

Abstract
The Bcl-2 family is responsible for regulating cell death pathways in neurons during development, after injury and in disease. The activation of the pro-death family member BAX is often the final step before cell death in neurons. Pro-survival family members such as BCL-X (BCL2L1) act to inhibit BAX activation. Overexpression studies have suggested that BCL-X could play an important physiological role in mediating neuronal viability. Loss-of-function studies performed in vivo have implicated BCL-X as a mediator of neuronal survival during the early stages of neurodevelopment. To assess whether BCL-X is needed to promote the survival of neurons in the central nervous system throughout life, Bcl-x was conditionally removed from the optic cup or throughout the adult mouse. During development BCL-X was required for the survival of differentiating retinal ganglion cells (RGCs) leading up to their normal window of developmental death. Despite its expression in adult RGCs, BCL-X was not required for maintaining RGC viability in adult retinas. However, the loss of BCL-X in adult RGCs did significantly increase the rate of death of RGCs after axonal injury. Thus, in developing and injured RGCs there appears to be an active cell survival program preventing neuronal death.
Introduction
The Bcl-2 family of genes mediates the intrinsic pathway of apoptosis, which significantly contributes to neuronal death during development, after injury, and in disease. For instance, the pro-death Bcl-2 family member BAX is required for retinal ganglion cell (RGC) death during development, after acute axonal injury, and in ocular hypertensive glaucoma (Li et al., 2000; Libby et al., 2005; Mosinger Ogilvie et al., 1998; Qin et al., 2004; White et al., 1998). BAX activation is controlled by the opposing actions of pro-death and pro-survival members of the Bcl-2 family. During development and after injury RGC apoptosis requires upstream pro-death Bcl-2 family members (Harder and Libby, 2011; McKernan and Cotter, 2007). The physiological role of the pro-survival Bcl-2 family members is less well understood than their pro-death counterparts. Importantly, while Bcl2 was shown to not have a role in maintaining RGC survival after axonal injury (Dietz et al., 2001), it does help maintain RGC viability in maturing RGCs (Cellerino et al., 1999). Thus, pro-survival Bcl-2 family members can play critical roles in maintaining RGC viability.
There are five pro-survival members of the Bcl-2 family (Bcl2, Bcl-x, Bcl-w, Bcl2a1a, and Mcl1). Their main function is to prevent BAX (or BAK1) activation and, therefore, cell death (Puthalakath and Strasser, 2002; Strasser, 2005; Willis et al., 2005; Willis et al., 2007). Most of the pro-survival Bcl-2 family members have important roles in neuronal development (Arbour et al., 2008; Crosio et al., 2006; Lukiw et al., 2005; Middleton et al., 2001; Mori et al., 2004; Motoyama et al., 1995; Shacka and Roth, 2006). In fact, antagonization of pro-survival Bcl-2 family members with small molecule inhibitors is enough to trigger neuronal death in vitro (Young et al., 2010). BCL-X has been specifically implicated as an important pro-survival factor in neuronal development and disease. Germline deletion of Bcl-x leads to death of neurons in the developing central nervous system and embryonic lethality (Motoyama et al., 1995). Conditional deletion of Bcl-x in dopaminergic neurons showed that Bcl-x is required for the survival of all but a few catecholaminergic cells in the developing substantia nigra (Savitt et al., 2005). Numerous neuroprotective treatments are reported to increase the intracellular ratio of BCL-X to pro-apoptotic members (Kilic et al., 2005; Koh, 2009; Ma et al., 2005; Pike, 1999; Wang et al., 2000) and in injured neurons overexpressing BCL-X can increase survival and sustain neuronal function (Garrity-Moses et al., 2005; Parsadanian et al., 1998; Wiessner et al., 1999). In RGCs Bcl-x transcript and protein expression is regulated after injury (Isenmann et al., 1997; Levin et al., 1997; McKernan and Cotter, 2007; Pelzel et al., 2010) and overexpression of BCL-X or BCL2 protects RGCs after axonal injury (Bonfanti et al., 1996; Cenni et al., 1996; Chierzi et al., 1999; Malik et al., 2005). Together these studies suggest that BCL-X may play a necessary physiological role in maintaining survival of adult and developing neurons. However, despite the importance of apoptotic cell death during development and in disease, to date there is limited knowledge of how critical physiological levels of pro-survival Bcl-2 family members are in maintaining neuronal survival throughout life (Isenmann et al., 2003). To test the function of an endogenous pro-survival Bcl-2 family member in the central nervous system, the role of Bcl-x (Bcl2l1) was assessed in developing and adult RGCs in vivo.
Methods
Animals
A floxed allele of Bcl2l1tm1.1Mam (Bcl-xfl ; Rucker et al., 2000) was removed from the developing retina using the Six3-cre allele (Furuta et al., 2000) and from the adult mouse using a ubiquitously expressed, tamoxifen inducible cre (Cre-ERTM; Hayashi and McMahon, 2002). A tamoxifen dose equivalent to 5mg/40g mouse was administered by intraperitoneal injection to 45-75 day old mice for 5 consecutive days. Experiments were performed either 15 days (controlled optic nerve crush) or 60 days (assessing long term survival in the absence of Bcl-x) following the first tamoxifen injection. Control mice were either heterozygous or wild type for the floxed allele. In addition, control mice containing cre were compared to control mice without cre to rule out effects of cre toxicity or Bcl-x heterozygosity in the retina using at least 3 mice with and without cre for comparison. No differences were noted between any control genotype (Bcl-x+/? Six3-cre? or Bcl-x+/? Cre-ER™?) and control genotypes were combined and are referred to throughout the manuscript as control. Note that the ? in the control genotypes is to denote that two types of genotypes are combined to make the control group: for the Bcl-x gene the ? reflects the fact that that allele could be wildtype or floxed; for Cre locus the ? reflects the fact that the allele could be present or absent. Morning vaginal plug checks were used to establish age E0.5 for embryonic stages. All experiments were conducted in accordance with the Association for Research in Vision and Ophthalmology’s statement on the use of animals in ophthalmic research and were approved by the University of Rochester’s University Committee on Animal Resources. Significant differences were determined by comparing control and Bcl-x knockout groups with Student t test at each time point during development and after CONC. At least 4 animals are used for each group in each comparison. Specific animal numbers for each experiment are detailed in the relevant figure legend.
