

Abstract
MECP2, an X-linked gene encoding the epigenetic factor methyl-CpG-binding protein-2, is mutated in Rett syndrome (RTT) and aberrantly expressed in autism. Most children affected by RTT are heterozygous Mecp2 −/+ females whose brain function is impaired postnatally due to MeCP2 deficiency. While prior functional investigations of MeCP2 have focused exclusively on neurons and have concluded the absence of MeCP2 in astrocytes, here we report that astrocytes express MeCP2, and MeCP2 deficiency in astrocytes causes significant abnormalities in BDNF regulation, cytokine production, and neuronal dendritic induction, effects that may contribute to abnormal neurodevelopment. In addition, we show that the MeCP2 deficiency state can progressively spread at least in part via gap junction communications between mosaic Mecp2 −/+ astrocytes in a novel non-cell-autonomous mechanism. This mechanism may lead to the pronounced loss of MeCP2 observed selectively in astrocytes in mouse Mecp2 −/+ brain, which is coincident with phenotypic regression characteristic of RTT. Our results suggest that astrocytes are viable therapeutic targets for RTT and perhaps regressive forms of autism.
Introduction
RTT and the more prevalent regressive type autism share a substantial phenotypic overlap, including autistic behaviors. The affected children start to lose developed skills after a period of apparently normal growth and development. This regressive clinical course has prompted categorization of both as “postnatal neurodevelopmental disorders” (Zoghbi, 2003). RTT is caused by loss-of-function mutations in the X-linked MECP2 encoding methyl-CpG-binding protein 2 (MeCP2). It affects predominantly heterozygous Mecp2 −/+ females who are mosaics of MECP2 mutant-expressing cells because of X chromosome inactivation (XCI) (Chahrour and Zoghbi, 2007). In addition to RTT, MECP2 mutations or MeCP2 deficiency are found in attention deficit hyperactive disorder, Down syndrome, mild learning-disability, neonatal encephalopathy, X-linked mental retardation, and notably, autism (Samaco et al., 2004, 2005; Moretti and Zoghbi, 2006; Nagarajan et al., 2006). Understanding the neurobiological mechanism of RTT, a monogenic disorder, may help elucidate the complex mechanism leading to autism.
Neuropathologically, both RTT and regressive type autism show reduced neuronal size, reduced dendritic arborization, and sparse and short dendritic spines in selected brain regions (Zoghbi, 2003; Armstrong, 2005). To understand how MeCP2 deficiency impairs brain function, much effort has been focused on cell-autonomous effects of neuronal MeCP2 deficiency because MeCP2 is highly expressed in mature neurons. Previous data support that MeCP2 deficiency in neurons is sufficient to cause a RTT-like neurologic phenotype in mouse (Chen et al., 2001). However, at least two lines of evidence indicate a role of non-neuronal cells in RTT. First, a CamK-Cre-mediated deletion of Mecp2 in postnatal neurons results in a milder and delayed RTT-like phenotype compared with that resulting from a germline or a Nestin-Cre mediated deletion (Chen et al., 2001). Second, widespread neuron-specific reintroduction of Mecp2 in Mecp2-null mice by two strictly neuronal promoters failed to show any phenotypic improvement (Alvarez-Saavedra et al., 2007), unlike the endogenous Mecp2 promoter that served to rescue the RTT phenotype upon removal of a stop codon (Guy et al., 2007).
Astrocytes actively control dendritic growth, synaptogenesis, synapse number, synapse function, and synaptic plasticity (Barres, 2008). The peak period of gliogenesis coincides with the elaboration of dendritic arbors and the establishment of synapses, in postnatal periods. In addition, astrocytes continue to be generated in the adult CNS. Therefore, functional abnormalities of astrocytes may contribute to neurodevelopmental disorders with a postnatal onset, such as RTT. We hypothesize that the inherent astrocytic abnormalities due to MeCP2 deficiency may contribute to the RTT phenotype.
However, the possible contribution of astrocytic dysfunction to RTT has not been examined, mainly due to previously concluded absence of MeCP2 in astrocytes (Shahbazian et al., 2002). Here, we provide in vitro and in vivo evidence to show that astrocytes express MeCP2, albeit at a lower level than neurons. Furthermore, MeCP2 deficient astrocytes are abnormal in several key functions, which might amount to decreased dendritic arborization and impaired immune regulation in RTT brain. Finally, we provide evidence to support a novel gap junction (GJ)-mediated spread of MeCP2 deficiency state among astrocytes in Mecp2 −/+ tissue, which might contribute to phenotypic regression typical of RTT.
Materials and Methods
Mouse model of RTT
Mecp2 tm1.1Bird/+ mice originated from Dr. Adrian Bird's laboratory were obtained from The Jackson Laboratory. Mice were mated with C57BL/6J mice (The Jackson Laboratory). Pups were immediately genotyped to determine the Mecp2 deletion according to the protocol provided by The Jackson Laboratory. The gender was determined using primers for the Sry gene on Y chromosome, which were 5′-TGG GAC TGG TGA CAA TTG TC-3′ and 5′-GAG TAC AGG TGT GCA GCT CT-3′. The University of California Davis Institutional Animal Care and Use Committee approved all animal experiments.
Cultures of astrocytes and neurons.
Primary astrocyte cultures were prepared from 1-d-old mouse cerebral cortex according to previously described methods (Maezawa et al., 2006). In most experiments, 2–4 weeks in vitro, confluent astrocyte cultures were used, which showed no differences in cell numbers, total RNA and protein quantities between different genotypes. The hippocampal neuronal cultures were prepared from E18 wild-type C57BL/6J mice according to the method of Xiang et al. (1998).
Quantitative RT-PCR.
Total RNA was isolated by homogenization of cell cultures in TRIzol reagent (Invitrogen) according to the manufacturer's instruction. Purified RNA was resuspended in RNase-free water and stored at −70°C until use. The forward and reverse primer sequences for Wt Mecp2 were 5′-GGT AAA GAC CCA TGT GAC CC-3′ and 5′-GGC TTG CCA CAT GAC AA-3′.
To determine the relative levels of Wt and mutant Mecp2 transcripts in mice, we used previously designed primer pairs (Miralvès et al., 2007). The forward primer sequence for Wt transcript was 5′-GAC CCC TTG GGA CTG AAG TT-3′ and that for mutant transcript was 5′-CCA TGC GAT AAG CTT GAT GA-3′. The following reverse primer was used to pair with both forward primers: 5′-CCA CCC TCC AGT TTG GTT TA-3′. Because these primer sets may also amplify the chromosomal DNA, the DNA content of the samples was further minimized by purifying RNA using the RNeasy MinElute Cleanup Kit (Qiagen) according to the manufacturer's instructions. Using the resulting RNA samples and the above primers, PCRs without prior reverse transcription yielded minimal detectable products.
We used a primer pair previously designed to quantify the murine Bdnf transcript in astrocytes (Koyama et al., 2005). The forward sequence was 5′-ATG ACC ATC CTT TTC CTT ACT ATG GT-3′ and the reverse sequence was 5′-TCT TCC CCT TTT AAT GGT CAG TGT AC-3′.
The cDNA was synthesized from 2 μg of total RNA using SuperScript First-stranded synthesis system (Invitrogen). Quantitative PCR was performed using the SYBR Green master mix in an ABI 7900HT Sequence Detection System (Applied Biosystems). The result was normalized to β-actin.
