

Abstract
Motivation: Next-generation sequence analysis has become an important task both in laboratory and clinical settings. A key stage in the majority sequence analysis workflows, such as resequencing, is the alignment of genomic reads to a reference genome. The accurate alignment of reads with large indels is a computationally challenging task for researchers.
Results: We introduce SeqAlto as a new algorithm for read alignment. For reads longer than or equal to 100 bp, SeqAlto is up to 10 × faster than existing algorithms, while retaining high accuracy and the ability to align reads with large (up to 50 bp) indels. This improvement in efficiency is particularly important in the analysis of future sequencing data where the number of reads approaches many billions. Furthermore, SeqAlto uses less than 8 GB of memory to align against the human genome. SeqAlto is benchmarked against several existing tools with both real and simulated data.
Availability: Linux and Mac OS X binaries free for academic use are available at http://www.stanford.edu/group/wonglab/seqalto
Contact: whwong@stanford.edu
1 INTRODUCTION
Resequencing with next-generation sequencers has become a popular method for characterizing genetic variation between individuals. A critical step in a typical resequencing workflow is accurate alignment of genomic reads to a reference genome. The resulting variants called from the aligned reads strongly depend on the accuracy of this initial alignment stage. In this article, we introduce SeqAlto, a fast and accurate read alignment tool that reliably align reads with large insertions and deletions (indels).
Next generation sequencers, such as HiSeq2000® from Illumina, are generating ever increasing volumes of data. Simply aligning these reads to a reference genome is a daunting task. The computational cost of sequencing is rapidly approaching the experimental cost. In the recently published 1000 genomes pilot article (The 1000 Genomes Project Consortium, 2010), it was stated that a 199 node cluster was used to align all of the reads to the reference genome with MAQ (Li et al., 2008). This kind of computational resource is not available to most laboratories or is not feasible in a clinical setting. At over 1 billion reads generated per run for a Illumina HiSeq2000 sequencer, the computational capacity required to align such large datasets in a reasonable amount of time far exceeds the capability of small laboratories or clinics. This is especially true if any analysis needs to be repeated with different parameters.
In addition to increases in the throughput of each sequencer, maximum read lengths of all sequencing platforms are steadily increasing beyond 100 bp. This poses a problem for many popular short-read aligners as shown in Section 3. On the other hand, SeqAlto is able to exploit this increased read length to improve the speed as well as accuracy of the alignment process. In fact, SeqAlto is not suitable for read lengths much less than 100 bp when aligning to the human genome. This is not a significant limitation as the majority of future sequencing platforms are able to output read lengths >100 bp and we expect this length to only increase in the future.
There are numerous read aligners available to researchers today. They can be broadly grouped into four classes: hashing the genome, hashing the reads, FM-index (Ferragina and Manzini, 2000) or sorting. Snap (Zaharia et al., 2011), Stampy (Lunter and Goodson, 2011), Novoalign (http://novocraft.com/), BFAST (Homer et al., 2009), GASSST (Rizk and Lavenier, 2010) and SHRiMP2 (David et al., 2011) index the genome with a hashtable of k-mers. BFAST and SHRiMP2 use large spaced seeds to improve sensitivity. GASSST uses a large array of filters to improved alignment speed. Snap follows a similar approach to SeqAlto by using large continuous seeds. MAQ (Li et al., 2008) and SeqMap (Jiang and Wong, 2008) hash the reads. This approach is useful when the reference is small such as a transcriptome. Bowtie2 (Salzberg, 2012), BWA (Li and Durbin, 2009), BWA-SW (Li and Durbin, 2010) and SOAP2 (Li et al., 2009) rely on the Burrows–Wheeler Transform (BWT) (Burrows and Wheeler, 1994) and FM–index. This approach has been shown to be very efficient for short reads and large genomes. However, we will see that it is problematic for longer reads (Section 3). BWA-SW stands out as it finds local alignments of the read rather than global alignments. Slider (Malhis et al., 2009) and Syzygy (Konagurthu et al., 2010) use a sorting and merging approach. This approach places a high demand on the storage system to be fast.
A selection of these read aligners is compared to SeqAlto in this article. Both real and simulated sequencer reads are used for the comparison. In addition to comparing the alignments generated, the variants called from the alignments are also compared.
2 APPROACH
SeqAlto takes the genome hashing approach but uses large contiguous seeds and adaptive stopping to achieve efficiencies much greater than all read aligners tested in this article. Hashing of the reference genome is a common approach also used in Snap, Stampy, Novoalign, BFAST etc. SeqAlto differs from all aligners surveyed except Snap as it is possible for all overlapping seeds to be indexed from the reference genome. SeqAlto also improves upon Snap by aligning reads with a banded affine-gap penalty Needlenam–Wunsch. As a result, SeqAlto is able to make use of longer reads to improve the alignment of reads with larger indels. We will show that the large seeds do not hurt alignment sensitivity when the targeted read lengths are greater than about 100 bp. In general, using large contiguous seeds requires huge amounts of memory to store the index. In order to overcome this difficulty, a novel sub-sampling approach is designed to fit the entire genome into under 8 GB of memory. The other feature of SeqAlto, adaptive stopping, declares each read to be aligned based on the current best alignment and the number of seeds examined. This approach works well for Illumina reads where the majority of errors are due to substitutions rather than gaps. It also offers users the ability to trade alignment accuracy for efficiency. Overall, the approach taken by SeqAlto targets high-quality reads with low number of gaps as errors. When aligning reads satisfying these criteria, SeqAlto is extremely fast and is able to accurately place reads with large insertions or deletions.