Histology and Immunocytochemistry
For retinal whole mounts and sectioning (plastic and frozen), tissue was processed as previously described (Fernandes et al., 2012; Harder and Libby, 2011). Plastic sections were used for retinal cross-section thickness measurements. Four areas were averaged per retina, each within 50 μm of the optic nerve. The measurement was taken from the nerve fiber layer to the tips of the photoreceptor outer segments. For retinal area measurements, whole retina images were reconstructed from 5X images of flat mounted retinas and area measurements were made using Image J. For immunohistochemistry, the following primary antibodies and dilutions in blocking solution were used: rabbit anti-cCASP3 marks cells with activated caspase 3, R&D Systems, 1:1000; goat anti-POU4F2, also known as BRN3B, labels RGCs (Gan et al., 1999), Santa Cruz, 1:300; mouse anti-TUJ1 immunolabels βIII-tubulin which is expressed in RGCs (Cui et al., 2003), Covance, 1:1000; rabbit anti-BCL-X, Cell Signaling, 1:500. Alexafluor-conjugated secondary antibodies (Invitrogen) were used at a dilution of 1:1000. For all cell counts identical retinal areas were assessed. For adult anti-CASP3 and anti-TUJ1 counts flat mounted retinas were counted over 20X fields and 40X fields respectively. 8 peripheral fields, 2 from each quadrant, were counted per eye. For embryonic cell counts four comparable central horizontal sections were counted per eye (cCASP3+ cells were counted along the entire section and POU4F2+ cells were counted per section at E12.5 and over 250 μm at E18.5).
Controlled optic nerve crush (CONC)
CONC was performed 15 days after the first tamoxifen injection, as previously described (Fernandes et al., 2012; Harder and Libby, 2011). In anesthetized mice, the optic nerve was exposed and clamped using self-closing forceps (Roboz RS-5027) for 3-4 seconds just behind the eye. Unmanipulated or sham surgery (all procedures performed except clamping the nerve) eyes were used as controls.
Results
Immature RGCs require BCL-X for survival
BCL-X is expressed throughout the neuroblastic retina (Fig. 1A). At E12.5, RGCs are being born at the ventricular surface (VS) and migrating to the presumptive RGC layer at the inner surface of the retina. BCL-X and the RGC marker βIII-tubulin (TUJ1; Cui et al., 2003) are coexpressed in the GCL at E12.5 and E18.5. The expression pattern of BCL-X indicates it may affect RGC birth and survival. Since germline deletion of Bcl-x results in embryonic lethality (Motoyama et al., 1995; Savitt et al., 2005), Six3-cre was used to delete a floxed allele of Bcl-x (Bcl-xf) from the developing optic cup. RGCs are among the earliest retinal neurons to get specified, represent a large proportion of the first wave of differentiation (Gan et al., 1999), and undergo a significant amount of programmed cell death during development (Mosinger Ogilvie et al., 1998). Based on POU4F2 immunolabeling (an early marker of RGC differentiation; Gan et al., 1999), Bcl-x deletion does not alter RGC generation at E12.5 (Fig. 2A,B). Also, the loss of BCL-X does not significantly increase cell death at E12.5, suggesting that both retinal progenitors and newly born RGCs do not require BCL-X for survival (Fig. 2C). Substantial naturally occurring developmental death of RGCs begins around E18.5 (Pequignot et al., 2003). However, without BCL-X large numbers of RGCs are prematurely lost between E12.5 and E18.5. During this time period, ectopic CASP3 activation and thinning of the retina indicate cell death is coincident with the loss of RGCs (Fig. 2C,D). These data indicate that in differentiating RGCs BCL-X is required to prevent apoptotic cell death.
Figure 1. BCL-X is expressed in differentiating RGCs.
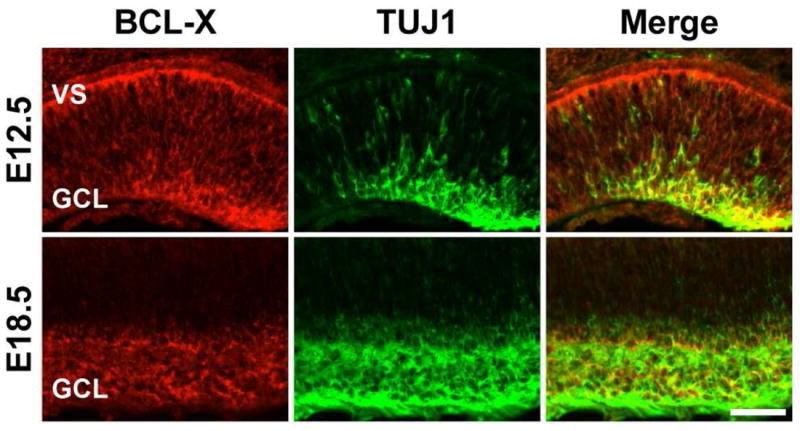
BCL-X (red) is expressed by differentiating RGCs (TUJ1+, green) in the retina. At E12.5, RGCs are being born at the ventricular surface (VS) and migrating to the presumptive RGC layer (GCL). BCL-X and TUJ1 are coexpressed (yellow) in the GCL at both E12.5 and E18.5. 4 retinas were examined for each genotype and time point.
Figure 2. BCL-X is required for the survival of differentiating RGCs.