Western blot analysis.
To obtain cell lysates, cells were washed with ice-cold PBS and incubated with a buffer containing 50 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 2% SDS, proteinase inhibitor cocktail (Sigma), and phosphatase inhibitor cocktail (Sigma). Lysates were briefly sonicated and cleared by centrifugation at 50,000 rpm for 10 min. Equivalent amounts of protein were analyzed by Tris/HCl gel electrophoresis. Proteins were transferred to polyvinylidene difluoride membranes and probed with antibodies. Visualization was performed using enhanced chemiluminescence (ECL, GE Healthcare).
To quantify and compare the secreted NGF and BDNF, the astrocyte conditioned medium (ACM) from confluent cultures was first concentrated and desalted by using a Centricon YM-10 concentrator (Millipore). Equal portions of samples were then analyzed by Western blot. For apolipoprotein E quantification, the equal volumes of ACM from confluent cultures without prior concentration were analyzed.
The following primary antibodies (dilutions) were used: rabbit anti-MeCP2 IgG (1:1000, Millipore, for amino acids 465–478 of mouse MeCP2, detecting both MeCP2 isoforms, e1 and e2), chicken anti-MeCP2 IgY (1:5000, for the C terminal of MeCP2, detecting both e1 and e2 isoforms) (Yasui et al., 2007), anti-BDNF antibody (1:1000, Millipore Bioscience Research Reagents), anti-NGF antibody (1:500, Abcam), anti-mouse apoE (1:2000, Biodesign International), anti-p38MAPK (1:1000, Cell Signaling Technology), anti-phospho-p38MAPK (1:1000, Cell Signaling Technology), anti-phospho-p42/44 MAPK(1:1000, Cell Signaling Technology), anti-phospho-SPAK/JNK(1:1000, Cell Signaling Technology), anti-Connexin43 (1:1000, BD Transduction Laboratories), and anti-β-actin (1:3000, Sigma). Secondary antibodies were horseradish peroxidase-conjugated anti-rabbit, anti-goat, anti-chicken, or anti-mouse antibodies (1:3000, GE Healthcare).
BrdU incorporation assay.
Astrocytes were plated onto 24-well culture plates at an initial density of 3 × 105 cells/well in DMEM10 and incubated for 6 h. Cells were washed with serum-free Opti-MEM medium three times and cultured in Opti-MEM containing 10 μm BrdU (Sigma) for additional 2 h. Cells with BrdU incorporation were demonstrated by immunocytochemical stain for BrdU using anti-BrdU antibody conjugated with Alexa594 (Invitrogen) following the manufacturer's protocol (Millipore Bioscience Research Reagents).
Gene silencing by RNA interference.
Wt astrocytes were plated onto 24-well plates at 3 × 105 cells per well and cultured in DMEM10 for 24 h. Cells were transfected with siRNAs using HiPerfect Transfection reagent according to the manufacturer's instructions (Qiagen). Two sets of siRNA duplexes (Qiagen) homologous to mouse Mecp2 sequence were designed using the HiPerformance Design Algorithm (Norvatis AG) and were purchased from Qiagen (catalog #: siRNA#1, SI00195594; siRNA#2, SI02737714). The siRNA homologous to mouse Cx-43 sequence was also purchased from Qiagen (catalog # SI02712423). As a transfection control, an All Stars Negative Control siRNA (Qiagen) was used.
Sandwich ELISA for cytokines.
To induce the innate immune response, confluent astrocyte cultures were washed three times with serum-free Opti-MEM and cultured in Opti-MEM for 24 h. Cultures were then treated with 100 ng/ml lipopolysaccharide (LPS, EMD Biosciences) in Opti-MEM for another 24 h. In this condition, LPS induced negligible cell proliferation and there was no significant difference in cell number or total quantities of cellular proteins and secreted proteins between cultures of different genotypes. The activation was evaluated by measuring cytokines TNF-α, IL-1β, and IL-6 in ACM using cytokine ELISA kits according to the manufacturer's instruction (R&D Systems and RayBiotec).
Neurite outgrowth analysis.
Hippocampal neurons freshly prepared from E18 Wt mice were plated evenly onto the confluent monolayer of astrocytes at 1 × 105 neurons per well in six-well plates. Neurites were allowed to grow for 24 and 72 h before fixation. Dendrites were stained with anti-MAP2 antibody (1:1000, Millipore Bioscience Research Reagents). MAP2-stained neurons were observed under a Nikon Eclipse E600 microscope and photographed by a digital camera (SPOT RTke, SPOT Diagnostics). All dendrites were traced using the Neurolucida software (MicroBrightField) and dendritic length was calculated by Neuroexplorer (MicroBrightField). In all experiments, neurons were randomly selected for photograph and the photography, tracing and analysis of neuritis were performed in an investigator-blinded manner. Dendritic complexity was quantified by measuring the length of dendrites and counting the number of branch points in each branch order. The primary dendrites were defined as those directly emanating from the soma. Higher-order branches were those arising from previous order's processes. The dendritic length in a branch order was calculated by averaging the lengths of all dendrites in that branch order. The number of branch points in a branch order was calculated by averaging the number of branch points that give rise to the dendrites in that branch order.
Study for the non-cell-autonomous effect between astrocytes.
Astrocytes derived from Wt C57BL/6 mice were transfected with a green fluorescence protein (GFP) expression vector using the Effectene Transfection Reagent (Qiagen). GFP-labeled astrocytes were mixed with unlabeled astrocytes either from Wt or Mecp2 −/+ mice at a ratio of 1 GFP labeled to 3 unlabeled astrocytes. The cell mixture was plated onto cover glass in DMEM10 and cultured for 24–48 h. Cells were fixed, immunostained with anti-MeCP2, and counterstained with the nuclear dye DAPI. Fluorescence photomicrographs of GFP-positive cells (all of which were wild type) were taken and their MeCP2 expression was analyzed by the Image J program. The images were transformed to 8 bit gray scale, the nuclear contours of GFP-positive cells were manually defined, and the maximal focal pixel values in individual nuclei were recorded. The MeCP2 immunoreactivities of cells were classified into three tiers: high, low, and none. The high immunoreactivity was defined by a pixel value larger than or equal to half of the maximal pixel value recorded from the highest MeCP2 expresser in the coculture of GFP-labeled Wt cells and unlabeled Wt cells. The low immunoreactivity was defined by a pixel value smaller than the half maximal value. The nonexpressers showed no detectable immunoreactivity above background. The photography and analysis of MeCP2 levels of GFP-labeled cells were conducted in an investigator-blinded manner.
To test whether the gap junction inhibitors block the non-cell-autonomous effect on MeCP2 expression, the above mixed cultures were treated with 100 μmol/L carbenoxolone (CBX, Sigma), 50 μmol/L 18α-glycyrrhetinic acid (GA, Sigma), or vehicle only for 24–48 h in serum-free Opti-MEM. Cells were fixed with 4% paraformaldehyde and immunostained with anti-MeCP2 (1:1000, Millipore). The MeCP2 expression was subsequently analyzed as described above. In additional groups, cells were pretreated with 400 μmol/L tolbutamide (TB, Sigma) for 30 min before the addition of CBX or GA.
Mouse tissue microarray and immunofluorescence staining.