3 RESULTS
SeqAlto is compared to Snap, Bowtie2, SOAP2 BWA, Novoalign and Stampy in terms of alignment accuracy and efficiency. Snap is a new read aligner that follows a similar approach to SeqAlto, but puts its emphasis on alignment speed. Bowtie2, SOAP2 and BWA are based on the BWT and are among the most popular tools for read alignment. Novoalign is a commercial read alignment package not based on the BWT and is among the most accurate read aligners. Stampy is a relatively new alignment tool that is targeted towards resequencing with Illumina reads similar to SeqAlto. Stampy was run in hybrid mode, as recommended by the documentation, where a combination of BWA with a hash-based approach is used. SeqAlto is run in two modes, one with only a sub-set (Section 5.1) of seeds in the genome indexed (SeqAlto) and the other with all seeds indexed (SeqAlto-all).
A simulated dataset (Section 4) with single and paired-end reads is used to compare each algorithm. The algorithms supporting gapped alignment are compared at aligning reads with various indel sizes. The algorithms are also compared with real Illumina HiSeq 2000 data (Short Read Archive, accession number SRX100628). Alignment accuracy does not always indicate variants called will be accurate and comprehensive. For example, an aligner may place many reads very accurately, but fail to place a few polymorphic reads. This could result in increased confidence in already confident variants, while missing other variants. Hence, variants are called from simulated paired-end reads and also the real dataset to directly evaluate the effect of each aligner on the variants called. All simulations were run with a single thread on an 8-core 2.27 GHz Intel Xeon X7560 unless otherwise specified. An alignment is called correct if its start location is within 50 bp of the simulated location not counting bases inserted or deleted. The effect of varying this correctness criteria is discussed in the Supplementary Material. For SeqAlto, 22-bp and 28-bp seeds were used for 100-bp and 200-bp reads, respectively. For Snap, 20-bp and 22-bp seeds were used for 100-bp and 200-bp reads, respectively as suggested by the author. Exact parameters used for each program can be found in the Supplementary Material. The %Err given in each table refers to the percentage aligned incorrectly among the reads satisfying each uniqueness criteria. The elapsed time recorded for Novoalign and Stampy includes the index loading time since they do not provide an option to record only the alignment time. Empirically, the index loading time is independent of the number of reads aligned and not more than 100–200 s.
3.1 Simulated single-end reads
One million single-end reads with parameters as described in Section 4 were generated. The command-line parameters used for each tool can be found in the Supplementary Material. Novoalign is not able to align 200-bp reads so it was excluded from the simulation for that read length.
Table 1 shows the results for 100-bp and 200-bp single-end reads. For both read lengths, SeqAlto is significantly faster than all of the other tools except Snap while retaining excellent accuracy. Snap is very fast, but not as sensitive as SeqAlto. Stampy and Novoalign are both highly accurate. However, both take much longer to align the reads. SOAP2 and Snap do not report MAPQ. In addition, SOAP2 was not able to be run in gapped mode, hence it only outputted ungapped alignments.
Table 1.
Results for simulated Illumina single-end reads of length 100-bp and 200-bp
| Aligner | Time | %Aln | %Unique (%Err) | %Q10 (%Err) |
|---|---|---|---|---|
| (a) 100-bp single-end reads | ||||
| SeqAlto | 175 | 93.94 | 89.23 [0.167] | 88.83 [0.090] |
| SeqAlto-all | 195 | 94.38 | 89.67 [0.132] | 89.28 [0.066] |
| Snap | 51 | 94.07 | 87.09 [0.020] | * |
| Bowtie2 | 528 | 92.69 | 90.74 [3.477] | 83.91 [0.071] |
| SOAP2 | 635 | 91.39 | 86.18 [0.233] | * |
| BWA | 1182 | 90.37 | 85.96 [0.119] | 85.81 [0.068] |
| Novoalign | 2092 | 94.39 | 92.23 [1.620] | 88.84 [0.009] |
| Stampy | 2645 | 94.53 | 90.12 [0.403] | 89.75 [0.176] |
| (b) 200-bp single-end reads | ||||
| SeqAlto | 264 | 94.47 | 91.25 [0.033] | 90.93 [0.019] |
| SeqAlto-all | 282 | 94.48 | 91.28 [0.031] | 90.95 [0.017] |
| Snap | 92 | 92.97 | 87.03 [0.007] | * |
| Bowtie2 | 1116 | 94.02 | 93.11 [2.240] | 87.87 [0.020] |
| SOAP2 | 2767 | 75.68 | 72.68 [0.066] | * |
| BWA | 3165 | 89.15 | 86.24 [0.044] | 86.19 [0.028] |
| Stampy | 6890 | 94.55 | 91.64 [0.234] | 91.42 [0.082] |
Time measured is elapsed time (seconds) Unique refers to MAPQ ≥ 1 if MAPQ available. Q10 refers to MAPQ ≥ 10.
3.2 Simulated paired-end reads
1 million paired-end reads with the same parameters as in the single-end case were generated. The template size was chosen from a normal distribution with mean 250 bp and 450 bp for read lengths 100 bp and 200 bp, respectively. A standard deviation of 50 is used for both read lengths. A read is called correct if both pairs are within 50 bp of the simulated location.
