophys. Acta 1797, 846–855 [DOI] [PubMed] [Google Scholar]
- 11. Cronin S. R., Khoury A., Ferry D. K., Hampton R. Y. (2000) Regulation of HMG-CoA reductase degradation requires the P-type ATPase Cod1p/Spf1p. J. Cell Biol. 148, 915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cronin S. R., Rao R., Hampton R. Y. (2002) Cod1p/Spf1p is a P-type ATPase involved in ER function and Ca2+ homeostasis. J. Cell Biol. 157, 1017–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jakobsen M. K., Poulsen L. R., Schulz A., Fleurat-Lessard P., Møller A., Husted S., Schiøtt M., Amtmann A., Palmgren M. G. (2005) Pollen development and fertilization in Arabidopsis is dependent on the MALE GAMETOGENESIS IMPAIRED ANTHERS gene encoding a type V P-type ATPase. Genes Dev. 19, 2757–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vashist S., Frank C. G., Jakob C. A., Ng D. T. (2002) Two distinctly localized P-type ATPases collaborate to maintain organelle homeostasis required for glycoprotein processing and quality control. Mol. Biol. Cell 13, 3955–3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Façanha A. L., Appelgren H., Tabish M., Okorokov L., Ekwall K. (2002) The endoplasmic reticulum cation P-type ATPase Cta4p is required for control of cell shape and microtubule dynamics. J. Cell Biol. 157, 1029–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ticconi C. A., Lucero R. D., Sakhonwasee S., Adamson A. W., Creff A., Nussaume L., Desnos T., Abel S. (2009) ER-resident proteins PDR2 and LPR1 mediate the developmental response of root meristems to phosphate availability. Proc. Natl. Acad. Sci. U.S.A. 106, 14174–14179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tipper D. J., Harley C. A. (2002) Yeast genes controlling responses to topogenic signals in a model transmembrane protein. Mol. Biol. Cell 13, 1158–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki C., Shimma Y. I. (1999) P-type ATPase spf1 mutants show a novel resistance mechanism for the killer toxin SMKT. Mol. Microbiol. 32, 813–823 [DOI] [PubMed] [Google Scholar]
- 19. Suzuki C., Ando Y., Machida S. (2001) Interaction of SMKT, a killer toxin produced by Pichia farinosa, with the yeast cell membranes. Yeast 18, 1471–1478 [DOI] [PubMed] [Google Scholar]
- 20. Suzuki C. (2001) Immunochemical and mutational analyses of P-type ATPase Spf1p involved in the yeast secretory pathway. Biosci. Biotechnol. Biochem. 65, 2405–2411 [DOI] [PubMed] [Google Scholar]
- 21. Bernsel A., Viklund H., Hennerdal A., Elofsson A. (2009) TOPCONS. Consensus prediction of membrane protein topology. Nucleic Acids Res. 37, W465–W468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clough S. J., Bent A. F. (1998) Floral dip. A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 [DOI] [PubMed] [Google Scholar]
- 23. Murashige T., Skoog F. (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plant. 15, 473–497 [Google Scholar]
- 24. Curtis M. D., Grossniklaus U. (2003) A gateway cloning vector set for high throughput functional analysis of genes in planta. Plant Physiol. 133, 462–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Runions J., Brach T., Kühner S., Hawes C. (2006) Photoactivation of GFP reveals protein dynamics within the endoplasmic reticulum membrane. J. Exp. Bot. 57, 43–50 [DOI] [PubMed] [Google Scholar]
- 26. Ito H., Fukuda Y., Murata K., Kimura A. (1983) Transformation of intact yeast-cells treated with alkali cations. J. Bacteriol. 153, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sarkadi B., Enyedi A., Földes-Papp Z., Gárdos G. (1986) Molecular characterization of the in situ red cell membrane calcium pump by limited proteolysis. J. Biol. Chem. 261, 9552–9557 [PubMed] [Google Scholar]