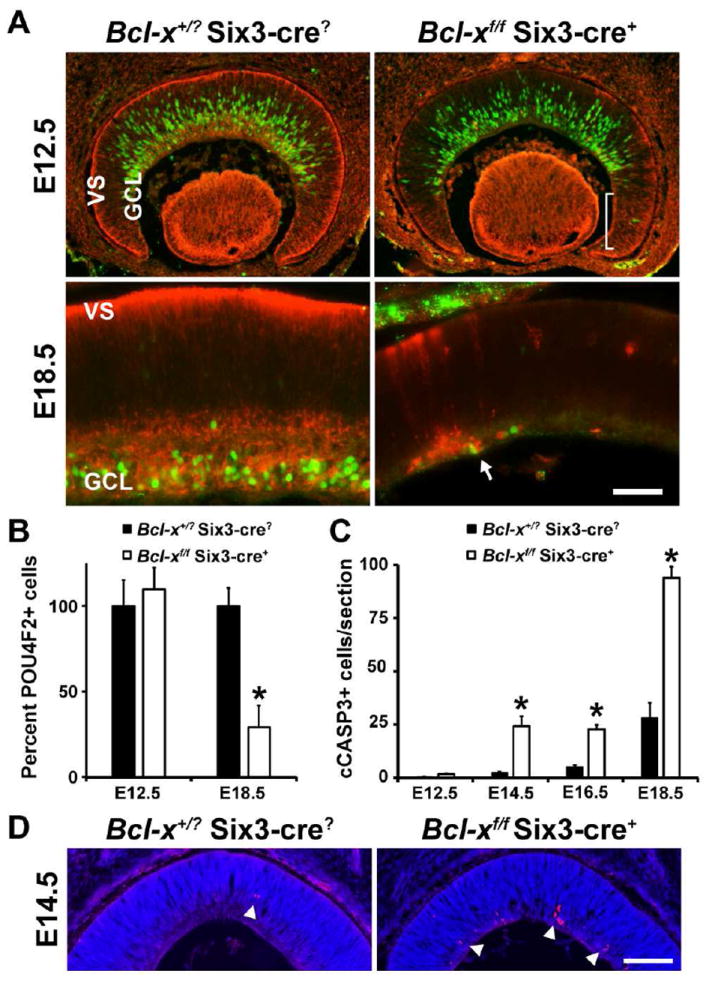
(A, B) Control (Bcl-x+/? Six3-cre?) RGCs express the RGC specific marker POU4F2 (green) soon after undergoing fate determination at the ventricular surface of the retina. Loss of BCL-X (red) in the retina (Bcl-xf/f Six3-cre+) does not alter the production or migration of RGCs as judged by their distribution and number. By E18.5, a time when all RGCs are born and reside in the GCL, there is a significant reduction in the number of RGCs (P<0.001), suggesting RGCs die during the differentiation process. Note, there is an incomplete deletion of Bcl-x by the Six3-cre allele (bracket) and surviving RGCs still express BCL-X (arrow). (C) The loss of RGCs by apoptosis is supported by an increase in the number of cleaved caspase 3 (cCASP3+) cells. This increase was statistically significant at each time point assessed (*, P<0.01) except for E12.5 (P=0.78). (D) Representative images of anti-cCASP3 immunostaining (red) with a nuclear counter stain (DAPI, blue) at E14.5 in the retinas of control and Bcl-xf/f Six3-cre+ mice. At least 4 different animals at each time point and for each genotype were assessed. Scale bar, A, 50μm; D, 100μm.
In addition, other types of retinal neurons appear to be susceptible to apoptotic death in the absence of Bcl-x. At E18.5, the few surviving RGCs are primarily cells in which Bcl-x was not deleted (Fig. 2A arrow), which is consistent with known mosaic expression of Six3-cre, particularly in the retinal margin (Cai et al., 2011; Fuhrmann et al., 2009; Poche et al., 2008). Thus, the large increase in cell death in Bcl-xf/f Six3-cre+ retinas at E18.5 is likely also associated with abnormal cell death of other types of retinal neurons.
The increase in cell death during development produced a smaller adult retina in the Bcl-xf/f Six3-cre+ mice compared to wild type. The Bcl-x knockout retina was reduced in thickness (Fig. 3A; Bcl-x+/? Six3-cre? 180±7 μm, Bcl-xf/f Six3-cre+ 96±7 μm; P<0.001, N=5 for each genotype) and surface area (Fig. 3B; Bcl-x+/? Six3-cre? 18±1 mm2, Bcl-xf/f Six3-cre+ 13±2 mm2; P<0.001, N=5 for each genotype). The Bcl-xf/f Six3-cre+ retina consisted of all major cell types, albeit reduced in number, and retained normal gross morphology (Fig. 3A and data not shown). In wild type adult retinas all RGCs express BCL-X, as determined by colabeling with the RGC marker TUJ1 (Fig. 3C). In 6 out of 6 Bcl-x knockout retinas all surviving RGCs expressed BCL-X (the Six3-cre is not a complete retinal deleter (Cai et al., 2011; Fuhrmann et al., 2009; Poche et al., 2008)). These results indicate that RGCs require BCL-X for survival during development and suggest that the survival of RGCs in the Bcl-xf/f Six3-cre retinas is the result of incomplete deletion of Bcl-x in the developing retina.
Figure 3. BCL-X is required for retinal neuron survival during development.
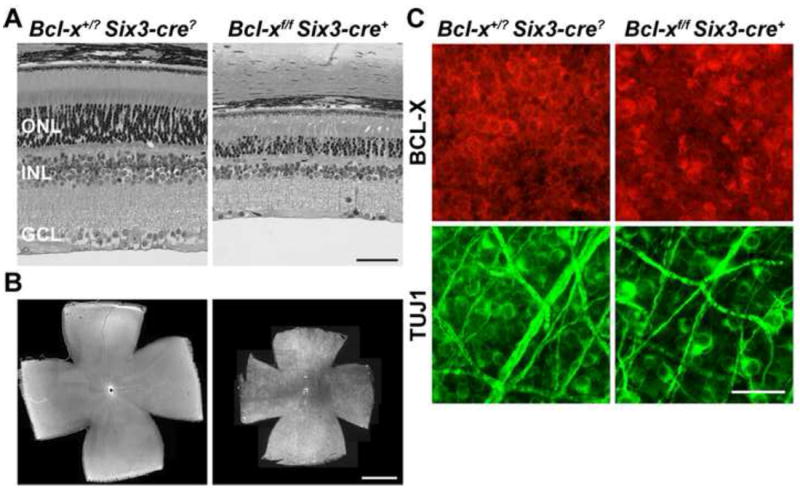
(A) Bcl-xf/f Six3-cre+ retinas at P45 have normal retinal layer organization, but a significant loss in retinal neuron number compared to control retinas (both thickness and surface area were significantly smaller; P<0.001 for each measurement; N=5 for each genotype). (B) Whole mount retinas, ganglion cell layer up, from control animals shows that BCL-X is expressed across the retina. In the smaller Bcl-xf/f Six3-cre retinas BCL-X appears to still be expressed in the ganglion cell layer (N=5 for each genotype). (C) Colabeling of the RGC marker TUJ1 and BCL-X shows that all surviving RGCs contained BCL-X (6 retinas, with at least 8, 40X fields examined per retina). Thus, BCL-X is required for RGC survival during development and the remaining RGCs in Bcl-xf/f Six3-cre+ eyes likely survive because Bcl-x is still present. ONL, Outer Nuclear Layer; INL, Inner Nuclear Layer; GCL, Ganglion Cell Layer; Scale bar: A, 50μm; B, 1mm; C, 25μm.
