

Abstract
Naïve CD4+T cells differentiate into various effector T helper subsets depending on the antigens and cytokine microenvironment they encounter. IL-9-secreting Th9 cells are the most recent T helper subset to be described. PU.1, one of the transcription factors required for the development of Th9 cells, binds to the Il9 gene. In this study we show that PU.1 increases histone acetylation at the Il9 locus through direct interactions with histone acetyltransferases (HAT). In the absence of PU.1, there is decreased association of Gcn5 and PCAF, and increased association of HDACs at the Il9 locus in Th9 cells. Inhibiting histone deacetylase activity augments PU.1-dependent IL-9 production. PU.1 forms a complex with Gcn5, and inhibiting the expression of Gcn5 results in reduced IL-9 production. Moreover, the effects of Gcn5 on IL-9 production are specific as the production of IL-10 and IL-21, two additional cytokines produced by Th9 cells, is not altered following decreased Gcn5 expression. Together, these data define a PU.1-dependent mechanism for altered histone acetylation and expression of the Il9 locus in Th9 cells.
Introduction
Naive CD4+T cells give rise to distinct T helper subsets in response to environmental cues. T helper cells are characterized by the cytokines they secrete along with the lineage-specific transcription factors they express. Th1, Th2, and Th17 cells which secrete IFN-γ, IL-4, and IL-17 respectively, have T-bet, GATA3 and RORγt as lineage specific transcriptional regulators (1). Recently, IL-9-secreting Th9 cells have been described that differentiate in response to TGFβ and IL-4 (2–4). Our group has previously demonstrated that PU.1 is one of the transcription factors required for the development of Th9 cells downstream of the TGFβ signal (5, 6). Downstream of IL-4, STAT6 is required to activate IRF4 and potentially other factors as part of a transcription factor network in Th9 cells (6, 7).
PU.1 is an ETS family transcription factor that has multiple functional domains. The ETS homology DNA-binding domain is at the C-terminal end, and the acidic and glutamine rich domains in the N-terminal end act as transactivation domains (8, 9). In Th2 cells, PU.1 interferes with Th2 cytokine expression by binding to, and limiting the function of GATA3 and IRF4 through interactions with the ETS DNA-binding domain (10–12). PU.1 binds directly to the Il9 promoter in Th9 cells (5, 6). In mice with a conditional mutation of the Sfpi1 allele (encoding PU.1), T cell-specific deletion of PU.1 results in decreased IL-9 production in vitro, and in vivo coincident with diminished allergic inflammation (5). However, a mechanism for PU.1-dependent Il9 expression in Th9 cells has not been defined.
Global gene expression results from a balance between histone acetylation and deacetylation controlled by a group of enzymes, histone acetylatransferases (HATs)2 and histone deacetylases (HDACs), respectively. Acetylation of lysine residues reduces electrostatic interactions between histone and the phosphodiester backbone thereby facilitating the accessibility of trans-acting factors to the gene (13). HDACs reverse this process in creating repressive chromatin and restricting trans-acting factors access to a gene (14). Many transcription factors can act as activators based on their interaction with proteins having intrinsic HAT activity (14–16). Global hyperacetylated states can be generated through the use of HDAC inhibitors which induce expression of a subset of genes (17, 18). Gcn5, one of the HATs, was initially characterized as a positive regulator of amino acid biosynthesis in S. cerevisiae, before its role in histone acetylation and transcription was appreciated (19). Gcn5 and PCAF, another HAT molecule, are subunits of several multi-protein complexes including SAGA, ATAC and STAGA (20). Although considerable work examining PU.1 function has been described, and it is considered a pioneer factor in cellular differentiation (21, 22), only limited work has examined the interaction of PU.1 with HAT proteins (23).
In this report, we demonstrate that PU.1 regulates the association of HDACs and HATs, and as a consequence histone acetylation at the Il9 locus. PU.1 forms a complex with Gcn5, and inhibiting Gcn5 expression results in diminished IL-9 production. Together, the data documented here demonstrate that PU.1 transactivates Il9 by recruiting specific HAT proteins, and increasing histone acetylation at the locus.
Materials and Methods
Mice
Wild-type (WT) C57BL/6 female mice were purchased from Harlan Biosciences, Indianapolis, IN. Mice with conditional PU.1 gene deletion (Sfpi1lck−/−) on C57BL/6 background were previously described (24) and were mated to mice expressing Cre transgene under the control of Lck promoter. Mice were maintained in pathogen-free conditions and studies were approved by the Indiana University Institutional Animal Care and Use Committee.
Th cell differentiation
Naïve CD4+CD62L+ T cells were purified from spleen and lymph nodes by magnetic selection (Miltenyi Biotec). Naïve CD4+ T cells (1 × 106 cells per ml in complete RPMI-1640 medium) were cultured with plate-bound anti-CD3 (2μg/ml; 145-2C11; BioXcell) and soluble anti-CD28 (1μg/ml; 37.51; BD Biosciences) under Th9 conditions (IL-4 (20ng/ml; Peprotech), TGF-β (2ng/ml; R&D systems) and anti-IFN-γ (10μg/ml; XMG; BioXcell)) in a 6-well plate. After 3 days, cultures were expanded with fresh complete RPMI-1640 medium with IL-4 and TGF-β added to the Th9 cells. Th9 cultures treated with HDAC inhibitor had 20nM TSA (Sigma-Aldrich) being added on day 0 of the cultures. After 5 days of differentiation cells were harvested and stimulated with 2μg/ml plate-bound anti-CD3 for 1 day. Cell free supernatant was collected and the amount IL-9 was assessed by ELISA (anti-IL-9 capture antibody (D8402E8; BD Biosciences) and biotin-labeled anti-IL-9 (D9302C12; Biolegend).