For immunofluorescence staining of brain tissue, we used paraformaldehyde-fixed frozen sections, paraffin-embedded whole brain sections, or tissue microarray sections. The tissue microarray was used for quantification and comparison of MeCP2 expression between groups of mice, because it made it possible to process multiple brain samples simultaneously, reducing sample-to-sample variability. Mouse cerebral tissue microarrays were prepared as previously described (Samaco et al., 2004; Nagarajan et al., 2006). Briefly, triplicate 600 μm cores of mouse cerebrum including cores from five Mecp2 −/+, three Mecp2 +/+ controls (P20–27w); one Mecp2 −/y and one Mecp2 +/y littermate (p10w) were inserted into a recipient paraffin block and sectioned. The processing, immunostaining, and analysis of microarray sections were also performed as previously described (Samaco et al., 2004; Nagarajan et al., 2006).
Primary antibodies used were: 1:50 anti-GFAP (Oncogene, mouse monoclonal); 1:1000 anti-MeCP2 (Aves, C-terminal, chicken polyclonal) and 1:50 anti-NeuN (Millipore Bioscience Research Reagents, mouse monoclonal). Secondary antibodies used were: 1:100 goat anti-mouse IgG-Oregon Green (Invitrogen) and 1:100 donkey anti-chicken IgG-Cy5 (Jackson ImmunoResearch). Chicken IgY (Aves) and mouse IgG (Millipore) were used on control slides with the same secondary antibodies as those used on the experimental slides to test for background levels of staining or immunofluorescence. Replicate slides were stained and scanned by LSC to increase the power of our statistical analyses.
Laser-scanning cytometry.
Laser-scanning cytometry (LSC) was performed as we previously described (LaSalle et al., 2001). LSC is similar to flow cytometry in its ability to quantitate fluorescence of individual cells in an un-biased and automated manner. In particular, with LSC solid-phase samples including adherent cultured cells, tissue sections and cytology smears can be examined for genetic, biochemical or morphological properties. Slides to be compared were scanned using the same nonsaturating photomultiplier settings. Cells were contoured based upon the DNA counter stain, PI. Astrocytes and neurons in the cortical tissue array were identified by gating on high cytoplasmic expression (peripheral integral) of GFAP, a glial marker, or high nuclear expression of NeuN, a marker of mature neurons, respectively. High expression was determined by gating on the right half max, a vertical line tangent to the right side of the fluorescence histogram. As the primary antibodies for GFAP and NeuN were raised in the same species, a separate set of slides was used for each marker. Once a population of glia or neurons was identified and gated, Mecp2 fluorescence in the same cell population was measured as the integrated total of all pixel values (integral)/cell area. Mecp2 fluorescence values for the Mecp2 −/+, Mecp2 +/+, and Mecp2 +/y mouse cores were normalized by subtracting fluorescence values for the Mecp2 −/y male. The mean and SEM for Mecp2 fluorescence (in astrocytes and neurons) within each relevant genotype was calculated from the normalized fluorescence values for individual cortical cores on 2–3 replicate slides.
Determination of gap junction coupling.
Gap junction activity was analyzed by the scrape-loading-dye transfer method (el-Fouly et al., 1987). Astrocytes were plated onto cover glass and cultured in DMEM10 till confluence. Cells were washed with serum-free Opti-MEM and incubated with 100 μmol/L CBX, 50 μmol/L GA, or vehicle only in Opti-MEM for 2 h. After cells were washed with PBS, 0.5% Lucifer yellow (Molecular probes) was loaded onto cell surface and the cells were immediately horizontally scraped using a razor blade. After incubation for 2 min in 37°C, cells were washed with PBS and Fixed with 4% paraformaldehyde. Cells in the immediate vicinity of the scrape were observed under a Nikon Eclipse E600 microscope and photographed by a digital camera (SPOT RTke, SPOT Diagnostics). Junctional permeability was evaluated by the spread of Lucifer yellow from the scrape to the adjacent coupled cells.
Statistical analysis.
ANOVA or repeated-measures ANOVA when applicable was used to compare quantitative values from cultures across groups. Tukey's studentized range test was used to adjust for multiple comparisons in post hoc pairwise tests.
Results
Astrocytes express MeCP2
To determine whether astrocytes express MeCP2, we first examined the cerebral sections of 1-month-old littermates of an established RTT model with the Cre-mediated deletion of Mecp2 exons 3 and 4 (Mecp2 tm1.1Bird/+ mice) (Guy et al., 2001). This Mecp2-deletion model shows RTT-like neuropathology and an earlier onset of neurological symptoms in males (Mecp2 −/y, null) than females (Mecp2 −/+, mosaic heterozygous). As expected, immunofluorescence demonstrated that virtually all neurons in the wild-type (Wt; Mecp2 +/+ and Mecp2 +/y) mice but none in the male null (Mecp2 −/y) mice were MeCP2 immunoreactive (Fig. 1 A). A large proportion of neurons in the female heterozygous (Mecp2 −/+) mice expressed MeCP2, consistent with the 60–80% level predicted from previous analyses of XCI in mouse RTT models (Braunschweig et al., 2004; Young and Zoghbi, 2004). Notably, GFAP-immunoreactive astrocytes also showed low levels of MeCP2 immunoreactivity in Wt mice but not in Mecp2 −/y mice (Fig. 1 A), indicating astrocytic expression of MeCP2. Both MeCP2-negative and MeCP2-positive immunofluorescent patterns were observed in GFAP-immunoreactive cells from Mecp2 −/+ mice (Fig. 1 A). The same result was generated by using a second independent MeCP2 antibody (Yasui et al., 2007, data not shown).
Figure 1.

Astrocytes express MeCP2. A, Hippocampal sections from the indicated mice were coimmunostained for MeCP2 (red nuclear stain) and the astrocytic marker GFAP (green cytoplasmic stain). Photomicrographs of corresponding CA1 region are shown in the upper row, and magnified fields from the stratum radiatum are shown in the lower row. Arrows point to examples of MeCP2+ astrocytes and dash-lined squares enclose examples of apparently MeCP2− astrocytes. B, The levels of Wt and mutant Mecp2 transcripts expressed by astrocytes derived from mice of indicated Mecp2 genotype were measured by quantitative RT-PCR. Shown are PCR products in gel. The PCR products were not amplified from genomic DNA because PCRs without reverse transcriptase (−RT control) yielded minimal products. C, Western blot analysis of cell lysates from astrocytes derived from mice of indicated Mecp2 genotype, analyzed by two distinct antibodies. D, Western blot analysis of lysates of primary neurons and primary astrocytes, to compare their levels of MeCP2 protein. The MeCP2 band in the Mecp2 −/+ astrocyte sample was barely detectable due to the limited ECL development to adequately compare between Wt neurons and Wt astrocytes. E, Astrocytes derived from mice of indicated Mecp2 genotypes were cultured and immunostained as in A and counterstained with DAPI (blue). Arrows point to examples of MeCP2+ astrocytes and arrowheads point to examples of MeCP2− astrocytes.