Table 2 shows the results for 100-bp and 200-bp paired-end reads. SeqAlto takes advantage of the longer effective read length of paired-end reads to further improve alignment sensitivity and accuracy. For 100-bp reads, SeqAlto is on par with Stampy and Novoalign in terms of accuracy while being about 10 × faster. When aligning paired-end reads, BWA tends to output partial alignments more frequently compared to the other algorithms, hence its accuracy drops as the read length increases.
Table 2.
Results for simulated Illumina paired-end reads of length 100 bp and 200 bp
| Aligner | Time | %Aln | %Unique (%Err) | %Q10 (%Err) |
|---|---|---|---|---|
| (a) 100-bp paired-end reads | ||||
| SeqAlto | 473 | 94.57 | 91.01 [0.044] | 90.77 [0.034] |
| SeqAlto-all | 493 | 94.58 | 91.10 [0.041] | 90.86 [0.032] |
| Snap | 115 | 94.08 | 90.49 [0.030] | * |
| Bowtie2 | 852 | 93.36 | 92.45 [2.159] | 87.32 [0.015] |
| SOAP2 | 2753 | 88.77 | 85.42 [0.113] | * |
| BWA | 2470 | 93.89 | 90.82 [0.134] | 90.60 [0.111] |
| Novoalign | 3899 | 94.45 | 91.19 [0.033] | 90.85 [0.009] |
| Stampy | 6398 | 94.58 | 91.53 [0.294] | 91.02 [0.068] |
| (b) 200-bp paired-end reads | ||||
| SeqAlto | 529 | 94.55 | 92.15 [0.021] | 92.09 [0.020] |
| SeqAlto-all | 579 | 94.55 | 92.15 [0.021] | 92.09 [0.020] |
| Snap | 157 | 88.20 | 85.96 [0.014] | * |
| Bowtie2 | 1712 | 94.32 | 93.94 [1.850] | 89.25 [0.010] |
| SOAP2 | 13 219 | 46.55 | 45.43 [0.066] | * |
| BWA | 5691 | 93.81 | 91.68 [0.167] | 91.60 [0.160] |
| Stampy | 14 059 | 94.56 | 92.44 [0.255] | 92.08 [0.054] |
Time measured is elapsed time (seconds) Unique refers to MAPQ ≥ 1 if MAPQ available. Q10 refers to MAPQ ≥ 10.
3.3 Simulated large indel reads
Detection of large indels in the genome is of particular interest to researchers performing resequencing. Although indels larger than about 25 bp can potentially be detected from pairing information, indels that are moderate in size pose a problem to many aligners. One million 100-bp single-end reads with a single deletion or insertion of fixed size and a 1% substitution rate are generated from the human genome. Results for a 2% substitution rate and larger (up to 50 bp) indels can be found in the Supplementary Material. This test examines the ability of each algorithm to reliably place insertions and deletions of various sizes. A read is aligned correctly if it is within 50 bp of the simulated location.
Figures 1 and 2 show the alignment sensitivity and accuracy for insertions and deletions ranging from 1 to 25 bp. SeqAlto maintains consistent alignment accuracy and sensitivity across a wide range of indel sizes. Stampy is also highly sensitive to a wide range of indel sizes. However, its accuracy suffers for larger sized indels. This will become particularly evident when variant calling is performed on the aligned reads. The other tools are only able to align single-end reads with smaller indels. Although paired-end reads should improve the alignment of these reads for all algorithms, Section 3.4 shows that this is still inadequate for variant calling.
Fig. 1.
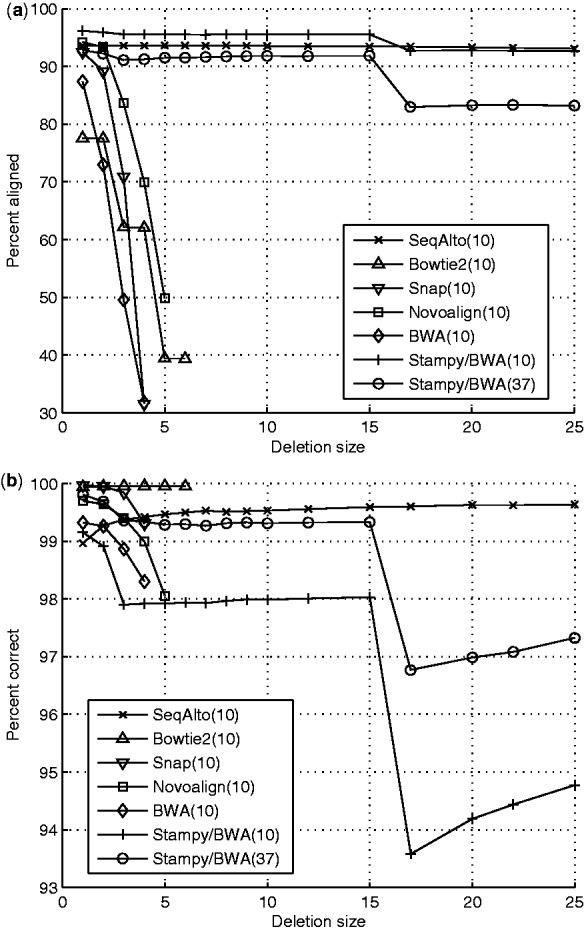
100-bp single-end reads with deletion in random location. Reported results are for MAPQ ≥ value in parentheses. Data points with < 30% of the reads aligned correctly were removed due to erratic behavior of the percent correct value, (a) percent of reads aligned (b) percent of aligned reads correct.