Bcl-x is not required for survival of adult RGCs
The continued expression of BCL-X in adult RGCs and its role as a required survival factor during development raises the question of whether adult RGC viability is also dependent on BCL-X. To address the importance of BCL-X in adult RGCs, Bcl-xf was removed in adult mice using a ubiquitously expressed, tamoxifen inducible cre-recombinase (Cre-ER™). Tamoxifen-induced recombination was highly effective, reducing the number of BCL-X positive cells in the RGC layer to 2% of control by 15 days following treatment (Fig 4A). Loss of BCL-X produced no noticeable change in retinal architecture and did not induce immediate cell death (Fig 4B, 5A,B) or signs of glial activation (data not shown). In fact, two months after Bcl-x deletion there was still normal retinal architecture (Fig 4B) and normal numbers of RGCs (Fig. 4C).
Figure 4. Adult RGCs do not require BCL-X for survival.

Bcl-xf was deleted in the adult using a ubiquitously expressed, tamoxifen inducible cre (Cre-ER™). (A) Assessment of BCL-X expression in Bcl-xf/f Cre-ER™+ mice with and without tamoxifen administration in retinal flat mounts (ganglion cell layer facing up) showed that Bcl-x was efficiently deleted in the adult using this Cre-ER™ (Top, low power composite image of entire retinas immunolabled for BCL-X; Bottom, high power image showing individual cells). (B) There was no apparent loss of retinal cells up to 60 days after deletion of Bcl-x (4 animals were assessed for each genotype). (C) Quantification of RGCs using the RGC specific marker TUJ1+ (Cui et al., 2003) confirmed that there was no loss of RGCs 60 days after deleting BCL-X (RGCs/mm2 in controls: 2828±203; Bcl-xf/f Cre-ER™+: 2695±221; P=0.67; N=4 for each genotype). ONL, Outer Nuclear Layer; INL, Inner Nuclear Layer; GCL, Ganglion Cell Layer. Scale bar: A top, 1mm, bottom, 25μm; B, 50μm; C, 25μm.
Figure 5. BCL-X is a pro-survival factor in injured RGCs.
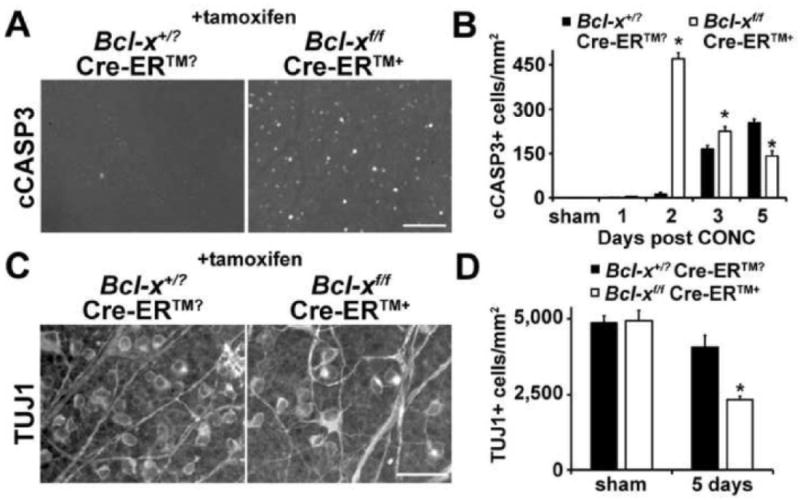
(A,B) After axonal injury (controlled optic nerve crush, CONC), RGC death begins around 2 days after injury, with only occasional cleaved caspase 3+ cells (cCASP3) being detected in wild type animals and the peak of RGC death occurring 5 days after injury (Harder and Libby, 2011). In the absence of Bcl-x (deleted in 45-75 day old mice using a ubiquitously expressed tamoxifen inducible cre, Cre-ER™) RGC cells undergo death at a much faster rate. In fact, RGC death peaks at 2 days after injury. Though by 5 days after injury, wild type mice have more cCASP3+ cells than Bcl-x knockout mice, presumably because there are less RGCs remaining to die in the mutants. (C,D) The increase in RGC death was confirmed by counting RGCs 5 days after injury. Eyes deficient in Bcl-x (Bcl-xf/f Cre-ER™+) had a significantly reduced number of TUJ1+ RGCs (TUJ1+ is an RGC specific marker ; Cui et al., 2003). At least 5 different animals at each time point and for each genotype were assessed. *, P<0.01; Scale bar: A, 50μm; C, 25μm.
BCL-X is a pro-survival factor in injured RGCs
Adult RGCs die by apoptotic pathways regulated by the Bcl-2 family after axonal injury (either mechanical or glaucomatous axonal injury; Bahr, 2000; Isenmann et al., 2003; Li et al., 2000; Libby et al., 2005). This cell death involves activation of BAX by other pro-death Bcl-2 family members. Over-expression of BCL-X can protect against cell death following axonal injury (Malik et al., 2005), but it is unknown whether endogenous BCL-X prevents death in injured adult neurons. After acute axonal injury there is near immediate pro-death injury signaling in RGCs (Agudo et al., 2009; Fernandes et al., 2012; Lukas et al., 2009), but RGC death does not begin until three days after injury and RGC dropout continues over the course of weeks. Bcl-x expression has been reported to transiently increase in retinas after axonal injury by several groups (Isenmann et al., 1997; Levin et al., 1997; Pelzel et al., 2010). Thus there is evidence supporting the possibility of an active pro-survival pathway in adult RGCs.