Retroviral transduction
Naïve CD4+CD62L+ T cells from WT mice were cultured under Th9 cell conditions, and on day 2, were transduced with control, intact PU.1 (PU.1), and mutant PU.1 (R232G, and Δ33–100) expressing retroviral vectors as previously described (11) in the presence of polybrene. On day 5, transduced cells (EGFP+ cells) were analyzed by flow cytometry. In some experiments Th9 cells were transduced with control retroviral vector or intact PU.1, or mutant PU.1-expressing retroviral vectors before EGFP+ cells were sorted on day 5, and sorted cells were either stimulated with plate-bound anti-CD3 for ELISA or they were prepared for chromatin immunoprecipitation.
Intracellular cytokine staining
Differentiated Th9 cells were stimulated with PMA (phorbol 12-myristate 13-acetate) and ionomycin for a period of 5h. Monensin was added to the cells for the last 3h of stimulation. After fixing the cells with paraformaldehyde and permeabilizing with saponin, cells were stained with fluorochrome conjugated anti-mouse IL-9 (RM9A4, Biolegend), and anti-mouse IL-4 (11B11, Biolegend). Cells were analyzed by flow cytometry with FACSCalibur (Beckton Dickinson) and were analyzed by WinMDI.
Chromatin Immunoprecipitation
Unstimulated Th9 cells were crosslinked with formaldehyde and immunocomplex was precipitated with protein A-agarose beads at 4°C overnight. The supernatant was incubated in the presence of rabbit polyclonal antibodies (anti-PU.1, anti-Gcn5 (Santa Cruz Biotechnology), anti-acetyl-H3 lysine 9/18, anti-acetyl-H3 lysine 14, anti-acetyl-H3 lysine 27, anti-acetyl-H4 lysine 5, anti-acetyl-H4 lysine 8, anti-acetyl-H4 lysine 16 (Millipore), anti-Histone H3, anti-acetyl-H3 lysine 36 (Upstate), anti-histone H4 (Abcam)) and rabbit IgG (Millipore), mouse monoclonal anti-HDAC1 and mouse IgG1, or goat polyclonal anti-IRF4, anti-HDAC2 and goat IgG (Santa Cruz Biotechnology). After reverse crosslinking, DNA was purified by phenol-chloroform extraction and ethanol precipitation. DNA quantification was performed with SYBR Green Fast PCR Master Mix via ABI 7500 Fast Real-time PCR system. A standard curve was generated from serial dilutions of input DNA to quantify immunoprecipitated DNA. Data for ChIP samples are presented with percent input from control Ig subtracted from percent input of the specific antibody.
Immunoblot
Total cell lysates were prepared from the indicated cell types. The lysates were resuspended in SDS-PAGE loading buffer, boiled at 100°C for 5 min and electrophoresed on SDS-PAGE gels (Invitrogen Life Technologies) before immunoblot with PU.1, IRF4, Gcn5, PCAF, p300, or β-actin.
Gcn5 small interfering RNA (siRNA) transfection
Differentiated Th9 cells from wild-type mice were transiently transfected with control or Gcn5-specific siRNA (0.2μM, Santa Cruz Biotechnology) using Amaxa Nucleofector (Lonza). Cells were rested overnight in 5% CO2 incubator with hIL-2 (50U/ml). Cells were either kept unstimulated for ChIP assay or stimulated with 2 μg/ml plate bound anti-CD3 for 6h for RNA isolation or for 24h to collect cell free supernatant for ELISA to assess IL-9, IL-10, and IL-21 production.
Quantitative RT-PCR
Total RNA was isolated from unstimulated or stimulated (plate-bound anti-CD3, 2μg/ml) cells using TRIzol and reverse transcribed according to manufacturer's instructions (Invitrogen Life Technologies). Taqman Fast Universal Master Mix and commercially available primers (ABI Biosystems) for mouse were used for Il9, Il10, Il21, Irf4, Sfpi1, and Gcn5. RNA expression was normalized to the expression of β2-microglobulin and the relative expression was calculated by the change-in-threshold (−ΔΔCT) method.
DNA-affinity precipitation assay (DAPA)
Nuclear lysates were prepared from wild-type and Sfpi1lck−/− Th9 cells. Lysates were incubated with streptavidin-agarose and consensus PU.1-binding oligonucleotides (WT oligonucleotide). The consensus PU.1 binding site was previously described (5). Protein-DNA complexes were separated by SDS-PAGE and immunoblots were probed with anti-Gcn5, anti-PCAF, and anti-PU.1.