Fig. 2.
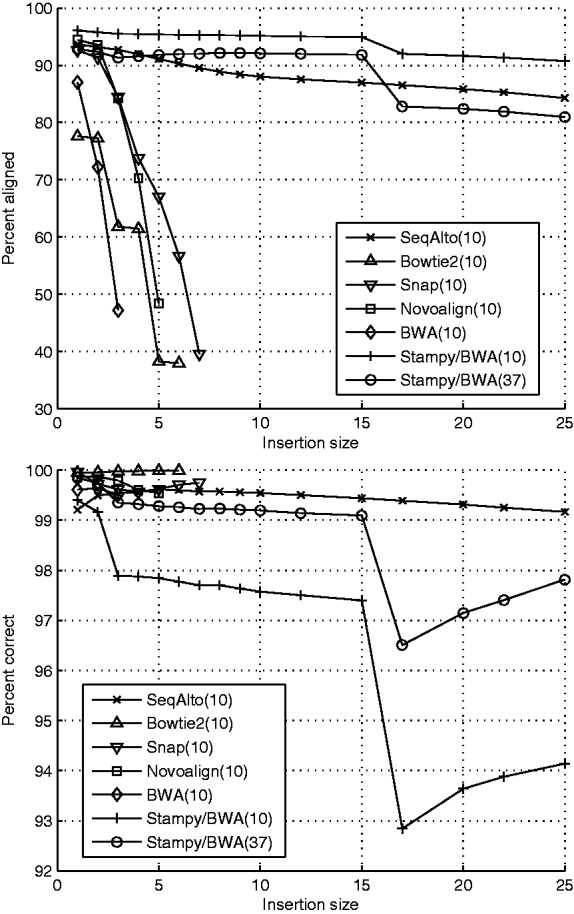
100-bp single-end reads with insertion in random location. Reported results are for MAPQ ≥ value in parentheses. Data points with < 30% of the reads aligned correctly were removed due to erratic behavior of the percent correct value, (a) percent of reads aligned (b) percent of aligned reads correct.
3.4 Simulated variant calling
The end results of resequencing are the variants called. SeqAlto, Snap, Bowtie2, BWA, Stampy and Novoalign are evaluated as the aligner for the GATK (DePristo et al., 2011) variant caller. Variants with a distribution as described in Section 4 are simulated on Chromosome 1 of the human genome. The location of these variants are recorded. Forty million 100 bp paired-end read pairs (about 32× coverage) are then simulated from this perturbed Chromosome 1 with a 2% error rate. In total, there are 422 120 Single Nucleotide Polymorphisms (SNPs) and 22 255 indel variants. The reads are aligned to Chromosome 1 then, the BAM file is passed to the GATK. Finally, each variant called is evaluated for correctness. The results for the Samtools mpileup (Li, 2011) variant caller and results with other parameter settings can be found in the Supplementary Material.
Table 3 shows the SNPs and indels called by the GATK. A SNP is called correct if it is in exactly the same location. An indel is called correct if is within 15-bp of the true location.
Table 3.
Summary of the SNPs and indels called by GATK after alignment with each algorithm
| Aligner | Called | %Correct | %Discovered |
|---|---|---|---|
| (a) GATK SNPs called | |||
| SeqAlto | 429 949 | 96.688 | 98.481 |
| Snap | 432 023 | 96.078 | 98.332 |
| Bowtie2 | 412 753 | 98.250 | 96.070 |
| BWA | 426 207 | 97.466 | 98.409 |
| Stampy | 427 137 | 97.290 | 98.446 |
| Novoalign | 430 906 | 96.674 | 98.686 |
| (b) GATK Indels called | |||
| SeqAlto | 22 057 | 99.941 | 98.477 |
| Snap | 25 563 | 93.319 | 90.303 |
| Bowtie2 | 20 750 | 99.918 | 91.809 |
| BWA | 21 228 | 99.915 | 95.174 |
| Stampy | 22 696 | 99.277 | 98.288 |
| Novoalign | 20 899 | 99.947 | 93.610 |
Overall, SeqAlto finds more real indels compared to all other algorithms. SeqAlto is more accurate than Stampy, which is the only algorithm that matches SeqAlto in number of variants called. For aligning SNPs, SeqAlto is competitive with all the other algorithms despite taking much less time to align the reads compared to Stampy and Novoalign. This simulation clearly highlights SeqAlto's ability to accurately align reads from complex variants.
3.5 Real illumina reads
All algorithms are used to align real Illumina sequencing data (Short Read Archive, accession number SRX100628). These reads came from an Illumina HiSeq 2000 sequencing run of HapMap individual NA19200. For the single-end benchmark, the first read in each pair was used. Since there is no ground truth for real data, the total number of reads aligned is used to judge the alignment sensitivity of each algorithm. For comparing alignment accuracy, 1 million reads were randomly sampled without replacement from the total reads and aligned to the human genome (hg19). The full dataset (75 630 683 read pairs) is also aligned to the human genome and used to call variants on Chromosome 1 with the GATK. The variants called are then compared to dbSNP (build 135) variants as a rough guide to the accuracy of variants discovered.