- 28. Vilsen B., Andersen J. P. (1998) Mutation to the glutamate in the fourth membrane segment of Na+,K+-ATPase and Ca2+-ATPase affects cation binding from both sides of the membrane and destabilizes the occluded enzyme forms. Biochemistry 37, 10961–10971 [DOI] [PubMed] [Google Scholar]
- 29. Baxter I., Tchieu J., Sussman M. R., Boutry M., Palmgren M. G., Gribskov M., Harper J. F., Axelsen K. B. (2003) Genomic comparison of P-type ATPase ion pumps in Arabidopsis and rice. Plant Physiol. 132, 618–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Villalba J. M., Palmgren M. G., Berberián G. E., Ferguson C., Serrano R. (1992) Functional expression of plant plasma membrane H+-ATPase in yeast endoplasmic reticulum. J. Biol. Chem. 267, 12341–12349 [PubMed] [Google Scholar]
- 31. Baekgaard L., Luoni L., De Michelis M. I., Palmgren M. G. (2006) The plant plasma membrane Ca2+ pump ACA8 contains overlapping as well as physically separated autoinhibitory and calmodulin-binding domains. J. Biol. Chem. 281, 1058–1065 [DOI] [PubMed] [Google Scholar]
- 32. Pedersen B. P., Buch-Pedersen M. J., Morth J. P., Palmgren M. G., Nissen P. (2007) Crystal structure of the plasma membrane proton pump. Nature 450, 1111–1114 [DOI] [PubMed] [Google Scholar]
- 33. Rand J. D., Grant C. M. (2006) The thioredoxin system protects ribosomes against stress-induced aggregation. Mol. Biol. Cell 17, 387–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ando A., Suzuki C. (2005) Cooperative function of the CHD5-like protein Mdm39p with a P-type ATPase Spf1p in the maintenance of ER homeostasis in Saccharomyces cerevisiae. Mol. Genet. Genomics 273, 497–506 [DOI] [PubMed] [Google Scholar]
- 35. Schmidt K., Wolfe D. M., Stiller B., Pearce D. A. (2009) Cd2+, Mn2+, Ni2+, and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem. Biophys. Res. Commun. 383, 198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lutsenko S., Kaplan J. H. (1995) Organization of P-type ATPases. Significance of structural diversity. Biochemistry 34, 15607–15613 [DOI] [PubMed] [Google Scholar]
- 37. Cid A., Perona R., Serrano R. (1987) Replacement of the promoter of the yeast plasma membrane ATPase gene by a galactose-dependent promoter and its physiological consequences. Curr. Genet. 12, 105–110 [DOI] [PubMed] [Google Scholar]
- 38. Post R. L., Kume S. (1973) Evidence for an aspartyl phosphate residue at the active site of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem. 248, 6993–7000 [PubMed] [Google Scholar]
- 39. Vara F., Serrano R. (1982) Partial purification and properties of the proton-translocating ATPase of plant plasma membranes. J. Biol. Chem. 257, 12826–12830 [PubMed] [Google Scholar]
- 40. Palmgren M. G. (2001) Plant plasma membrane H+-ATPases. Powerhouses for nutrient uptake. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 817–845 [DOI] [PubMed] [Google Scholar]
- 41. de Meis L., Martins O. B., Alves E. W. (1980) Role of water, hydrogen ion, and temperature on the synthesis of adenosine triphosphate by the sarcoplasmic reticulum adenosine triphosphatase in the absence of a calcium ion gradient. Biochemistry 19, 4252–4261 [DOI] [PubMed] [Google Scholar]
- 42. Toyoshima C., Nakasako M., Nomura H., Ogawa H. (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405, 647–655 [DOI] [PubMed] [Google Scholar]