To test the importance of BCL-X in injured adult neurons, RGC axons were mechanically injured using a controlled optic nerve crush (CONC, a standard technique used to induce axonal injury in RGCs) injury. Similar to wild type, the BCL-X negative retina had no observable cell death at one day following axonal injury. In distinct contrast to the negligible number of apoptotic RGCs at two days in the injured wild type retina, the BCL-X negative retina had a significant increase and an immediate peak of apoptotic death (Fig 5A,B). Cell death did not peak in the wild type retina until at least 5 days (Fig. 5B and Harder and Libby, 2011) and this peak was significantly less than the amount of death observed at 2 days in the BCL-X negative retina. The early loss of RGCs in Bcl-x deficient retinas was confirmed by counting surviving RGCs 5 days after injury (Fig. 5C,D). This striking pattern of early cell death in the mutant indicates that BCL-X acts as a survival factor allowing many RGCs to withstand the initial cell death signaling that occurs following axonal injury.
Discussion
Pro-survival Bcl-2 family members are powerful mediators of cell survival (Adams, 2003; van Delft and Huang, 2006; Willis et al., 2007). In fact, antagonizing pro-survival Bcl-2 family members can initiate the mitochondrial cell death pathway in at least one type of cultured neuron (Young et al., 2010). In addition pro-survival Bcl-2 family members prevent pro-apoptotic BH3-only proteins from activating BAX (or its subfamily members BAK1 and BOK) (Puthalakath and Strasser, 2002; Strasser, 2005; Willis and Adams, 2005). Different cell types, including neurons, appear to vary in their latent potential for BAX activation in the absence of pro-survival Bcl-2 family members (Young et al., 2010). Overexpression of both BCL-X and BCL2 in RGCs has been shown to protect RGCs from death during development and after insult in the adult (Bonfanti et al., 1996; Cenni et al., 1996; Chierzi et al., 1999; Malik et al., 2005). Different forms of stress can also variably alter the expression of pro-survival and pro-apoptotic Bcl-2 family members and induce cell death. Despite a potentially critical role in survival, the physiological function of pro-survival Bcl-2 family members in neurons throughout life is not well understood. Here, loss-of-function studies were used to determine at what stages RGCs require pro-survival signaling of BCL-X for survival.
Developing RGCs require Bcl-x for survival
Proper retinal development requires a significant amount of programmed cell death and this death is dependent on pro-apoptotic Bcl-2 family members (BAX and BBC3; Harder and Libby, 2011; Mosinger Ogilvie et al., 1998; White et al., 1998). The pro-survival Bcl-2 family member BCL-X is expressed in the retina throughout development and is present in RGCs suggesting it may be required to counteract pro-apoptotic signaling in developing RGCs. In the absence of Bcl-x, widespread ectopic cell death in the developing nervous system has been reported (Motoyama et al., 1995), although embryonic lethality at E13.5 precluded assessing the retinal phenotype or the absolute requirement for BCL-X for neuronal survival. Deleting Bcl-x with a retinal specific cre at the beginning of retinal development (Six3-cre; Furuta et al., 2000) led to ectopic death of developing RGCs. In the developing brain, based on cell position in the developing tissue, it has been suggested that the general role of Bcl-x is in maintaining survival of differentiating neurons (Motoyama et al., 1995; Shindler et al., 1997). Using an early, specific marker for RGCs, POU4F2 (Badea et al., 2009; Wang et al., 2002), it is clear that BCL-X is not required for RGC survival in the earliest stages of differentiation. RGCs were initially generated in normal numbers and migrated to/toward their proper position in the retina. Normal developmental death of RGCs occurs between E18.5 and P7 after RGC generation is complete and results in about half of all RGCs dying (Pequignot et al., 2003). Surprisingly, in the Bcl-x deficient retina, RGC death occurred prematurely between E14.5 and E18.5, corresponding with a period when few wild type RGCs die. The data presented here demonstrate that an active pro-survival factor is required for RGCs to survive soon after differentiation begins. This result implies that a pro-survival pathway that includes BCL-X antagonizes an active cell death signal that is normally present during the differentiation process. It will be interesting to determine if the downregulation of pro-survival factors contributes to RGC death during the normal death window because this change appears to be sufficient to induce apoptosis. Defining the extent and regulation of both the pro-survival and pro-death pathways will be needed to fully understand neuronal developmental death.
Based on the adult retinal morphology including decreased retinal size and decreased thickness of all retinal layers, other retinal neurons may also require BCL-X during development. Consistent with increased death of other developing neurons, after most RGCs have died at E18.5, there are significantly more activated CASP3+ cells in the Bcl-x deficient retinas. However, early born RGCs can help control later retinal neuron production by affecting retinal progenitor proliferation (Mu et al., 2005; Wang et al., 2005). Thus, the loss of RGCs may also contribute to the decrease in other cell types. The use of other cres will be needed to test the importance of Bcl-x in the development of later born retinal neurons.
Endogenous BCL-X delays adult RGC death after acute axonal injury
Similar to developing RGCs, adult RGCs robustly express BCL-X, raising the possibility that adult RGCs also require BCL-X in order to survive. To rule out developmental effects, Bcl-x was conditionally disrupted in the adult and RGC survival was assessed. Gross retinal morphology, including the number of RGCs, was still normal 60 days after Bcl-x deletion. Thus at some point during development RGCs lose their requirement for BCL-X as a survival factor. Other neurons in the central nervous system appear to exhibit a similar phenotype. For example, although BCL-X is normally expressed in adult catecholaminergic neurons, Savitt et al. observed a few surviving catecholaminergic neurons not expressing BCL-X in the adult (Savitt et al., 2005). One caveat of their observation was the possibility of developmental effects due to Bcl-x deletion since Bcl-x was conditionally deleted in these neurons during embryonic development. However in combination with results presented here, it appears that BCL-X is not required for the survival of normal adult neurons despite its wide expression pattern in the central nervous system. Interestingly, using a Bcl2 knockout mouse Cellerino and colleagues showed that Bcl2 was involved in RGC survival after the normal window of RGC death (Cellerino et al., 1999). These data suggest different Bcl-2 family members play key roles in maintaining RGC viability at various stages of maturation.