Table 4(a) shows the results for single-end reads. For single-end reads, SeqAlto is able to align more reads in much less time compared to the faster algorithms. Stampy is able to align significantly more reads compared to the other algorithms. However, the additional reads can potentially cause false positives in the variant calling as shown in Section 3.4. Table 4(b) shows the results for paired-end reads. Again, SeqAlto is faster than all aligners except Bowtie2 and Snap while being able to align more reads.
Table 4.
Results for real Illumina reads of length 101 bp
| Aligner | Time | %Align | %Unique | %Q10 |
|---|---|---|---|---|
| (a) 101-bp real single end reads | ||||
| SeqAlto | 545 | 94.970 | 88.927 | 88.339 |
| SeqAlto-all | 656 | 95.015 | 88.944 | 88.357 |
| Snap | 49 | 93.661 | 85.261 | * |
| Bowtie2 | 521 | 96.122 | 94.347 | 86.213 |
| SOAP2 | 1261 | 90.985 | 85.575 | * |
| BWA | 1056 | 91.826 | 86.803 | 86.655 |
| Novoalign | 3128 | 95.582 | 89.499 | 89.214 |
| Stampy | 1851 | 98.634 | 92.866 | 91.123 |
| (b) 101-bp real paired-end reads | ||||
| SeqAlto | 1394 | 96.122 | 91.117 | 90.794 |
| SeqAlto-all | 1627 | 96.136 | 91.127 | 90.803 |
| Snap | 172 | 88.692 | 88.692 | * |
| Bowtie2 | 1075 | 94.579 | 93.746 | 87.733 |
| SOAP2 | 2935 | 89.128 | 85.857 | * |
| BWA | 2024 | 93.815 | 90.494 | 90.109 |
| Novoalign | 4982 | 93.846 | 90.421 | 90.239 |
| Stampy | 5003 | 96.274 | 92.640 | 91.623 |
A 22-mer seed was used for SeqAlto Time measured is elapsed time (seconds). Unique refers to MAPQ ≥ 1 if MAPQ available. Q10 refers to MAPQ ≥ 10.
The entire dataset (5× coverage) was aligned to the human genome. The same GATK pipeline as in Section 3.4 was used to call variants with the alignment results. The variants called are compared to dbSNP (build 135). The results of the comparison are shown in Table 5. SeqAlto is slightly less accurate than the other methods when calling SNPs. For indels, SeqAlto calls more indels compared to all other algorithms except Stampy and does so with relatively good accuracy.
Table 5.
Summary of the SNPs and indels called by GATK on Chromosome 1 after alignment of real data with each algorithm
| Aligner | Called | %dbSNP | %Discovered |
|---|---|---|---|
| (a) GATK SNPs Called | |||
| SeqAlto | 252 965 | 91.212 | 87.780 |
| Snap | 305 281 | 75.623 | 87.830 |
| Bowtie2 | 203 508 | 95.973 | 74.304 |
| BWA | 241 084 | 93.324 | 85.595 |
| Stampy | 218 289 | 93.620 | 77.747 |
| Novoalign | 227 350 | 92.932 | 80.380 |
| (b) GATK Indels Called | |||
| SeqAlto | 9729 | 76.986 | 82.808 |
| Snap | 7399 | 70.915 | 58.010 |
| Bowtie2 | 8794 | 80.964 | 78.718 |
| BWA | 8838 | 80.878 | 79.027 |
| Stampy | 10 408 | 73.770 | 84.887 |
| Novoalign | 9386 | 78.329 | 81.282 |
%Discovered refers to percentage of the known variants in dbSNP found.
4 SIMULATION METHODS
Accurate simulation of high-throughput sequencing reads is a challenge. SeqAlto includes a simulation mode where a new haploid genome with indels and substitutions is generated from a reference genome. In order to determine the relationship between locations on this new genome to locations on the reference, an exact mapping between the locations is generated simultaneously. Reads are then generated from this new genome with an error profile that can be tailored to a particular sequencing platform. Each read is tagged with the location that it was generated from and after alignment to the reference genome, the stored mapping is used to determine whether each read is aligned to the correct location. Other approaches to simulating sequencing reads include (Huang et al., 2011).
Following publication of the 1000 genomes pilot project (The 1000 Genomes Project Consortium, 2010), there is a better understanding of the distribution of indel lengths in the human genome. Indels generated by the SeqAlto read simulator have lengths that follow a power law distribution similar to what was observed in the 1000 genomes project. The indel length is sampled from the exponent of an exponential distribution with mean one. This results in the largest indel simulated to be on the order of 100 000 bp to 1 000 000 bp. SNPs and indels are simulated independently. The SNP mutation rate is set to 0.25% and the indel mutation rate to be 0.01%. For Illumina reads, we used a uniform error rate across the read of 2%. The results for reads with other error rates can be found in the Supplementary Material.
5 METHODS
SeqAlto searches for the global alignment of sequencer reads to a reference using the commonly known seed and extend approach. There are three phases to the alignment process: index construction, ungapped alignment and Needleman–Wunsch extension.
Define a k-mer (seed) to be a sequence of nucleotides of length k. Each nucleotide must come from the set {A, C, T, G}. Hence, each k-mer is encoded as an unsigned integer with 2 bits per nucleotide. On a 64-bit system, this allows for k-mers of size of up to 32 bp. If the a k-mer has a nucleotide other than {A, C, T, G}, such as N, it is replaced with a uniform random nucleotide since there is only a 2-bit alphabet available. This encoding is the same approach used in BWA and many other tools.