We next investigated cultured primary astrocytes derived from newborn mice. To minimize the presence of nonastrocytic cells, especially neurons in culture, we typically examined astrocytes 2–4 weeks in vitro; these cultures contained minimal neurons and microglia (Maezawa et al., 2006) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Quantitative RT-PCR using primer pairs that specifically amplify Wt and mutant Mecp2 transcripts, respectively (Miralvès et al., 2007), showed that both Mecp2 +/y and Mecp2 +/+ astrocytes (Wt-As) expressed Wt Mecp2, while Mecp2 −/y astrocytes expressed only mutant Mecp2 transcript, as expected (Fig. 1 B). The mosaic Mecp2 −/+ astrocytes expressed equal amount of Wt and mutant Mecp2 transcripts (mean ± SE: 51.2 ± 0.3% for Wt and 48.7 ± 0.3% for mutant, n = 3), consistent with random XCI (Fig. 1 B). Western blot analysis using two independent MeCP2-specific antibodies detected the same 75 kDa MeCP2 protein band in Wt-As, but not in Mecp2 −/y astrocytes (Fig. 1 C). The MeCP2 protein level in Wt-As was ∼25% of that of Wt-neurons (Fig. 1 D). Surprisingly, the mosaic Mecp2 −/+ astrocyte cultures at 2–4 weeks in vitro generally showed an overall MeCP2 protein level 10–30% of the Wt-As (Fig. 1 C), despite the ∼50% transcript level (Fig. 1 B), not compatible to a theoretically predicted 50% reduction for balanced XCI. In contrast, cultured Mecp2 −/+ neurons generally showed a MeCP2 expression level at 70–80% of Wt neurons (Fig. 1 D) (Braunschweig et al., 2004; Young and Zoghbi, 2004), suggesting a unique astrocytic mechanism of reduced MeCP2 expression in mosaic cultures from Mecp2 −/+ mice.
To confirm that the MeCP2 detected in cultures was indeed from astrocytes, we performed immunocytochemical stains. Numerous GFAP-positive cells showed clear nuclear MeCP2 immunoreactivity in Wt cultures, but not in Mecp2 −/y cultures (Fig. 1 E). The Mecp2 −/+ cultures showed a mosaic of MeCP2+ and MeCP2− cells. However, the MeCP2+ cells in the Mecp2 −/+ cultures showed much weaker immunoreactivities compared with those in Wt (Mecp2 +/+ and Mecp2 +/y) cultures (Fig. 1 E). Quantification of four independent sets of astrocyte cultures showed that the average ratios of MeCP2 immunoreactivities of Mecp2 −/+ cultures over Mecp2 +/+ cultures were 25.7 ± 2.5% by immunocytochemistry and 18.4 ± 5.3 by Western blotting (Fig. 1 B), lower than the 50% value predicted by a balanced XCI. We attributed this observation to a non-cell-autonomous effect between MeCP2− and MeCP2+ astrocytes, the evidence for which will be discussed in the later sections.
MeCP2-deficient astrocytes are abnormal in growth and regulation of brain-derived neurotrophic factor, proinflammatory cytokines, and p38MAPK activity
To determine whether the absence of MeCP2 in Mecp2 −/y astrocytes and the reduction of MeCP2 in Mecp2 −/+ astrocytes have functional consequences, we examined a few key astrocytic functions. We first noticed that MeCP2-deficient astrocytes (MD-As) (referring to both Mecp2 −/y and Mecp2 −/+ astrocytes) grew substantially slower than Wt-As. The BrdU assay demonstrated that Mecp2 −/y astrocytes incorporated 41.0 ± 3.0% of the level of BrdU incorporated by the Wt-As in 2 h (n = 5, p < 0.001), consistent with our previous observation of MeCP2-deficient lymphocytes (Balmer et al., 2002). Interestingly, Mecp2 −/+ astrocytes showed a similar degree of growth retardation (39.8 ± 3.8% of Wt, n = 5, p < 0.001), consistent with their low MeCP2 expression.
Due to the growth disadvantage of MD-As, in the following experiments we used high density plating and compared between confluent cultures, which showed no differences in cell numbers, nor total RNA and protein quantities between different genotypes. To determine whether MeCP2 is a functional transcriptional regulator in astrocytes, we examined the expression of Bdnf (encoding brain-derived neurotrophic factor or BDNF), an established target of MeCP2 binding and transcriptional regulation (Chen et al., 2003; Martinowich et al., 2003; Chang et al., 2006). Mecp2 −/y and Mecp2 −/+ astrocytes expressed Bdnf at 216 ± 35% (n = 4, p < 0.05) and 165 ± 57% (n = 3, p = 0.06) (quantitative RT-PCR result), and released BDNF at 245 ± 14% and 223 ± 32% of Wt levels (n = 3, p < 0.05), respectively (Fig. 2 A,B). This regulation appeared specific, because the release of nerve growth factor and apolipoprotein E by MD-As was not altered (Fig. 2 B), suggesting that astrocytic MeCP2 deficiency does not result in a general defect in protein synthesis or secretion.
Figure 2.

MeCP2-deficient astrocytes are abnormal in regulation of Bdnf, cytokines, and p38MAPK. A, Bdnf mRNA levels were measured by quantitative RT-PCR and normalized to the expression of β-actin. Shown are PCR products in gel. B, Astrocyte conditioned media (ACM) were analyzed by Western blots for released BDNF, nerve growth factor (NGF), and Apolipoprotein E (ApoE). C, The indicated cytokines in ACM from cultures treated with LPS were measured by ELISA. n = 3, *p < 0.05 and **p < 0.001 compared with Wt. Without LPS stimulation, the basal levels of all three cytokines were minimal, and there were no significant differences between groups. D, The activation states of p38MAPK, ERK, and JNK in unstimulated astrocytes were evaluated by Western blot using antibodies for their respective phosphorylated epitopes. An antibody for p38MAPK was used to quantify the total p38MAPK level. The activation of p38MAPK is represented by the band intensity of phospho-p38MAPK normalized to that of total p38MAPK. The bar graph shows the fold increase of p38MAPK activation in Mecp2 −/+ and Mecp2 −/y astrocytes compared with Wt-As. n = 3, *p < 0.05 compared with Wt. E, The activation of p38MAPK in astrocytes following 24 h LPS stimulation. As in D, without stimulation, both Mecp2 −/y and Mecp2 −/+ astrocytes showed ∼1.5-fold increase in p38MAPK phosphorylation. After LPS stimulation, there was no difference of phosphorylation level between groups.
An important astrocytic function is to initiate and regulate immune responses through the release of proinflammatory cytokines (Farina et al., 2007). We determined whether the cytokine response after activation by LPS is altered by MeCP2 deficiency. We used confluent cultures in which LPS induced negligible cell proliferation. The activation was evaluated by measuring the levels of proinflammatory cytokines tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6 in astrocyte conditioned media (ACM) using ELISA. When the cultures were stimulated by LPS, the levels of TNF-α, IL-1β, and IL-6 were dramatically increased above baseline from all animals, but MD-As responded to LPS with significantly less IL-1β and IL-6 than Wt-As did (Fig. 2 C). MD-As also showed a trend for reduced TNF-α response, but this did not reach statistical significance.
We further studied the activation of mitogen activated protein kinases (MAPK) in unstimulated astrocytes because these kinases are implicated in the modulation of cytokine production as well as in glial responses to cytokines. The activation state of each kinase was evaluated by the level of protein with activating phosphorylation. In unstimulated MD-As, neither c-Jun N-terminal kinase (JNK) nor p42/p44 MAPK (ERK) was abnormally phosphorylated. In contrast, p38 MAPK was constitutively hyperphosphorylated (Fig. 2 D), indicating that this signaling pathway is partially activated in unstimulated MD-As. After LPS treatment, astrocytes of all genotypes had the same degree of phosphorylation of p38MAPK (Fig. 2 E). This result suggests that in MD-As, the p38MAPK pathways are already hyper-activated, and may make MD-As hyporesponsive to further stimulation with LPS.