RGC death following axonal injury, an important insult in glaucoma (Anderson and Hendrickson, 1974; Howell et al., 2007; Howell et al., 2012; Quigley et al., 1983; Schlamp et al., 2006), is another form of apoptotic death that requires pro-apoptotic Bcl-2 family members. The activation of an apoptotic death pathway suggests that endogenous pro-survival Bcl-2 family members may determine an RGC’s susceptibility to death after an axonal insult. In fact, RGC death occurred earlier after mechanical optic nerve injury in the Bcl-x deficient retina compared to controls. By 2 days following injury, an RGC’s resistance to apoptosis appears to depend in part on BCL-X. This may be attributed to its ability to interact with BAX (Adams, 2003; van Delft and Huang, 2006) and its role in linking cellular metabolism to apoptotic sensitivity (Yi et al., 2011). Endogenous expression levels of BCL-X have been reported to be upregulated and downregulated after axonal injury in RGCs (Isenmann et al., 1997; Levin et al., 1997; McKernan and Cotter, 2007; Pelzel et al., 2010) suggesting that to fully unravel the process by which RGCs die. Thus, RGCs need to be examined on an individual cell basis in order to isolate surviving versus dying cells. In pathological conditions, RGC death is likely achieved by the complex interplay between the pro-survival and pro-death Bcl-2 family members (Nickells, 2010). BCL-X clearly has a major role in this process, but pro-death members BAX, BIM, BBC3, and potentially others are required to activate the intrinsic pathways of apoptosis in RGCs after axonal injury (Harder and Libby, 2011; Li et al., 2000; Libby et al., 2005; McKernan and Cotter, 2007; Qin et al., 2004). The relative level of expression of all these molecules is likely critical to determining how much of an insult a cell can withstand before undergoing apoptosis. Finally, due to the specific importance of BCL-X in resisting pro-apoptotic signaling in RGCs, fluctuations in BCL-X expression may also affect susceptibility to chronic or variable insults. Thus it may be important to identify how BCL-X and other Bcl-2 family members are regulated in adult neurons. Manipulating these endogenous pathways may increase a neurons ability to withstand an insult and therefore, be a potentially powerful therapeutic target.
Acknowledgments
The authors would like to thank Drs. Furuta (Six3-cre) and Hennighausen (Bcl-xf) for generously providing mouse strains and Thurma McDaniel and Donna Shannon for technical help. This work was supported by EY018606 (RTL), T32 EY007125 (JMH), David Bryant Trust (RTL), Research to Prevent Blindness Career Development Award (RTL) and a Research to Prevent Blindness unrestricted grant to the Department of Ophthalmology.
Footnotes
The Authors declare no conflicts of interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev. 2003;17:2481–2495. doi: 10.1101/gad.1126903. [DOI] [PubMed] [Google Scholar]
- Agudo M, Perez-Marin MC, Sobrado-Calvo P, Lonngren U, Salinas-Navarro M, Canovas I, Nadal-Nicolas FM, Miralles-Imperial J, Hallbook F, Vidal-Sanz M. Immediate upregulation of proteins belonging to different branches of the apoptotic cascade in the retina after optic nerve transection and optic nerve crush. Invest Ophthalmol Vis Sci. 2009;50:424–431. doi: 10.1167/iovs.08-2404. [DOI] [PubMed] [Google Scholar]
- Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol. 1974;13:771–783. [PubMed] [Google Scholar]
- Arbour N, Vanderluit JL, Le Grand JN, Jahani-Asl A, Ruzhynsky VA, Cheung EC, Kelly MA, MacKenzie AE, Park DS, Opferman JT, Slack RS. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci. 2008;28:6068–6078. doi: 10.1523/JNEUROSCI.4940-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea TC, Cahill H, Ecker J, Hattar S, Nathans J. Distinct roles of transcription factors brn3a and brn3b in controlling the development, morphology, and function of retinal ganglion cells. Neuron. 2009;61:852–864. doi: 10.1016/j.neuron.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr M. Live or let die - retinal ganglion cell death and survival during development and in the lesioned adult CNS. Trends Neurosci. 2000;23:483–490. doi: 10.1016/s0166-2236(00)01637-4. [DOI] [PubMed] [Google Scholar]
- Bonfanti L, Strettoi E, Chierzi S, Cenni MC, Liu XH, Martinou JC, Maffei L, Rabacchi SA. Protection of retinal ganglion cells from natural and axotomy-induced cell death in neonatal transgenic mice overexpressing bcl-2. J of Neurosci. 1996;16:4186–4194. doi: 10.1523/JNEUROSCI.16-13-04186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Simons DL, Fu XY, Feng GS, Wu SM, Zhang X. Loss of Shp2-mediated mitogen-activated protein kinase signaling in Muller glial cells results in retinal degeneration. Mol Cell Biol. 2011;31:2973–2983. doi: 10.1128/MCB.05054-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A, Michaelidis T, Barski JJ, Bahr M, Thoenen H, Meyer M. Retinal ganglion cell loss after the period of naturally occurring cell death in bcl-2-/- mice. Neuroreport. 1999;10:1091–1095. doi: 10.1097/00001756-199904060-00034. [DOI] [PubMed] [Google Scholar]
- Cenni MC, Bonfanti L, Martinou JC, Ratto GM, Strettoi E, Maffei L. Long-term survival of retinal ganglion cells following optic nerve section in adult bcl-2 transgenic mice. Eur J Neurosci. 1996;8:1735–1745. doi: 10.1111/j.1460-9568.1996.tb01317.x. [DOI] [PubMed] [Google Scholar]