- 43. Clausen J. D., Vilsen B., McIntosh D. B., Einholm A. P., Andersen J. P. (2004) Glutamate 183 in the conserved TGES motif of domain A of sarcoplasmic reticulum Ca2+-ATPase assists in catalysis of E2/E2P partial reactions. Proc. Natl. Acad. Sci. U.S.A. 101, 2776–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lustoza A. C., Palma L. M., Façanha A. R., Okorokov L. A., Okorokova-Façanha A. L. (2011) P5A-type ATPase Cta4p is essential for Ca2+ transport in the endoplasmic reticulum of Schizosaccharomyces pombe. PLoS One 6, e27843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Courchesne W. E., Ozturk S. (2003) Amiodarone induces a caffeine-inhibited, MID1-dependent rise in free cytoplasmic calcium in Saccharomyces cerevisiae. Mol. Microbiol. 47, 223–234 [DOI] [PubMed] [Google Scholar]
- 46. Ekberg K., Pedersen B. P., Sørensen D. M., Nielsen A. K., Veierskov B., Nissen P., Palmgren M. G., Buch-Pedersen M. J. (2010) Structural identification of cation binding pockets in the plasma membrane proton pump. Proc. Natl. Acad. Sci. U.S.A. 107, 21400–21405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kinoshita T., Nishimura M., Shimazaki K. (1995) Cytosolic concentration of Ca2+ regulates the plasma membrane H+-ATPase in guard cells of fava bean. Plant Cell 7, 1333–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Strayle J., Pozzan T., Rudolph H. K. (1999) Steady-state free Ca2+ in the yeast endoplasmic reticulum reaches only 10 microm and is mainly controlled by the secretory pathway pump pmr1. EMBO J. 18, 4733–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sorin A., Rosas G., Rao R. (1997) PMR1, a Ca2+-ATPase in yeast Golgi, has properties distinct from sarco/endoplasmic reticulum and plasma membrane calcium pumps. J. Biol. Chem. 272, 9895–9901 [DOI] [PubMed] [Google Scholar]
- 50. Gupta S. S., Ton V. K., Beaudry V., Rulli S., Cunningham K., Rao R. (2003) Antifungal activity of amiodarone is mediated by disruption of calcium homeostasis. J. Biol. Chem. 278, 28831–28839 [DOI] [PubMed] [Google Scholar]
- 51. Buch-Pedersen M. J., Rudashevskaya E. L., Berner T. S., Venema K., Palmgren M. G. (2006) Potassium as an intrinsic uncoupler of the plasma membrane H+-ATPase. J. Biol. Chem. 281, 38285–38292 [DOI] [PubMed] [Google Scholar]
- 52. Picard M., Jensen A. M., Sørensen T. L., Champeil P., Møller J. V., Nissen P. (2007) Ca2+ versus Mg2+ coordination at the nucleotide-binding site of the sarcoplasmic reticulum Ca2+-ATPase. J. Mol. Biol. 368, 1–7 [DOI] [PubMed] [Google Scholar]
- 53. Paroutis P., Touret N., Grinstein S. (2004) The pH of the secretory pathway. Measurement, determinants, and regulation. Physiology 19, 207–215 [DOI] [PubMed] [Google Scholar]
- 54. Lenoir G., Williamson P., Puts C. F., Holthuis J. C. (2009) Cdc50p plays a vital role in the ATPase reaction cycle of the putative aminophospholipid transporter Drs2p. J. Biol. Chem. 284, 17956–17967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Baekgaard L., Mikkelsen M. D., Sørensen D. M., Hegelund J. N., Persson D. P., Mills R. F., Yang Z., Husted S., Andersen J. P., Buch-Pedersen M. J., Schjoerring J. K., Williams L. E., Palmgren M. G. (2010) A combined zinc/cadmium sensor and zinc/cadmium export regulator in a heavy metal pump. J. Biol. Chem. 285, 31243–31252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Poulsen L. R., López-Marqués R. L., Palmgren M. G. (2008) Flippases. Still more questions than answers. Cell. Mol. Life Sci. 65, 3119–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Usenovic M., Knight A. L., Ray A., Wong V., Brown K. R., Caldwell G. A., Caldwell K. A., Stagljar I., Krainc D. (2012) Identification of novel ATP13A2 interactors and their role in α-synuclein misfolding and toxicity. Hum. Mol. Genet., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lingrel J. B. (2010) The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu. Rev. Physiol. 72, 395–412 [DOI] [PMC free article] [PubMed] [Google Scholar]