To exclude the possibility that the above abnormalities of MD-As were secondary to neuronal influences in vivo before we established the culture, we used two Mecp2-specific small interference RNAs (siRNAs) to knockdown the expression of Mecp2 in Wt-As to ∼20% of control levels. Mecp2 knockdown resulted in the same abnormalities (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), indicating that MD-A defects are due to cell-autonomous effects of astrocytic MeCP2 deficiency.
MD-As induce less dendritic outgrowth from hippocampal neurons
A prominent neuropathological feature of RTT brain is reduced dendritic arborization (Zoghbi, 2003, Armstrong, 2005). It has not been established whether cell-autonomous effects of neuronal MeCP2 deficiency affect dendritic arborization; at least a previous study did not reveal differences in dendritic arborization between Wt and MeCP2-deficient neurons when they were cultured alone (Chao et al., 2007). Because contact with astrocytes promotes neuronal dendritic growth (van den Pol and Spencer, 2000), we tested whether dendritic arborization of hippocampal neurons is affected by MeCP2 deficiency in cocultured astrocytes. We plated Wt hippocampal neurons on confluent astrocytes of different genotypes and found that although astrocytes of all genotypes promoted dendritic growth, MD-As promoted significantly smaller total dendritic length in cocultured neurons, measured at both 24 and 72 h after neuronal plating (Fig. 3 A–C). The difference in dendritic length was due to a combination of fewer branch points and shorter dendrites in each branch order (Fig. 3 D,E). No differences in the numbers of neurons were observed between cultures, eliminating a possible explanation of differences in plating efficiency or neuron survival (Fig. 3 F). siRNA-mediated knockdown of MeCP2 in Wt-As also resulted in reduced dendritic outgrowth from cocultured hippocampal neurons (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), indicating that the reduced dendritic maturation in neurons is a direct consequence of astrocytic MeCP2 deficiency.
Figure 3.

MeCP2-deficient astrocytes support less dendritic growth from Wt neurons. E18 Wt hippocampal neurons were plated onto culture dishes without astrocytes or onto confluent monolayers of astrocytes of indicated genotype. The dendrites were allowed to grow for 24 and 72 h. A, Representative phase-contrast images of 72 h cocultures. B, Dendrites were demonstrated by immunostaining for dendritic marker MAP2 (red). Shown are representative photomicrographs of 72 h cocultures. C, Total dendritic length per neuron in each culture condition was quantified by Neurolucida and Neuroexplorer software. Dendrites from the numbers of individual neurons (indicated in the bars) over four independent experiments were imaged and analyzed. Average dendritic length was significantly different between the four groups (p < 0.001). In post hoc pairwise comparisons adjusted for multiple comparisons, the mean dendritic length induced by Wt-As was significantly higher than each of the other groups (*), but there was no significant difference between groups with Mecp2 −/+ and Mecp2 −/y. D, The 24 h dendritic length data were further stratified into average length in each branch order. E, Branch point complexity was evaluated by counting the number of branch points per order. Mecp2 −/+ and Mecp2 −/y astrocytes induced smaller dendrite length and branch point number than Wt-As in branch order 1–4 (p < 0.02), but there was no significant difference between Mecp2 −/+ and Mecp2 −/y (p = 0.1 for dendritic length and p = 0.86 for branch point number). F, The number of neurons was obtained by counting NeuN-immunoreactive nuclei and normalized by the neuron number in the “neuron-only” group in each experiment.
The MeCP2 deficiency state spreads between astrocytes via a non-cell-autonomous mechanism
The above characterization of MD-As reveals abnormalities that may lead to impaired astrocytic growth, neurotrophic regulation, immune modulation, and dendritic arborization, supporting a detrimental non-cell-autonomous influence of MD-As on neurons. However, an intriguing finding was that cultured astrocytes of Mecp2 −/+ mosaic females expressed very low levels of MeCP2 protein and showed astrocytic abnormalities comparable to those of Mecp2-null-derived astrocytes (Figs. 1 –3). This was not due to preferential growth of MeCP2-deficient astrocytes, which grew slowly as shown above. Notably, this unexpectedly low MeCP2 expression was not present in newly established Mecp2 −/+ astrocyte cultures, but the MeCP2 protein levels became significantly lower than 50% of Wt levels upon prolonged culture (>2 weeks) (Fig. 4 A). This decrease was not a result of altered transcriptional regulation because quantitative RT-PCR revealed the same total transcript levels and balanced XCI pattern throughout the culture period up to 8 weeks (Fig. 1 B and data not shown). These results suggest a spread of the MeCP2 deficiency state in Mecp2 −/+ astrocyte cultures by a posttranscriptional mechanism.
Figure 4.

A non-cell-autonomous effect reduces the MeCP2 expression of Wt-As in Mecp2 −/+ cultures. A, Western blot analysis of MeCP2 levels in cultured astrocytes with indicated time in vitro. B, GFP-labeled Wt-As (green) were cocultured with unlabeled Wt-As or Mecp2 −/+ astrocytes and subsequently stained with anti-MeCP2 (red) and DAPI (blue). The arrowhead in the left panel points to a GFP-labeled Wt-As that retained a high level of MeCP2 expression in the vicinity of MeCP2+ astrocytes. The arrow in the right panel points to a GFP-labeled Wt-As that lost its MeCP2 expression in the vicinity of MeCP2− astrocytes. C, MeCP2 immunoreactivities in GFP-labeled Wt-As were classified into three tiers: high, low, and none. Shown are average percentages of cells in these three tiers of MeCP2 expression. Totally ∼350 GFP-labeled cells per group in four independent experiments were analyzed. A shift in the distribution of high versus low expression (see Material and Methods) was observed between the groups (p < 0.01). D, The time-dependent changes in the MeCP2 expression of GFP-labeled Wt-As cocultured with Mecp2 −/+ astrocytes. n = 3.
We have previously hypothesized a non-cell-autonomous mechanism via which the MeCP2-nonexpressing (MeCP2−) cells may negatively influence the MeCP2 expression of MeCP2 expressing (MeCP2+) cells (Braunschweig et al., 2004). To examine this possibility in Mecp2 −/+ astrocyte cultures, we directly tested whether the existence of MeCP2− astrocytes in Mecp2 −/+ cultures would affect the MeCP2 level of cocultured Wt astrocytes identified by the green fluorescent protein (GFP) label. To evaluate MeCP2 expression in individual GFP-labeled Wt-As, we semiquantitatively classified them into high, low, and none MeCP2 expressers according to preset criteria (see Material and Methods) and counted them in each culture. GFP-labeled Wt-As, when cultured alone or seeded onto cultures of unlabeled Wt-As, stably showed ∼80% high expressers, 20% low expressers, and 0% nonexpressers. However, we found that the MeCP2 expression in GFP-labeled WT-A was reduced when cocultured with Mecp2 −/+ astrocytes (Fig. 4 B,C) in a time-dependent manner (Fig. 4 D), indicating a non-cell-autonomous influence from the MeCP2− astrocytes that appears to progressively spread throughout the culture.