- Chierzi S, Strettoi E, Cenni MC, Maffei L. Optic nerve crush: axonal responses in wild-type and bcl-2 transgenic mice. J Neurosci. 1999;19:8367–8376. doi: 10.1523/JNEUROSCI.19-19-08367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio C, Casciati A, Iaccarino C, Rotilio G, Carri MT. Bcl2a1 serves as a switch in death of motor neurons in amyotrophic lateral sclerosis. Cell Death Differ. 2006;13:2150–2153. doi: 10.1038/sj.cdd.4401943. [DOI] [PubMed] [Google Scholar]
- Cui Q, Yip HK, Zhao RC, So KF, Harvey AR. Intraocular elevation of cyclic AMP potentiates ciliary neurotrophic factor-induced regeneration of adult rat retinal ganglion cell axons. Mol Cell Neurosci. 2003;22:49–61. doi: 10.1016/s1044-7431(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Dietz GP, Kilic E, Bahr M, Isenmann S. Bcl-2 is not required in retinal ganglion cells surviving optic nerve axotomy. Neuroreport. 2001;12:3353–3356. doi: 10.1097/00001756-200110290-00041. [DOI] [PubMed] [Google Scholar]
- Fernandes KA, Harder JM, Fornarola LB, Freeman RS, Clark AF, Pang IH, John SW, Libby RT. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis. 2012;46:393–401. doi: 10.1016/j.nbd.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann S, Riesenberg AN, Mathiesen AM, Brown EC, Vetter ML, Brown NL. Characterization of a transient TCF/LEF-responsive progenitor population in the embryonic mouse retina. Invest Ophthalmol Vis Sci. 2009;50:432–440. doi: 10.1167/iovs.08-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, Lagutin O, Hogan BL, Oliver GC. Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis. 2000;26:130–132. [PubMed] [Google Scholar]
- Gan L, Wang SW, Huang Z, Klein WH. POU domain factor Brn-3b is essential for retinal ganglion cell differentiation and survival but not for initial cell fate specification. Dev Biol. 1999;210:469–480. doi: 10.1006/dbio.1999.9280. [DOI] [PubMed] [Google Scholar]
- Garrity-Moses ME, Teng Q, Liu J, Tanase D, Boulis NM. Neuroprotective adeno-associated virus Bcl-xL gene transfer in models of motor neuron disease. Muscle Nerve. 2005;32:734–744. doi: 10.1002/mus.20418. [DOI] [PubMed] [Google Scholar]
- Harder JM, Libby RT. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol Neurodegener. 2011;6:50. doi: 10.1186/1750-1326-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–1537. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Soto I, Libby RT, John SW. Intrinsic axonal degeneration pathways are critical for glaucomatous damage. Exp Neurol. 2012 doi: 10.1016/j.expneurol.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenmann S, Kretz A, Cellerino A. Molecular determinants of retinal ganglion cell development, survival, and regeneration. Prog Retin Eye Res. 2003;22:483–543. doi: 10.1016/s1350-9462(03)00027-2. [DOI] [PubMed] [Google Scholar]
- Isenmann S, Wahl C, Krajewski S, Reed JC, Bahr M. Up-regulation of Bax protein in degenerating retinal ganglion cells precedes apoptotic cell death after optic nerve lesion in the rat. Eur J Neurosci. 1997;9:1763–1772. doi: 10.1111/j.1460-9568.1997.tb01534.x. [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005;19:2026–2028. doi: 10.1096/fj.05-3941fje. [DOI] [PubMed] [Google Scholar]
- Koh PO. Gingko biloba extract (EGb 761) prevents increase of Bad-Bcl-XL interaction following cerebral ischemia. Am J Chin Med. 2009;37:867–876. doi: 10.1142/S0192415X0900734X. [DOI] [PubMed] [Google Scholar]
- Levin LA, Schlamp CL, Spieldoch RL, Geszvain KM, Nickells RW. Identification of the bcl-2 family of genes in the rat retina. Invest Ophthalmol Vis Sci. 1997;38:2545–2553. [PubMed] [Google Scholar]
- Li Y, Schlamp CL, Poulsen KP, Nickells RW. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000;71:209–213. doi: 10.1006/exer.2000.0873. [DOI] [PubMed] [Google Scholar]
- Libby RT, Li Y, Savinova OV, Barter J, Smith RS, Nickells RW, John SW. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas TJ, Wang AL, Yuan M, Neufeld AH. Early cellular signaling responses to axonal injury. Cell Commun Signal. 2009;7:5. doi: 10.1186/1478-811X-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Hossain M, Chow A, Arshad M, Battson RM, Sanders RD, Mehmet H, Edwards AD, Franks NP, Maze M. Xenon and hypothermia combine to provide neuroprotection from neonatal asphyxia. Ann Neurol. 2005;58:182–193. doi: 10.1002/ana.20547. [DOI] [PubMed] [Google Scholar]
- Malik JM, Shevtsova Z, Bahr M, Kugler S. Long-term in vivo inhibition of CNS neurodegeneration by Bcl-XL gene transfer. Mol Ther. 2005;11:373–381. doi: 10.1016/j.ymthe.2004.11.014. [DOI] [PubMed] [Google Scholar]
- McKernan DP, Cotter TG. A Critical role for Bim in retinal ganglion cell death. J Neurochem. 2007;102:922–930. doi: 10.1111/j.1471-4159.2007.04573.x. [DOI] [PubMed] [Google Scholar]
- Middleton G, Wyatt S, Ninkina N, Davies AM. Reciprocal developmental changes in the roles of Bcl-w and Bcl-x(L) in regulating sensory neuron survival. Development. 2001;128:447–457. doi: 10.1242/dev.128.3.447. [DOI] [PubMed] [Google Scholar]
- Mori M, Burgess DL, Gefrides LA, Foreman PJ, Opferman JT, Korsmeyer SJ, Cavalheiro EA, Naffah-Mazzacoratti MG, Noebels JL. Expression of apoptosis inhibitor protein Mcl1 linked to neuroprotection in CNS neurons. Cell Death Differ. 2004;11:1223–1233. doi: 10.1038/sj.cdd.4401483. [DOI] [PubMed] [Google Scholar]