Let the index be defined as a list of tuples recording the k-mer and the location in the reference of that k-mer. The index is sorted by the numeric value of each k-mer for fast searching. SeqAlto achieves its high speed first by using much larger k-mer sizes compared to existing approaches, examining less repetitive k-mers first and by adaptively stopping the k-mer search. Large contiguous k-mers greatly reduce the number of locations of each k-mer in the reference to mostly unique hits. In general, large k-mers would reduce the sensitivity of the alignment. However, for longer reads this choice of k-mer size does not reduce sensitivity as shown in the simulation results. Furthermore, a novel sub-sampling approach is used to reduce the amount of memory required for the index to under 8 GB. Compared to BWT-based approaches for large k-mer search, our approach requires less random access to memory per index lookup resulting in significantly improved performance.
5.1 Index construction
A genome index ideally contains the location of all overlapping k-mers in the genome. Naively storing all k-mers of size between 17 bp and 32 bp in one strand of the human genome requires about 36 GB of memory. Using a combination, of two strategies, we can reduce the size of the index to under 8 GB with only a minor penalty on the sensitivity. This enables SeqAlto to be run on almost all desktop computers. These strategies can be tuned for more powerful computers as they become available.
At the first stage of the index construction, each overlapping k-mer together with its location is extracted from the genome and stored in an array W. This array of tuples is then sorted by the k-mer value. Then the leading m bits are removed from each k-mer such that (2k – m) is less than 32. These m bits are stored in a prefix array P that records the start and end locations in W of k-mers tuples prefixed with the same m bits. Since W is sorted, k-mers with the same prefix exist next to each other. Now, only the (2k – m) remaining bits of each k-mer needs to be stored in W. This array of tuples W together with the prefix array P form the genome index. Considering only genomes with less than 4 billion nucleotides, this two-level index reduces the size of each (k-mer, location) tuple in the index to 8 Bytes instead of 12 Bytes. Hence, for small prefix table sizes the entire human genome index will only take about 24 G to store. This index construction also greatly improves the lookup speed over a naive binary search.
SeqAlto has the option to not store every k-mer in the reference. In order to reduce memory consumption without greatly impacting alignment, quality and speed of alignment we only store a k-mer if it satisfies a hash function f(x), where x is the numeric value of the k-mer. The hash function f(x) is chosen so that there is approximately uniform coverage of the genome. In SeqAlto, (1) is used.
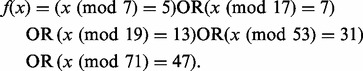 |
(1) |
Equation (1) was selected so that memory usage for the human genome is under 8 GB and for Chromosome 1, the median gap size is 3 bp and the largest gap apart from the centromere is 131 bp. Empirically, there is little loss in sensitivity from using this approach as seen in the Section 3. In principle, this expression can also be easily tuned for machines with more memory, as any expression of similar form is adequate. On machines with 32 GB or more memory, this second step is not required and a larger index can be constructed for improved sensitivity and accuracy.
The index is essentially a large sorted list. Hence, it is easy to add arbitrary additional k-mers to the index with negligible cost to both the construction and search. k-mers overlapping known SNPs and indels could easily be added to the index to improve the sensitivity of alignment. This change would only moderately increase the size of the index.
5.2 Ungapped alignment
The first stage of the alignment process is the ungapped alignment stage. This stage is separate from the gapped stage since we expect that most gapped reads will not align anywhere in the genome as high-quality ungapped alignments. This separation also allows for easy interfacing with a hardware accelerator for the gapped alignment.
Each read is treated separately in the alignment process. All overlapping k-mers are extracted from each read. k-mers satisfying the function f(x) as defined in (1) are retained and the other k-mers are discarded. Highly repetitive k-mers are also discarded.
Each k-mer is then searched for in the index according to the following order. Starting with the non-overlapping k-mers, the number of locations the k-mer exists in the genome is computed from the index. If this number is less than a predefined variable MAX_LOC, all of the locations are examined for an ungapped alignment of the read. Once all k-mers with number of locations less than MAX_LOC have been examined, the other k-mers are examined. Due to the large k-mer size, many k-mers either align uniquely to the genome or do not align at all. Hence, the ungapped alignment stage proceeds very quickly.
Similar to the idea in Baeza-yates and Perleberg (1992), it is not necessary to examine all k-mers in a read. For instance, reads with no mismatches should be unambiguously identified after examining just one non-overlapping k-mer. By the pigeon hole principle, reads with m mismatches only require examination of (m + 1) non-overlapping k-mers. Additional k-mers are examined in order to determine if a read is from a repetitive region (see Section 5.6). Hence, SeqAlto decides to stop examining k-mers according to the following boolean function 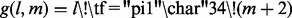 , where m is the number of mismatches and gaps of the best alignment so far and l is the number of k-mers examined. For reads with high number of mismatches, this early stopping does not guarantee finding the correct hit. This procedure is described in Algorithms 1 and 2. Overall, this procedure results in SeqAlto spending less time on high-quality reads.
, where m is the number of mismatches and gaps of the best alignment so far and l is the number of k-mers examined. For reads with high number of mismatches, this early stopping does not guarantee finding the correct hit. This procedure is described in Algorithms 1 and 2. Overall, this procedure results in SeqAlto spending less time on high-quality reads.