Our previous assessment of the MeCP2 expression in mouse and human RTT brains demonstrated that the MeCP2+ cells from the Mecp2 −/+ tissues showed lower MeCP2 expression than the MeCP2+ cells from Mecp2 +/+ tissues (Braunschweig et al., 2004). A part of this reduction in Mecp2 −/+ brain may be explained by the non-cell-autonomous influence from MeCP2− astrocytes on MeCP2+ astrocytes, as suggested by above culture results. We speculate that since our cultures consist of a highly dense population of astrocytes, the non-cell-autonomous effect observed may reflect a process that happens in vivo, but at an accelerated pace. To further examine the hypothesis of a non-cell-autonomous effect on astrocytes in vivo, we examined the levels of MeCP2 expression in neurons and astrocytes in the hippocampus of Mecp2 −/+ female mice at 5–7 months of age, when they were in the immediately presymptomatic or early symptomatic stage (Guy et al., 2001). We noticed that in these mice, the MeCP2 reduction was particularly pronounced in astrocytes compared with neurons. Most astrocytes in 7-month-old Mecp2 −/+ mice contained minimal MeCP2 (Fig. 5 A,B), unlike those in the 1-month-old Mecp2 −/+ mice (Fig. 1 A). Quantification by laser scanning cytometry revealed that the Mecp2 −/+, NeuN-immunoreactive neurons expressed MeCP2 at 56.5 ± 12.9% of age-matched Wt levels. In contrast, the Mecp2 −/+, GFAP-immunoreactive astrocytes expressed MeCP2 at 16.2 ± 10.2% of Wt levels (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), substantially lower than the 50% level for balanced XCI and in accordance with the level of reduction observed in cultured astrocytes. This result is consistent with a pathological role of astrocytic MeCP2 deficiency in RTT and suggests an astrocytic mechanism in reducing the overall level of MeCP2 in the female Mecp2 −/+ mosaic brain.
Figure 5.
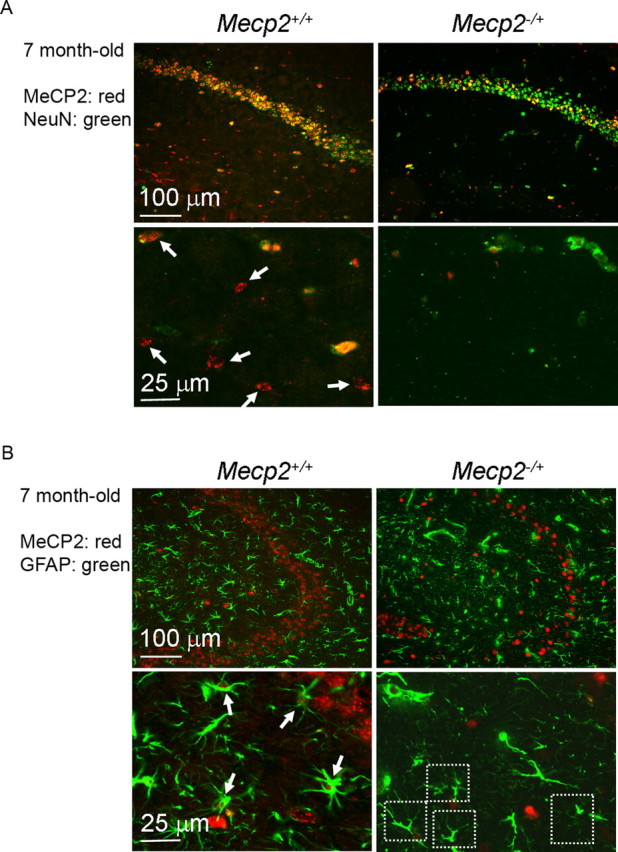
Mecp2 −/+ mice in the immediately presymptomatic or early symptomatic stage show a pronounced reduction of astrocytic MeCP2. Hippocampal sections from indicated mice were coimmunostained for either MeCP2 (red nuclear stain)/NeuN (green nuclear stain) (A) or MeCP2/GFAP (green cytoplasmic stain) (B). In A, arrows point to examples of MeCP2+ non-neuronal cells in the stratum radiatum, which are readily found in 7-month-old Mecp2 +/+ mice (two left panels) but rarely found in 7-month-old Mecp2 −/+ mice (two right panels). In B, arrows point to examples of MeCP2+ astrocytes and dash-lined squares enclose examples of apparently MeCP2− astrocytes. A large majority of astrocytes in 7-month-old Mecp2 −/+ mice appeared to be MeCP2−.
Gap junctions mediate at least in part the non-cell-autonomous effect between astrocytes
We next investigated potential communication mechanisms that could mediate the non-cell-autonomous effect of astrocytic Mecp2 deficiency. A role for soluble factors transferable in the astrocyte conditioned medium was not favored because cell-free medium transferred from the MeCP2− cultures failed to affect the MeCP2 levels of the MeCP2+ cultures (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Next, we examined intercellular gap junctions (GJs), which allow passage of small molecules between cells and play an important role in synchronizing functions of neighboring astrocytes (Dermietzel et al., 1991). We first showed that MD-As express Wt levels of connexin (Cx)-43, the major subunit component of GJs in astrocytes, as well as the minor GJ components Cx-26 (Altevogt and Paul, 2004). MD-As form functional GJ couplings to allow normal intercellular transfer of Lucifer yellow, which could be blocked by two specific GJ inhibitors, carbenoxolone (CBX) and 18α-glycyrrhetinic acid (GA) (Takeuchi et al., 2006) (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Therefore the MeCP2 deficiency appears not to impair GJ communications between astrocytes.
To determine whether GJ communications mediate the non-cell-autonomous effect on MeCP2 expression, we compared the MeCP2 expression patterns of GFP-labeled Wt-As cocultured with Mecp2 −/+ astrocytes in the presence or absence of GJ inhibitors. These GJ inhibitors did not affect the viability or MeCP2 level of Wt astrocytes (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). Inhibition of GJ communication by CBX and GA significantly reduced the spread of MeCP2 deficiency and reversed the MeCP2 expression pattern to that of Wt (Fig. 6). Furthermore, when the inhibition of GJ communication was specifically reduced by tolbutamide (Tabernero et al., 2006), the spread of MeCP2 deficiency was recovered (Fig. 6), confirming the participation of GJs. In addition to pharmacological maneuvers, we also used a Cx-43 siRNA that downregulated the Cx-43 level to ∼30% and attenuated GJ dye coupling (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). Although the transfection procedure by itself appeared to mildly reduce the overall MeCP2 level, the Cx-43 siRNA transfection significantly reduced the spread of MeCP2 deficiency (Fig. 6). Together, results from pharmacological and genetic manipulations suggest that GJs, especially the Cx-43-containing GJs, mediate at least in part the spread of MeCP2 deficiency in Mecp2 −/+ astrocyte cultures. Notably, because the tools used above to block whole GJ channels also act upon unopposed GJ hemichannels, our experiments did not exclude the role of factors released by GJ hemichannels (Takeuchi et al., 2006). However, this possibility is not supported by the above conditioned media transfer experiment (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Experiments using siRNA-mediated knockdown of Mecp2 in astrocytes showed the same results as astrocytes derived from Mecp2 −/+ mice, suggesting that the GJ-mediated spread of the MeCP2 deficiency state in astrocytes is a direct effect of MeCP2 deficiency (supplemental Fig. 9, available at www.jneurosci.org as supplemental material).