Results
Histone deacetylase inhibitor enhances IL-9 production
Histone acetylation of lysine residues by HAT domain-containing proteins relaxes the condensed chromatin resulting in greater levels of gene transcription. This process is reversed by histone deacetylases (HDACs). To determine if HDAC association at the Il9 locus was dependent on PU.1, we examined HDAC1 and HDAC2 binding in wild-type and PU.1-deficient Th9 cells at two previously described conserved non-coding sequences (CNS) 5-prime of the Il9 gene (25)(Fig. 1A). There was significantly enhanced HDAC1 and HDAC2 binding at Il9 promoter (CNS1) in the absence of PU.1 (Fig. 1B). We then wanted to define if HDAC inhibition reverses repressive chromatin and leads to enhanced IL-9 production. Wild-type Th9 cells were treated with TSA an antifungal antibiotic that selectively inhibits class I and II HDACs. Th9 cells treated with TSA had increased percentages of IL-9-secreting cells and enhanced Il9 gene expression (Fig. 1C and D). However, TSA does not induce the expression of Il10 and Il21, cytokines produced by Th9 cells (5, 26)(Fig. 1D). This observation is consistent with studies showing that HDAC inhibition only alters the expression of a subset of genes (17, 18). To examine whether TSA treatment affects expression of Th9-specific transcription factors, we measured the expression of Sfpi1, the gene encoding PU.1, and Irf4 after TSA treatment. We observed an increase in Sfpi1 expression, but no change in Irf4 expression in Th9 cells (Fig. 1E). This was confirmed by immunoblot of protein extracts prepared from Th9 cells treated with TSA (Fig. 1F). To determine if TSA-mediated enhancement of IL-9 was PU.1-dependent, Th9 cells from both wild-type and PU.1-deficient mice were treated with TSA. PU.1-deficient Th9 cells secreted significantly lower amounts of IL-9 than wild-type Th9 cells (Fig. 1G). Although TSA was able to significantly increase IL-9 production from wild-type Th9 cells, addition of TSA in PU.1-deficient Th9 cultures did not increase IL-9 production (Fig. 1G).
Figure 1.

Inhibiting histone deacetylases augment IL-9 production
A, Schematic diagram of the CNS of the Il9 gene. B, Naïve CD4+T cells from wild-type and Sfpi1lck−/− mice were cultured under Th9 cell conditions for 5d. Chromatin immunoprecipitation was performed for HDAC1 and HDAC2 before quantitative PCR for two Il9 CNS sites. C–D, Naïve CD4+T cells from wild-type mice were cultured under Th9 conditions for 5d in the presence or absence of TSA. On day 5, cells were stimulated with PMA and ionomycin before intracellular staining for IL-9 and IL-4 was performed (C). Differentiated Th9 cells were stimulated with anti-CD3 for 6h before RNA isolation. Expression of the indicated genes was assessed using quantitative PCR (D). E, Th9 cells cultured and treated as in C were examined for Sfpi1 and Irf4 expression. F, Total cell extracts were immunoblotted for PU.1, IRF4, and β-actin as a control (left), blots were scanned and presented in densitometry units (right). G, Naïve CD4+T cells from wild-type and Sfpi1lck−/− mice were cultured under Th9 cell conditions for 5d in the presence or absence of TSA. On day5, cells were harvested and stimulated with plate bound 2 μg/ml anti-CD3 for 24h. IL-9 was assessed by ELISA. H–I, Naïve CD4+T cells from wild-type mice were cultured as in (C) for 5d. Chromatin immunoprecipitation was performed for AcH3K14, AcH4K5, PU.1 and IRF4 before quantitative PCR for Il9 CNS1. ChIP data are presented with control Ig values subtracted from the specific Ig percent input values. Data are average ± S.D of 4–6 mice from 2–3 experiments (B–E), or replicates representative of 3 independent experiments (F, H, I) or average ± S.D of 3 mice (G). * p<0.05, **p<0.01 (C–E, paired t-test; G, one way ANOVA post hoc Bonferroni correction).
We then tested whether TSA-induced Il9 expression correlated with altered histone acetylation at the Il9 gene or altered transcription factor binding at the Il9 promoter. Acetylation of H3K14 and H4K5 was increased after wild-type Th9 cells were treated with TSA (Fig. 1H). There was increased PU.1 and IRF4 binding to Il9 promoter after TSA treatment (Fig. 1I), despite TSA not altering Irf4 expression. Overall, these data demonstrate that HDAC inhibition leads to increased IL-9 production, and this correlates with increased histone acetylation and increased PU.1 and IRF4 binding to the Il9 promoter.
The Activation domains of PU.1 induce IL-9 in Th9 cells
Since both PU.1 and induced histone acetylation are required for Il9 expression in Th9 cells (5, 27), we next explored how PU.1 promotes histone acetylation. Transcription factors are composed of distinct functional modules. To define the structural requirements for PU.1-mediated IL-9 induction, we transduced developing Th9 cells with control retrovirus, intact PU.1-expressing retrovirus (PU.1), and PU.1 mutant-expressing retroviruses that lacked the transactivation domain (Δ33–100) or had a point mutation in the DNA binding domain (R232G), that results in diminished DNA binding (8). Full length PU.1 enhances both the percentage and the intensity of IL-9-secreting T cells (Fig. 2A and B). In contrast, cells transduced with either the transactivation mutant or with the DNA-binding mutant retroviruses failed to induce IL-9 (Fig. 2A and B). Overall, these data suggest that the activation domain and DNA-binding domain of PU.1 are required for the induction of IL-9.
Figure 2.