Algorithm 2 Examine k-mer.
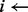 index of k-mer to be examined
index of k-mer to be examined
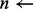 number of locations k-mer exists
number of locations k-mer exists
if
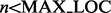 then
then
Mark k-mer i as examined
5: Check all locations of k-mer i for ungapped alignment
return number of mismatches in best alignment
else if
 all k-mer with n < MAX-LOC examined then
all k-mer with n < MAX-LOC examined then
Mark k-mer i as examined
Check all locations of k-mer i for ungapped alignment
10: return number of mismatches in best alignment
else
return-1
end if
Algorithm 1 Ungapped Alignment.
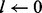

while not g(l, m) and (exists k-mers unexamined) do
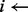 index of next unexamined k –mer
index of next unexamined k –mer
5: 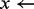 examine(ith
k-mer)
examine(ith
k-mer)
if
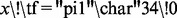 then
then
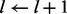
end if
if
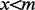 then
then
10: 
end if end while
If the ungapped alignment outputs a best alignment with penalty score greater than a single gap open, the read is then examined for a single gap alignment. At this stage, the location of each k-mer is normalized by subtracting the location of the k-mer in the read. For each pair of normalized locations with distance smaller than the maximum gap size, the locations are extended to find single gap alignments with at most one mismatch on each side. This is able to resolve some indels in repetitive regions.
5.3 Needleman–Wunsch extension
All reads that do not align in the ungapped alignment stage are passed to the Needleman–Wunsch extension stage. The order of examining k-mers is the same as for the ungapped alignment. Each k-mer location is searched with a banded Needleman–Wunsch with affine gap penalty to find gapped alignments. The width of the band can be chosen by the user and by default it finds up to at least 50 bp indels. The same function g(l, m) is used to determine if the program should continue examining k-mers. Single instruction multiple data (SIMD) acceleration through SSE2 instructions is used to improve the performance of this stage.
5.4 Paired-end alignment
Paired-end information is both used to help search for alignments and also resolve discordant read pairs. If one read in a pair aligns and the other read does not, SeqAlto searches for the remaining read within the maximum insert size of the pair with a combination of Smith–Waterman and Needleman–Wunsch. These alignments are flagged in the output giving the user additional flexibility in post-processing. If both pairs align with one or more equally good hits, all possible combinations are checked for a concordant pair. If all possible combinations are discordant, SeqAlto searches for a concordant alignment within a region around each alignment defined by the maximum insert size.
5.5 Hybrid mode
For reads much less than 100 bp, SeqAlto has some trouble aligning reads in repeat regions and also reads with high numbers of mismatches due to an inability to locate enough seeds on the reads. In hybrid mode, SeqAlto outputs reads with few valid seeds as a FASTQ file that can be then aligned with BWA or any other alignment tool. The two resulting SAM files can then be merged to produce the final alignment.
5.6 Mapping quality score
Mapping quality (MAPQ) scores provide a way for users to judge the reliability of each alignment. They were first introduced in MAQ (Li et al., 2008) as an estimation of the probability a read was aligned incorrectly. Accurate calculation of mapping quality considerably slows down the alignment process since all k-mers should be visited. Despite not visiting all k-mers, SeqAlto also provides an estimate of mapping quality. The mapping quality reported by SeqAlto is not as specific as the mapping quality reported by other tools. However, in practice there is little difference.
As discussed in the Mapping and Assembly with Quality (MAQ) article (Li et al., 2008), there are mainly two sources of alignment error. The first is when the best alignment fails to be reported by the alignment algorithm. The second is when a read aligns optimally to an incorrect location due to genetic variation or read errors. Similar to BWA, SeqAlto ignores the first source of error and assumes either the best alignment will be reported or no alignment will be reported at all. SeqAlto estimates the second source of error by recording all alignments that differ from the best by at most one mismatch and also the percentage of the read covered by highly repetitive k-mers. The formula used is similar to that of BWA for compatibility with downstream tools.
5.7 Choice of k-mer size
The size of the k-mer seed needs to be selected prior to index construction. A single size is clearly not optimal for all read lengths. Smaller k-mers will be too repetitive and larger k-mers will reduce sensitivity. Table 6 displays the performance with various k-mer sizes.
Table 6.
Alignment performance with various k-mer sizes for 100-bp single-end reads
| k-mer | Time | %Aln | %Unique [%Err] | %Q10 [%Err] |
|---|---|---|---|---|
| 18 | 218 | 94.26 | 89.53 [0.151] | 89.15 [0.074] |
| 20 | 197 | 94.16 | 89.44 [0.152] | 89.04 [0.077] |
| 22 | 178 | 93.94 | 89.23 [0.167] | 88.83 [0.090] |
| 25 | 151 | 93.22 | 88.52 [0.202] | 88.12 [0.119] |
| 28 | 146 | 91.95 | 87.30 [0.266] | 86.90 [0.167] |
Time measured in seconds.
As indicated in Table 6, 22-mer seeds are optimal for 100 bp reads. For longer reads, using 28-mer seeds is probably a better choice.
5.8 Multi-threading
SeqAlto is able to take advantage of modern multi-core computers by distributing the alignment of each read to multiple threads. There is little communication between the threads except when reading and writing files. SeqAlto is run in default mode on 1 million 101-bp paired-end reads from the real Illumina dataset and the alignment time is recorded.