Figure 6.

The non-cell-autonomous effect between MeCP2− and MeCP2+ astrocytes is mediated at least partly by GJs. GFP-labeled Wt-As were cocultured with unlabeled Wt-As, Mecp2 −/+ astrocytes (−/+ A), and Mecp2 −/+ astrocytes with GJ inhibitors CBX and GA, respectively, and cultured for 24 h. In additional two groups, the inhibition of GJs by inhibitors was specifically prevented by tolbutamide (TB). In further two groups, the Cx-43-specific siRNA to achieve GJ inhibition and the control siRNA was used, respectively. The MeCP2 immunoreactivities in GFP-labeled Wt-As in each culture were analyzed and presented as described in Figure 4 C. The average percentage of cells with high (and therefore low) MeCP2 expression differed between the groups (p < 0.01). Post hoc pairwise comparisons, adjusted for multiple comparisons indicated that there was a significant shift from high to low expression in the following: (1) from “w/Wt-A” to “w/−/+A” to “w/−/+A+CBX+TB” and to “w/−/+A+GA+TB”; (2) from “w/−/+A+GA” to “w/−/+A” and to “w/−/+ A+GA+TB”; (3) from “w/−/+A+CBX” to “w/−/+A” and to “w/−/+ A+CBX+TB”; (4) from “w/Wt-A” to “w/−/+A+control siRNA”; and (5) from “w/−/+A+Cx43 siRNA” to “w/−/+A+control siRNA.”
Discussion
RTT is often considered to be the result of cell-autonomous neuronal Mecp2 mutations, and astrocytes have been excluded from prior investigations because of reported absence of MeCP2 (Shahbazian et al., 2002). Here we demonstrate that astrocytes express MeCP2, and that MeCP2 deficiency in astrocytes directly causes functional abnormalities, which may adversely affect immune regulation and neuronal dendritic maturation. In addition, we demonstrate the intriguing finding that Mecp2 −/+ astrocytes exhibit a more severe functional deficit than predicted by balanced XCI. Last, we demonstrate a non-cell-autonomous effect exerted by MeCP2− astrocytes that negatively influence MeCP2 levels of MeCP2+ astrocytes and that this effect is at least partly mediated by GJ communication.
Our results showing MeCP2 expression in astrocytes are consistent with a recent in vivo study in which a knock-in expression of MeCP2-enhanced green fluorescent protein fusion protein in mice showed astrocytic expression of MeCP2 (Schmid et al., 2008). The recently published transcriptome database of acutely isolated, highly pure astrocytic populations from both developing and mature mice also confirms the expression of MeCP2 in astrocytes (Cahoy et al., 2008). In contrast, prior conclusions that astrocytes were negative for MeCP2 expression were based on the lack of detectable immunoreactivity in contrast to neighboring neurons (Shahbazian et al., 2002), a result that can be explained by a lack of sensitivity to the relatively lower levels of MeCP2 in astrocytes compared with neurons. Together, ours and others' data clearly demonstrate astrocytic expression of MeCP2. In addition, culturing astrocytes does not artificially enrich the level of MeCP2 expression (Cahoy et al., 2008), therefore can be used to study astrocytic MeCP2 function in vitro.
Using the strategy of reexpression of Wt Mecp2 in Mecp2-null mice by endogenous or non-neuron-specific promoters, it has been demonstrated that the RTT phenotype is reversible in mice (Giacometti et al., 2007; Guy et al., 2007; Jugloff et al., 2008). Perhaps the strongest evidence supporting the “neuron-centered” hypothesis in RTT came from a partial reversal of RTT phenotype by the transgenic reexpression of Wt Mecp2 in postmitotic neurons using the presumed neuron-specific tau promoter (Luikenhuis et al., 2004). However, tau is also expressed in glia and tau accumulation in astrocytes is a prominent feature in a group of neurological diseases called tauopathies (Shin et al., 1991; LoPresti et al., 1995; Togo and Dickson, 2002; Forman et al., 2005). In contrast, reintroduction of Mecp2 in mutant mice by two strictly neuronal promoters failed to show any phenotypic improvement despite widespread neuron-specific expression (Alvarez-Saavedra et al., 2007). These mouse studies, together with our demonstration of abnormalities of MD-As, raise an interesting possibility that neuronal function may recover only if the astrocytic function also recovers. This instructive/supportive role played by astrocytes in modifying neuronal plasticity in postnatal brain has been well demonstrated in several other experimental models (Müller and Best, 1989; Pascual et al., 2005; Bacaj et al., 2008).
Interestingly, an analysis of brain metabolites in Mecp2-null mice by magnetic resonance spectroscopy uncovered a reduction of the astrocyte marker myo-inositol, suggesting astrocytic aberration in vivo (Viola et al., 2007). Our characterization of MD-As revealed abnormalities that may impair neuronal plasticity by failure to support dendritic maturation, and may also impair brain's response to noxious stimuli due to reduced astrocytic growth and cytokine secretion. The mechanisms underlying these observations remain to be determined. Our data strongly suggest a “lost of function” mechanism involving disrupted cellular signaling pathways resulting in slow growth, reduced cytokine responses, and reduced dendritic induction. However, a “gain of function” mechanism via which the MD-As generate toxic soluble factors or surface molecules that retard the dendritic growth cannot be ruled out.
We found approximately twofold increases of both Bdnf transcript and protein levels in MD-As, consistent with what has been observed in cultured primary neurons (Chen et al., 2003). However, the regulation of Bdnf in brain likely involves activation as well as repression (Yasui et al., 2007; Chahrour et al., 2008), because overall BDNF levels are lower in Mecp2 −/y brain compared with controls (Chang et al., 2006). This may be attributed to decreased overall brain activity in Mecp2 −/y mice, and may involve abnormal regulations by microRNA (Klein et al., 2007) and p38MAPKactivity as shown here.
Although female Mecp2 −/+ models would best represent human RTT, which is mainly a disease of girls, most studies have used Mecp2 −/y mice due to their early onset and better replication of some RTT symptoms. However, our previous and current studies on Mecp2 −/+ tissues have revealed non-cell-autonomous effects between MeCP2− and MeCP2+ astrocytes and neurons that cannot be revealed by exclusively studying Mecp2 −/y mice. We found that the non-cell-autonomous effect was more pronounced in astrocytes compared with neurons in brain, and MeCP2 deficiency was perpetuated with increased time of astrocytic culture. By exploring unique intercellular communication mechanisms used by astrocytes, we found a novel role for gap junctions (in particular, those mediated by Cx-43) in transmitting the MeCP2 deficient state between astrocytes. The most likely explanation for our observation is that negative regulators controlling MeCP2 protein levels are transferred from MeCP2− to MeCP2+ astrocytes via GJs over time. The candidate regulators may include Ca2+, inositol trisphosphate, glutamate, or small regulatory miRNAs, such as miR132 that posttranscriptionally downregulates MeCP2 (Klein et al., 2007).