Activation and DNA-binding domains of PU.1 induce IL-9
A–B, Naïve CD4+T cells from wild-type mice were activated with anti-CD3 and anti-CD28 and cultured under Th9 cell conditions for 48h before being transduced with control or full-length PU.1 (PU.1) or PU.1 mutant-expressing retroviruses. After 5d in culture cells were stimulated with PMA and ionomycin. Percentages of IL-9-secreting cells (A) and IL-9 mean fluorescence intensity (MFI) (B) was analyzed based on EFGP+ cells. C–E, Naïve CD4+T cells from wild-type and Sfpi1lck−/− mice were cultured under Th9 conditions and transduced with control, full-length or PU.1 mutant-expressing retroviruses as indicated. On day 5 of culture, EGFP+ cells were activated with PMA and ionomycin for flow cytometry based on EGFP+ cells (C, D) or sorted and activated with 2 μg/ml plate-bound anti-CD3 for 24h. Cell free supernatant was collected to measure IL-9 by ELISA (E). Data are average ± S.D of 6–8 mice from 3–4 experiments (A, B) or average ± S.D of 3 mice (C–E). *p<0.05, ** p<0.01 (one way ANOVA with post hoc Bonferroni correction).
To further demonstrate the requirement of the activation domain in IL-9 expression, we transduced retroviruses expressing full-length PU.1 or activation domain-mutant PU.1 into PU.1-deficient Th9 cultures (cells from Sfpi1lck−/− mice). PU.1-deficient Th9 cells transduced with control retrovirus had significantly reduced IL-9-secreting cells and IL-9 production compared to wild-type Th9 cells, and increased production of IL-4, as previously described (Chang et al, 2009) (Fig. 2C–E). However, full-length PU.1 was able to recover IL-9 production in PU.1-deficient Th9 cells to a level similar to wild-type Th9 cells transduced with control retrovirus, and repress production of IL-4 (Fig. 2C–E). In contrast, the activation domain mutant had a significantly diminished ability to rescue IL-9 in PU.1-deficient Th9 cells. The DNA binding mutant did not induce IL-9 when transduced into PU.1-deficient Th9 cultures (data not shown). Together, these data suggest that the activation domain of PU.1 is critical for the induction of IL-9.
Altered histone acetylase binding at the Il9 locus
Since PU.1 can bind to the Il9 promoter in Th9 cells (5), we hypothesized that PU.1 is recruiting other factors such as histone modifying enzymes including histone acetyltrasnferases (HATs) to the Il9 locus. We examined the recruitment of HATs including Gcn5, PCAF, and p300 in wild-type and PU.1-deficient Th9 cells to two previously described Il9 conserved non-coding sequences (25). We observed decreased recruitment of both Gcn5 and PCAF at Il9 CNS1 and CNS0 in the absence of PU.1 (Fig. 3A). In contrast to previous reports showing that PU.1 recruited p300 to the Igκ locus in B cells (23), there was increased binding of p300 at the Il9 promoter in PU.1-deficient Th9 cells (Fig. 3A). To rule out the possibility that in the absence of PU.1 there is difference in the expression of HAT molecules, we prepared nuclear extracts from WT and PU.1-deficient Th9 cells and examined the expression of Gcn5, PCAF, and p300 using immunoblot. Loss of PU.1 did not alter the expression of these HATs (Fig. 3B). Thus, PU.1 is required for the association of specific HATs to the Il9 promoter.
Figure 3.

Altered histone acetyltransferases binding at Il9 gene
A, Naïve CD4+T cells from wild-type and Sfpi1lck−/− mice were activated with anti-CD3 and anti-CD28 and cultured under Th9 cell conditions for 5d. Cells were then fixed and chromatin immunoprecipitation was performed for Gcn5, PCAF, and p300 before quantitative PCR for two Il9 CNS sites. ChIP data are presented with control Ig values subtracted from the specific Ig percent input values. B, Total cell lysates were prepared from WT and PU.1-deficient Th9 cells and were immunoblotted for Gcn5, PCAF, and p300. β-actin was used as loading control. Data are average ± S.D of 6–8 mice from 3–4 experiments (A) or representative of two independent experiments (B). * p<0.05, ** p<0.01.
Altered histone acetylation at the Il9 locus
We then defined the functional consequences of altered HAT binding to Il9 gene. We had previously observed decreased total H3 acetylation at the Il9 promoter in PU.1-deficient Th9 cells (5). We therefore examined the acetylation of specific histone residues in WT and PU.1-deficient Th9 cells. We observed significantly attenuated histone acetylation of histone H3K14 at the Il9 promoter in PU.1-deficient Th9 cells (Fig. 4A). Acetylation of H3K9/18, H4K5, H4K8, and H4K16 was decreased significantly at the Il9 CNS1 and CNS0. However, there was no difference in acetylation of H3K27 and H3K36 between wild-type and PU.1-deficient Th9 cells (Fig. 4A). As a control we compared total H3 and H4 at Il9 promoter between WT and PU.1-deficient Th9 cells and observed no difference in total histone content at the locus (Fig. 4B). Overall, these data indicate that PU.1 regulates specific histone acetylation at the Il9 locus.
Figure 4.