Table 7 shows that SeqAlto scales well up to at least eight threads while BWA is limited by its non-parallelized pairing step. An Illumina HiSeq2000 generates about 600 gigabases of data per run. This translates to about 3 billion 100-bp pairs. On a modern octa-core workstation computer, SeqAlto is able to align the 3 billion pairs in about 6 days compared to about 15 days with BWA on the same computer. Considering one Illumina HiSeq 2000 takes 11 days to generate the sequence data, this allows one inexpensive desktop workstation to service several large sequencers. As read lengths and the quality of sequencer output improves, these results will become even more significant.
Table 7.
Speed-up due to multi-threading of SeqAlto and BWA
| SeqAlto |
BWA |
|||||
|---|---|---|---|---|---|---|
| Threads | Time | Speed-up | MPH | Time | Speed-up | MPH |
| 1 | 1395 | 1.0× | 2.58 | 2010 | 1.0× | 1.79 |
| 2 | 700 | 2.0× | 5.14 | 1121 | 1.8× | 3.21 |
| 4 | 360 | 3.9× | 10.00 | 612 | 3.3× | 5.88 |
| 8 | 184 | 7.6× | 19.57 | 436 | 4.6× | 8.26 |
Time measured in seconds. MPH stands for millions of pairs aligned per hour.
6 DISCUSSION
Overall, SeqAlto is an efficient alignment tool that targets the future of resequencing where reads are long and high quality. When aligning longer reads, SeqAlto is faster than most existing tools while retaining the ability to accurately align complex variants. This is particularly important for the high volume of resequencing that will be required as sequencing enters clinical applications. For convenience, SeqAlto outputs in the common SAM format that is compatible with numerous downstream analysis tools.
The comparison of alignment algorithms is primarily performed with simulated reads as real reads do not have a clear ground truth available. On simulated reads SeqAlto is shown to be faster than most current tools and able to accurately align reads with large indels. When aligning real Illumina reads, SeqAlto maintains its efficiency advantage over most of the tools. This advantage is expected to increase as read quality improves. The ability to align reads with large indels is particularly evident when variants are called from the aligned reads. Both GATK and Samtools were able to correctly call more indel variants from SeqAlto's aligned reads from both real and simulated data compared to all the other tools except Stampy. In the cases where less variants were called, SeqAlto was more accurate.
There is a clear trend towards longer read lengths among high-throughput sequencers. SeqAlto has been shown to be ideal for aligning long high-quality reads for the purpose of resequencing. We expect that the large seed approach taken by SeqAlto to be the standard approach for read alignment as the volume and length of reads increase.
Supplementary Material
ACKNOWLEDGEMENT
Many thanks to the help and comments from the team at Bina Technologies Inc. Especially, AJ Minich and Jian Li for help with running the real variant comparisons.
Funding: John C. Mu, Hui Jiang and Wing H. Wong were partially supported by National Institutes of Health grants R01HG004634 and R01HG005717 to Wing H Wong.
Conflict of Interest: All authors are hold stock in Bina Technologies inc. John C. Mu partially completed the work as an intern at Bina. Hui Jiang and Wing H. Wong are scientific advisors for Bina. Amirhossein Kiani, Marghoob Mohiyuddin and Narges Bani Asadi are currently employed at Bina. Narges Bani Asadi and Wing H. Wong are co-founders of Bina.
REFERENCES
- Baeza-yates RA, Perleberg CH. Combinatorial Pattern Matching, Third Annual Symposium. Springer-Verlag; 1992. Fast and practical approximate string matching; pp. 185–192. [Google Scholar]
- Burrows M, Wheeler DJ. A block-sorting lossless data compression algorithm. HP Labs Technical Reports, SRC-RR-124. 1994 [Google Scholar]
- David M, et al. SHRiMP2: sensitive yet practical short read mapping. Bioinformatics. 2011;27:1011–1012. doi: 10.1093/bioinformatics/btr046. [DOI] [PubMed] [Google Scholar]
- DePristo MA, et al. A framework for variation discovery and genotyping using next-generation dna sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferragina P, Manzini G. In Proc. 41st IEEE Symposium on Foundations of Computer Science (FOCS). Redondo, Beach, CA, USA. Redondo Beach, CA, USA: 2000. Opportunistic data structures with applications. [Google Scholar]
- Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS One. 2009;4:e7767. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Li L, Myers JR, Marth GT. Art: a next-generation sequencing read simulator. Bioinformatics. 2012;28:593–594. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wong WH. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008;24:2395–2396. doi: 10.1093/bioinformatics/btn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konagurthu AS, et al. Proceedings of the 10th International Conference on Algorithms in Bioinformatics. 2010. Design of an efficient out-of-core read alignment algorithm; pp. 189–201. [Google Scholar]
- Langmead BS, Steven L. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of illumina sequence reads. Genome Res. 2011;21:936–939. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhis N, et al. Slidermaximum use of probability information for alignment of short sequence reads and snp detection. Bioinformatics. 2009;25:6–13. doi: 10.1093/bioinformatics/btn565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizk G, Lavenier D. GASSST: global alignment short sequence search tool. Bioinformatics. 2010;26:2534–2540. doi: 10.1093/bioinformatics/btq485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaharia M, et al. Faster and more accurate sequence alignment with snap. arXiv. 2011 arXiv:1111.5572v1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.