It remains difficult to explain the phenotypic regression in RTT patients and Mecp2 −/+ mice after months of apparently normal development and maturation of the brain. It appears that in MECP2 heterozygotes the MeCP2-related functions are well compensated in early life until disruption by certain accumulative events. Our in vitro and in vivo data may lend support to a mechanism by which astrocytic abnormalities resulting from MeCP2 deficiency can progressively spread through the GJ-connected astrocytic network in brain. We showed that in the relatively pure, highly dense astrocytic cultures, the spread of MeCP2 deficiency occurs within days to weeks. In in vivo conditions, however, this spread may occur at a slower pace, taking months. This is conceivable because the relatively lower astrocytic density in brain compared with culture and that GJ communication mediated by connexins is tightly regulated by neuronal activity in vivo and is focally restricted by the regional topographical neuron-glia organization (Houades et al., 2008). This progressive loss of MeCP2 in astrocytes might eventually generate a decompensation state in which astrocytes can no longer adequately support brain function. Indeed, using the objective, highly sensitive LSC method to quantify neuronal and astrocytic MeCP2 respectively, we detected around the time of phenotypic regression a pronounced, astrocyte-selective decline of MeCP2, which was much larger than expected from random XCI but in accordance with our astrocytic culture results. In contrast, neuronal MeCP2 levels in Mecp2 −/+ mice remained at ∼50% of Wt. We hypothesize that in Mecp2 −/+ brain, the progressive loss of MeCP2 selectively in astrocytes might induce in a non-cell-autonomous manner the functional decompensation of neurons which triggers phenotypic regression, despite the fact that neurons may continue to maintain well compensated cell-autonomous function of neuronal MeCP2.
Our results, therefore, suggest a modification of the current “neuron-centered” hypothesis regarding RTT pathogenesis to include astrocytes as an integral component of the neuron-glia functional units that are impaired due to MeCP2 deficiency. Therapeutic approaches that recover Wt MeCP2 expression in astrocytes and decrease the astrocytic spread of MeCP2 deficiency should be considered for RTT and perhaps a subset of autism with MeCP2 deficiency.
Footnotes
This work was funded by the University of California Davis M.I.N.D. (Medical Investigation of Neurodevelopmental Disorders) Institute and by National Institutes of Health Grants HD048799, HD041462, and HD055143.
References
- Altevogt BM, Paul DL. Four classes of intercellular channels between glial cells in the CNS. J Neurosci. 2004;24:4313–4323. doi: 10.1523/JNEUROSCI.3303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Saavedra M, Sáez MA, Kang D, Zoghbi HY, Young JI. Cell-specific expression of wild-type MeCP2 in mouse models of Rett syndrome yields insight about pathogenesis. Hum Mol Genet. 2007;16:2315–2325. doi: 10.1093/hmg/ddm185. [DOI] [PubMed] [Google Scholar]
- Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- Bacaj T, Tevlin M, Lu Y, Shaham S. Glia are essential for sensory organ function in C. elegans . Science. 2008;322:744–747. doi: 10.1126/science.1163074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer D, Arredondo J, Samaco RC, LaSalle JM. MECP2 mutations in Rett syndrome adversely affect lymphocyte growth, but do not affect imprinted gene expression in blood or brain. Hum Genet. 2002;110:545–552. doi: 10.1007/s00439-002-0724-4. [DOI] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X-Chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet. 2004;13:1275–1286. doi: 10.1093/hmg/ddh142. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Hertberg EL, Kessler JA, Spray DC. Gap junctions between cultured astrocytes: immunocytochemical, molecular, and electrophysiological analysis. J Neurosci. 1991;11:1421–1432. doi: 10.1523/JNEUROSCI.11-05-01421.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Fouly MH, Trosko JE, Chang CC. Scrape-loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Exp Cell Res. 1987;168:422–430. doi: 10.1016/0014-4827(87)90014-0. [DOI] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM, Trojanowski JQ. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci. 2005;25:3539–3550. doi: 10.1523/JNEUROSCI.0081-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A. 2007;104:1931–1936. doi: 10.1073/pnas.0610593104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houades V, Koulakoff A, Ezan P, Seif I, Giaume C. Gap junction-mediated astrocytic networks in the mouse barrel cortex. J Neurosci. 2008;28:5207–5217. doi: 10.1523/JNEUROSCI.5100-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugloff DG, Vandamme K, Logan R, Visanji NP, Brotchie JM, Eubanks JH. Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Hum Mol Genet. 2008;17:1386–1396. doi: 10.1093/hmg/ddn026. [DOI] [PubMed] [Google Scholar]
- Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci. 2007;10:1513–1514. doi: 10.1038/nn2010. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Baba A, Matsuda T. Endothelins stimulate the expression of neurotrophin-3 in rat brain and rat cultured astrocytes. Neuroscience. 2005;136:425–433. doi: 10.1016/j.neuroscience.2005.08.004. [DOI] [PubMed] [Google Scholar]
- LaSalle JM, Goldstine J, Balmer D, Greco CM. Quantitative localization of heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum Mol Genet. 2001;10:1729–1740. doi: 10.1093/hmg/10.17.1729. [DOI] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci U S A. 1995;92:10369–10373. doi: 10.1073/pnas.92.22.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Maeda N, Montine TJ, Montine KS. Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice. J Neuroinflammation. 2006;3:10. doi: 10.1186/1742-2094-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Miralvès J, Magdeleine E, Joly E. Design of an improved set of oligonucleotide primers for genotyping MeCP2tm1.1Bird KO mice by PCR. Mol Neurodegener. 2007;2:16. doi: 10.1186/1750-1326-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Müller CM, Best J. Ocular dominance plasticity in adult cat visual cortex after transplantation of cultured astrocytes. Nature. 1989;342:427–430. doi: 10.1038/342427a0. [DOI] [PubMed] [Google Scholar]
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:e1–e11. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, Tsujimoto N, Qu Q, Lei H, Li E, Chen T, Blaustein CS. A methyl-CpG-binding protein 2-enhanced green fluorescent protein reporter mouse model provides a new tool for studying the neuronal basis of Rett syndrome. Neuroreport. 2008;19:393–398. doi: 10.1097/WNR.0b013e3282f5661c. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Shin RW, Iwaki T, Kitamoto T, Tateishi J. Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer's disease brain tissues. Lab Invest. 1991;64:693–702. [PubMed] [Google Scholar]
- Tabernero A, Sánchez-Alvarez R, Medina JM. Increased levels of cyclins D1 and D3 after inhibition of gap junctional communication in astrocytes. J Neurochem. 2006;96:973–982. doi: 10.1111/j.1471-4159.2005.03623.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- Togo T, Dickson DW. Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol (Berl) 2002;104:398–402. doi: 10.1007/s00401-002-0569-x. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Spencer DD. Differential neurite growth on astrocyte substrates: interspecies facilitation in green fluorescent protein-transfected rat and human neurons. Neuroscience. 2000;95:603–616. doi: 10.1016/s0306-4522(99)00430-3. [DOI] [PubMed] [Google Scholar]
- Viola A, Saywell V, Villard L, Cozzone PJ, Lutz NW. Metabolic fingerprints of altered brain growth, osmoregulation and neurotransmission in a Rett syndrome model. PLoS ONE. 2007;2:e157. doi: 10.1371/journal.pone.0000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JI, Zoghbi HY. X-chromosome inactivation patterns are unbalanced and affect the phenotypic outcome in a mouse model of Rett syndrome. Am J Hum Genet. 2004;74:511–520. doi: 10.1086/382228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]