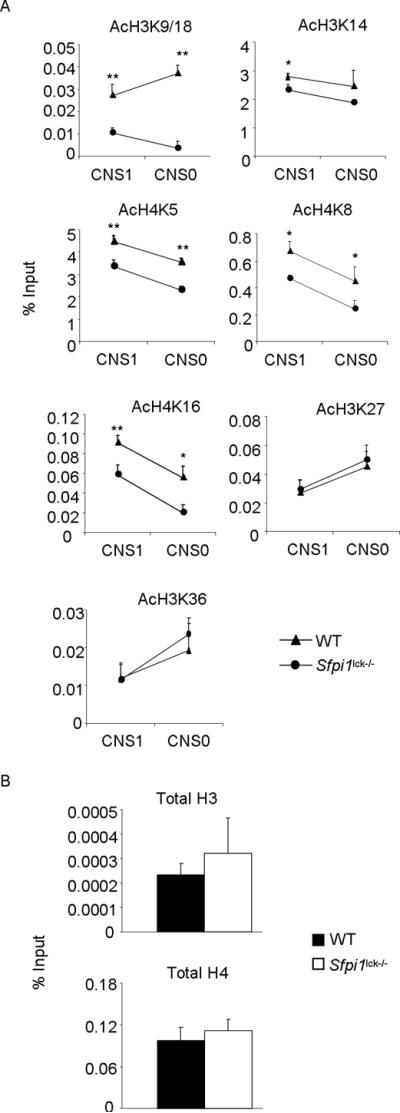
Altered histone acetylation at Il9 gene
A–B, Naïve CD4+T cells from wild-type and Sfpi1lck−/− mice were cultured under Th9 cell conditions for 5d. The differentiated cells were used for chromatin immunoprecipitation for AcH3K9/18, AcH3K14, AcH4K5, AcH4K8, AcH4K16, AcH3K27, and AcH3K36 before quantitative PCR for Il9 CNS1 and Il9 CNS0 (A) or for total H3 and total H4 before quantitative PCR for Il9 CNS1 (B). ChIP data are presented with control Ig values subtracted from the specific Ig percent input values. Data are average ± S.D of 6–8 mice from 3–4 experiments. * p<0.05, ** p<0.01.
PU.1 associates with Gcn5
As we observed decreased Gcn5 and PCAF recruitment to the Il9 promoter in the absence of PU.1 (Fig. 3), we wanted to determine if PU.1 was able to directly associate with either of these HAT proteins. We first tested an association of PU.1 with each HAT by co-immunoprecipitation, but were unable to observe significant association (data not shown). We speculated that PU.1 interacts with HATs more efficiently when bound to DNA. To test this, a DNA-affinity precipitation assay (DAPA) was performed with nuclear lysates prepared from wild-type and PU.1-deficient Th9 cells incubated with biotinylated WT oligonucleotide (consensus PU.1 binding site). DNA-bound PU.1 was able to precipitate Gcn5 from wild-type lysates in the presence of WT oligonucleotide (Fig. 5A). However, Gcn5 was not precipitated when consensus oligonucleotide was used with PU.1-deficient Th9 extracts (Fig. 5A). PCAF was not precipitated from either wild-type or PU.1-deficient Th9 lysates (Fig. 5A). These results suggest that DNA-bound PU.1 is able associate directly with Gcn5, but not PCAF. To determine if the requirement for the activation domain in the induction of IL-9 correlated with a requirement for the activation domain to interact with Gcn5, we transfected cells with full-length or Δ33–100 mutant PU.1 and performed DAPA. DNA-bound PU.1 requires the activation domain to efficiently precipitate Gcn5 (Fig. 5B).
Figure 5.

PU.1 associates with Gcn5
A, Naïve CD4+T cells from wild-type and Sfpi1lck−/− mice were cultured under Th9 conditions for 5d. Th9 nuclear lysates were incubated with WT oligonucleotide and streptavidin-agarose. Lysates were then subjected to immunoblot for Gcn5, PCAF and PU.1. Input samples were prepared from untreated nuclear lysates. B, Phoenix-GP cells were transfected with control (MIEG), full-length PU.1 (PU.1) or activation domain mutant PU.1 (Δ33–100)-expressing plasmids. Nuclear extracts were then prepared and subjected to DAPA using WT oligonucleotide before immunoblot for Gcn5 (left panel). Densitometry of the immunoblots was performed where the background signal (MIEG) was subtracted from the full-length or activation domain mutant PU.1 immunoblot signal and data are presented as arbitrary densitometry units (right panel). C, Naïve CD4+T cells from wild-type and Sfpi1lck−/− mice were cultured under Th9 conditions and transduced as Fig. 1C. On day 5, EGFP+ cells were sorted and used for Gcn5 chromatin immunoprecipitation before quantitative PCR for Il9 CNS1. ChIP data are presented with control Ig values subtracted from the specific Ig percent input values. Data are average ± S.D. of 3 mice. ** p<0.01 (One way ANOVA with post hoc Bonferroni correction).
To further demonstrate that PU.1 recruited Gcn5 to the Il9 promoter, we tested whether ectopic expression of PU.1 recovered Gcn5 binding at the Il9 promoter in PU.1-deficient Th9 cells. PU.1-deficient Th9 cells transduced with control retrovirus had significantly attenuated Gcn5 binding at the Il9 promoter compared to wild-type cells (Fig. 5C). Full-length PU.1 was able to rescue Gcn5 binding in PU.1-deficient Th9 cells (Fig. 5C). However, cells transduced with the activation domain mutant failed to rescue Gcn5 binding (Fig. 5C), further confirming the importance of the activation domain. Together, these data demonstrate that PU.1 associates with Gcn5 and that the activation domain is critical for Gcn5 binding to Il9 promoter.