- Mosinger Ogilvie J, Deckwerth TL, Knudson CM, Korsmeyer SJ. Suppression of developmental retinal cell death but not of photoreceptor degeneration in Bax-deficient mice. Invest Ophthalmol Vis Sci. 1998;39:1713–1720. [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Mu X, Fu X, Sun H, Liang S, Maeda H, Frishman LJ, Klein WH. Ganglion cells are required for normal progenitor- cell proliferation but not cell-fate determination or patterning in the developing mouse retina. Curr Biol. 2005;15:525–530. doi: 10.1016/j.cub.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Nickells RW. Variations in the rheostat model of apoptosis: what studies of retinal ganglion cell death tell us about the functions of the Bcl2 family proteins. Exp Eye Res. 2010;91:2–8. doi: 10.1016/j.exer.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsadanian AS, Cheng Y, Keller-Peck CR, Holtzman DM, Snider WD. Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J Neurosci. 1998;18:1009–1019. doi: 10.1523/JNEUROSCI.18-03-01009.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelzel HR, Schlamp CL, Nickells RW. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010;11:62. doi: 10.1186/1471-2202-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pequignot MO, Provost AC, Salle S, Taupin P, Sainton KM, Marchant D, Martinou JC, Ameisen JC, Jais JP, Abitbol M. Major role of BAX in apoptosis during retinal development and in establishment of a functional postnatal retina. Dev Dyn. 2003;228:231–238. doi: 10.1002/dvdy.10376. [DOI] [PubMed] [Google Scholar]
- Pike CJ. Estrogen modulates neuronal Bcl-xL expression and beta-amyloid-induced apoptosis: relevance to Alzheimer’s disease. J Neurochem. 1999;72:1552–1563. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- Poche RA, Raven MA, Kwan KM, Furuta Y, Behringer RR, Reese BE. Somal positioning and dendritic growth of horizontal cells are regulated by interactions with homotypic neighbors. Eur J Neurosci. 2008;27:1607–1614. doi: 10.1111/j.1460-9568.2008.06132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath H, Strasser A. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002;9:505–512. doi: 10.1038/sj.cdd.4400998. [DOI] [PubMed] [Google Scholar]
- Qin Q, Patil K, Sharma SC. The role of Bax-inhibiting peptide in retinal ganglion cell apoptosis after optic nerve transection. Neurosci Lett. 2004;372:17–21. doi: 10.1016/j.neulet.2004.08.075. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Hohman RM, Addicks EM, Massof RW, Green WR. Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am J Ophthalmol. 1983;95:673–691. doi: 10.1016/0002-9394(83)90389-6. [DOI] [PubMed] [Google Scholar]
- Rucker EB, 3rd, Dierisseau P, Wagner KU, Garrett L, Wynshaw-Boris A, Flaws JA, Hennighausen L. Bcl-x and Bax regulate mouse primordial germ cell survival and apoptosis during embryogenesis. Mol Endocrinol. 2000;14:1038–1052. doi: 10.1210/mend.14.7.0465. [DOI] [PubMed] [Google Scholar]
- Savitt JM, Jang SS, Mu W, Dawson VL, Dawson TM. Bcl-x is required for proper development of the mouse substantia nigra. J Neurosci. 2005;25:6721–6728. doi: 10.1523/JNEUROSCI.0760-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006;7:66. doi: 10.1186/1471-2202-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Roth KA. Bcl-2 family and the central nervous system: from rheostat to real complex. Cell Death Differ. 2006;13:1299–1304. doi: 10.1038/sj.cdd.4401974. [DOI] [PubMed] [Google Scholar]
- Shindler KS, Latham CB, Roth KA. Bax deficiency prevents the increased cell death of immature neurons in bcl-x-deficient mice. Journal of Neuroscience. 1997;17:3112–3119. doi: 10.1523/JNEUROSCI.17-09-03112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Huang DC. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res. 2006;16:203–213. doi: 10.1038/sj.cr.7310028. [DOI] [PubMed] [Google Scholar]
- Wang C, Kaufmann JA, Sanchez-Ross MG, Johnson KM. Mechanisms of N-methyl-D-aspartate-induced apoptosis in phencyclidine-treated cultured forebrain neurons. J Pharmacol Exp Ther. 2000;294:287–295. [PubMed] [Google Scholar]
- Wang SW, Mu X, Bowers WJ, Kim DS, Plas DJ, Crair MC, Federoff HJ, Gan L, Klein WH. Brn3b/Brn3c double knockout mice reveal an unsuspected role for Brn3c in retinal ganglion cell axon outgrowth. Development. 2002;129:467–477. doi: 10.1242/dev.129.2.467. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dakubo GD, Thurig S, Mazerolle CJ, Wallace VA. Retinal ganglion cell-derived sonic hedgehog locally controls proliferation and the timing of RGC development in the embryonic mouse retina. Development. 2005;132:5103–5113. doi: 10.1242/dev.02096. [DOI] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiessner C, Allegrini PR, Rupalla K, Sauer D, Oltersdorf T, McGregor AL, Bischoff S, Bottiger BW, van der Putten H. Neuron-specific transgene expression of Bcl-XL but not Bcl-2 genes reduced lesion size after permanent middle cerebral artery occlusion in mice. Neurosci Lett. 1999;268:119–122. doi: 10.1016/s0304-3940(99)00392-4. [DOI] [PubMed] [Google Scholar]
- Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, Vander Heiden MG, Yang R, Li F, Locasale JW, Sharfi H, Zhai B, Rodriguez-Mias R, Luithardt H, Cantley LC, Daley GQ, Asara JM, Gygi SP, Wagner G, Liu CF, Yuan J. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell. 2011;146:607–620. doi: 10.1016/j.cell.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KW, Pinon LG, Dhiraj D, Twiddy D, Macfarlane M, Hickman J, Nicotera P. Mitochondrial fragmentation and neuronal cell death in response to the Bcl-2/Bcl-x(L)/Bcl-w antagonist ABT-737. Neuropharm. 2010;58:1258–1267. doi: 10.1016/j.neuropharm.2010.03.008. [DOI] [PubMed] [Google Scholar]