Gcn5 is required for IL-9 expression
Since PU.1 associates with and recruits Gcn5 to Il9, we wanted to determine if reduced Gcn5 expression affects Il9 gene expression. Gcn5-null embryos die during embryogenesis (28). Therefore, differentiated wild-type Th9 cells were transfected with control or Gcn5-specific siRNA. Th9 cells transfected with Gcn5-specific siRNA had significantly decreased Gcn5 mRNA, and decreased Gcn5 binding to the Il9 promoter (Fig. 6A, B). Concomitantly, IL-9 production was reduced significantly when Gcn5 expression was inhibited (Fig. 6C). To determine if altered Gcn5 expression affects the production of other cytokines we assessed the amount of IL-10 and IL-21 produced by Th9 cells with diminished Gcn5 expression. Gcn5 inhibition does not decrease IL-10 and IL-21 production (Fig. 6C). Decreased Gcn5 expression did not alter PU.1 or IRF4 binding to the Il9 promoter (Fig. 6D). Gcn5 acetylates H3K9/18, H4K8, and under some conditions H4K5 (29–31). We observed acetylation of H3K9/18, H4K5 and H4K8 at the Il9 promoter was attenuated when Gcn5 was inhibited (Fig. 6E). Together, these data suggest that Gcn5 inhibition leads to reduced IL-9 production concomitant with decreased histone acetylation of Gcn5-targeted lysine residues at the Il9 promoter.
Figure 6.

Gcn5 inhibition reduces IL-9 production
A–C, Naive CD4+T cells from wild-type mice were cultured under Th9 conditions for 5d. Differentiated cells were then transfected with control or Gcn5-specific siRNA. After transfection cells were rested overnight with IL-2 and were then stimulated with plate-bound anti-CD3 (2μg/ml) for 6h for isolating RNA or for 24h to harvest cell free supernatant for ELISA (A, C) or kept unstimulated for chromatin immunoprecipitation (B). D–E, ChIP assay was performed from Th9 cells transfected with Gcn5 siRNA as in (A) for Gcn5, PU.1, IRF4, AcH3K9/18, AcH4K5 and AcH4K8 followed by quantitative PCR with primer for Il9 CNS1. ChIP data are presented with control Ig values subtracted from the specific Ig percent input values. Data are average ± S.D of 4–6 mice from 2–3 experiments. * p<0.05, ** p<0.01.
Discussion
Transcription factors play pivotal roles in the development of distinct T helper subsets. Expression of lineage-specific transcription factors is the hallmark of effector T cells. Our previous work demonstrated that PU.1 promotes the development of Th9 cells (5). The present report addresses the mechanisms by which PU.1 induces IL-9 in Th9 cells.
PU.1 regulates the expression of various myeloid and lymphoid genes and its functional domains have been characterized. In the N-terminus, multiple functional domains have been identified (8, 32–34). The PEST domain, enriched in proline, glutamic acid, serine, and threonine, may be important for regulating protein stability (35, 36), though in most reports, the PEST domain was not required for transactivation (8, 32, 33, 37, 38). In this report, we document that the acidic transactivation domain was also required for IL-9 induction. The PU.1 ETS domain is located at the C-terminus and mediates DNA binding and interactions with multiple proteins (39–41). In Th9 cells, the ETS domain of PU.1 has two functions; it limits Th2 responses by interacting with GATA3, but also binds directly to Il9 to facilitate chromatin remodeling and transactivation (5, 11, 12). Thus, PU.1 uses multiple domains to regulate the Th9 phenotype.
One likely functional role of the activation domain is for PU.1-mediated recruitment of histone modifying enzymes to the Il9 gene. Th9 cells had the highest total acetylation of both histone H3 and H4 compared with that of other T helper subsets, and total H3 acetylation was particularly affected by the absence of PU.1 (5). The interactions of PU.1 with HAT proteins have not been extensively examined, although previous studies suggested that PU.1 interacts with the histone acetyltransferases p300, Tip60, and MOZ (23, 42, 43). We observed differential recruitment of HAT molecules to the Il9 promoter in the absence of PU.1 and diminished histone acetylation at specific residues (Fig. 3 and 4A). The specificity of PU.1 effects on histone acetylation at Il9 was evident in reduced acetylation of H3K9/18, H3K14, H4K5, H4K8 and H4K16 residues in PU.1-deficient Th9 cells, although acetylation of histone H3K27, and H3K36 remained unaltered (Fig. 4). It is possible that PU.1 can mediate other histone modifications including lysine methylation. PU.1 has been shown to repress GATA-1 to block the erythroid differentiation program by recruiting histone methyltransferase Suv39h thereby causing methylation of histone H3K9 (44). However, histone methylation of H3K27 was found to be PU.1-independent (5).
The balance between histone acetylation and histone deacetylation controlled by HATs and HDACs, respectively, contributes to regulated gene expression. HDAC activity has been shown to regulate T cell development and function (45). T cell-specific deletion of HDAC1 results in enhanced IL-4 production from in vitro cultured Th2 cells, and increased allergic inflammation (46). It is possible that enhanced HDAC binding to Il9 in the absence of PU.1 contributes to reduced Il9 transcription (Fig. 1B). HDAC inhibition creates a hyperacetylated state, however, not all gene expression is increased (17, 18). PU.1 is regulated by HDAC inhibition, but the effects may be cell-type specific. In macrophage and pro-T cell lines, HDAC inhibition resulted in loss of PU.1 expression (47). Consistent with our results, the HAT inhibitor, curcumin, reduced PU.1 expression in Th9 cultures (48). We observed enhanced IL-9 production after HDAC inhibition along with increased Sfpi1 expression and enhanced PU.1 binding to Il9 (Fig. 1). Irf4 expression remained unchanged, although enhanced accessibility of IRF4 to Il9 was observed (Fig. 1). These results suggest that by regulating histone acetylation, PU.1 also affects the binding of additional transcription factors to the Il9 locus.
In the absence of PU.1, the association of Gcn5 and PCAF, two proteins found in multi-protein chromatin remodeling complexes (20), with the Il9 locus was decreased or eliminated. Previous studies have shown that PU.1 interacts with p300 in B cells (23). We observed increased p300 association at the Il9 locus in the absence of PU.1, suggesting that PU.1 is not critical for p300 binding to Il9, and that other transcription factors may mediate p300 recruitment in Th9 cells. The expression of Gcn5 and PCAF do not change upon the loss of PU.1 (Fig. 3B), suggesting that the decreased association of Gcn5 and PCAF with the Il9 locus contributes to reduced Il9 transcription. We further demonstrate in this report that DNA-bound PU.1 associates directly with Gcn5, but not PCAF (Fig. 5). Gcn5 functionally contributed to Il9 expression, since IL-9 production was significantly diminished when Gcn5 expression was reduced (Fig. 6). However, diminished Gcn5 expression did not change IL-10 and IL-21 production demonstrating that, in Th9 cells, the Il9 locus is a specific target of Gcn5. This is also consistent with our previous data that Th9 production of IL-10 is independent of PU.1 (5). Inhibition of Gcn5 expression alone does not alter PU.1 and IRF4 binding to the Il9 gene (Fig. 6D).
The present study highlights the structural requirements of PU.1 in regulating the Il9 gene, the unique chromatin modifications associated with PU.1-dependent Il9 gene expression, and the histone acetyltransferases required for those modifications. Based on our data, we propose the following mechanism. PU.1 binds to the Il9 promoter in developing Th9 cells. This results in complex formation and recruitment of Gcn5-containing chromatin remodeling complexes, as well as an indirect recruitment of PCAF, leading to increased histone acetylation. In the absence of PU.1, there is decreased association with a subset of HATs, and increased HDAC association. Gcn5 is critical for regulating histone acetylation at the promoter, and although it is not required for PU.1 or IRF4 binding to Il9, Gcn5-dependent histone acetylation likely increases binding of T cell receptor-induced factors such as NF-κB or NFAT that are important for Il9 gene expression following antigen receptor stimulation (27) or may facilitate the function of general transcription factors including RNA polymerase II (49). Together, our data provide a mechanism for PU.1-dependent Il9 regulation in developing Th9 cells. Future work will define how PU.1 interacts with additional transcription factors at the Il9 locus, and how these interactions contribute to IL-9 production during inflammatory disease.
Acknowledgements
The authors thank Drs. J. Sun, Q. Yu, S. Goenka, and D. Pham for helpful comments on the manuscript. We also thank Stephen Nutt for providing Sfpi1 conditional mutant mice.
This work was supported by Public Health Service grant AI057459.
Footnotes
Abbreviations: HAT, histone acetyltransferase; HDAC, histone deacetylase; PCAF, p300/CBP associated factor.
Disclosures The authors have no financial interests related to this work.
REFERENCES
- 1.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, Khoury S, Oukka M, Kuchroo VK. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat Immunol. 2008;9:1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, Martin B, Wilhelm C, Stockinger B. Transforming growth factor-beta `reprograms' the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341–1346. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 4.Jabeen R, Kaplan MH. The symphony of the ninth: the development and function of Th9 cells. Curr Opin Immunol. 2012 doi: 10.1016/j.coi.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, Jabeen R, McKinley C, Ahyi AN, Han L, Nguyen ET, Robertson MJ, Perumal NB, Tepper RS, Nutt SL, Kaplan MH. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol. 2010;11:527–534. doi: 10.1038/ni.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goswami R, Jabeen R, Yagi R, Pham D, Zhu J, Goenka S, Kaplan MH. STAT6-dependent regulation of Th9 development. J Immunol. 2012;188:968–975. doi: 10.4049/jimmunol.1102840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Staudt V, Bothur E, Klein M, Lingnau K, Reuter S, Grebe N, Gerlitzki B, Hoffmann M, Ulges A, Taube C, Dehzad N, Becker M, Stassen M, Steinborn A, Lohoff M, Schild H, Schmitt E, Bopp T. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity. 2010;33:192–202. doi: 10.1016/j.immuni.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 8.Klemsz MJ, Maki RA. Activation of transcription by PU.1 requires both acidic and glutamine domains. Mol Cell Biol. 1996;16:390–397. doi: 10.1128/mcb.16.1.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher RC, Olson MC, Pongubala JM, Perkel JM, Atchison ML, Scott EW, Simon MC. Normal myeloid development requires both the glutamine-rich transactivation domain and the PEST region of transcription factor PU.1 but not the potent acidic transactivation domain. Mol Cell Biol. 1998;18:4347–4357. doi: 10.1128/mcb.18.7.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahyi AN, Chang HC, Dent AL, Nutt SL, Kaplan MH. IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J Immunol. 2009;183:1598–1606. doi: 10.4049/jimmunol.0803302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang HC, Zhang S, Thieu VT, Slee RB, Bruns HA, Laribee RN, Klemsz MJ, Kaplan MH. PU.1 expression delineates heterogeneity in primary Th2 cells. Immunity. 2005;22:693–703. doi: 10.1016/j.immuni.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 12.Chang HC, Han L, Jabeen R, Carotta S, Nutt SL, Kaplan MH. PU.1 regulates TCR expression by modulating GATA-3 activity. J Immunol. 2009;183:4887–4894. doi: 10.4049/jimmunol.0900363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 14.Cress WD, Seto E. Histone deacetylases, transcriptional control, and cancer. J Cell Physiol. 2000;184:1–16. doi: 10.1002/(SICI)1097-4652(200007)184:1<1::AID-JCP1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Lin Q, Wang W, Wade P, Wong J. Specific targeting and constitutive association of histone deacetylase complexes during transcriptional repression. Genes Dev. 2002;16:687–692. doi: 10.1101/gad.962502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Utley RT, Ikeda K, Grant PA, Cote J, Steger DJ, Eberharter A, John S, Workman JL. Transcriptional activators direct histone acetyltransferase complexes to nucleosomes. Nature. 1998;394:498–502. doi: 10.1038/28886. [DOI] [PubMed] [Google Scholar]
- 17.Luo RX, Dean DC. Chromatin remodeling and transcriptional regulation. J Natl Cancer Inst. 1999;91:1288–1294. doi: 10.1093/jnci/91.15.1288. [DOI] [PubMed] [Google Scholar]
- 18.Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- 19.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? Embo J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 21.Carotta S, Wu L, Nutt SL. Surprising new roles for PU.1 in the adaptive immune response. Immunol Rev. 2010;238:63–75. doi: 10.1111/j.1600-065X.2010.00955.x. [DOI] [PubMed] [Google Scholar]
- 22.Mak KS, Funnell AP, Pearson RC, Crossley M. PU.1 and Haematopoietic Cell Fate: Dosage Matters. Int J Cell Biol. 2011;2011:808524. doi: 10.1155/2011/808524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai Y, Srinivasan L, Perkins L, Atchison ML. Protein acetylation regulates both PU.1 transactivation and Ig kappa 3' enhancer activity. J Immunol. 2005;175:5160–5169. doi: 10.4049/jimmunol.175.8.5160. [DOI] [PubMed] [Google Scholar]
- 24.Dakic A, Metcalf D, Di Rago L, Mifsud S, Wu L, Nutt SL. PU.1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J Exp Med. 2005;201:1487–1502. doi: 10.1084/jem.20050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perumal NB, Kaplan MH. Regulating Il9 transcription in T helper cells. Trends Immunol. 2011;32:146–150. doi: 10.1016/j.it.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaplan MH, Glosson NL, Stritesky GL, Yeh N, Kinzfogl J, Rohrabaugh SL, Goswami R, Pham D, Levy DE, Brutkiewicz RR, Blum JS, Cooper S, Hangoc G, Broxmeyer HE. STAT3-dependent IL-21 production from T helper cells regulates hematopoietic progenitor cell homeostasis. Blood. 2011;117:6198–6201. doi: 10.1182/blood-2011-02-334367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jash A, Sahoo A, Kim GC, Chae CS, Hwang JS, Kim JE, Im SH. Nuclear Factor of Activated T Cells 1 (NFAT1)-induced Permissive Chromatin Modification Facilitates Nuclear Factor-kappaB (NF-kappaB)-mediated Interleukin-9 (IL-9) Transactivation. J Biol Chem. 2012;287:15445–15457. doi: 10.1074/jbc.M112.340356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu W, Edmondson DG, Evrard YA, Wakamiya M, Behringer RR, Roth SY. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat Genet. 2000;26:229–232. doi: 10.1038/79973. [DOI] [PubMed] [Google Scholar]
- 29.Kuo MH, Brownell JE, Sobel RE, Ranalli TA, Cook RG, Edmondson DG, Roth SY, Allis CD. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature. 1996;383:269–272. doi: 10.1038/383269a0. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Bone JR, Edmondson DG, Turner BM, Roth SY. Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. Embo J. 1998;17:3155–3167. doi: 10.1093/emboj/17.11.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Hagemeier C, Bannister AJ, Cook A, Kouzarides T. The activation domain of transcription factor PU.1 binds the retinoblastoma (RB) protein and the transcription factor TFIID in vitro: RB shows sequence similarity to TFIID and TFIIB. Proc Natl Acad Sci U S A. 1993;90:1580–1584. doi: 10.1073/pnas.90.4.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kominato Y, Galson D, Waterman WR, Webb AC, Auron PE. Monocyte expression of the human prointerleukin 1 beta gene (IL1B) is dependent on promoter sequences which bind the hematopoietic transcription factor Spi-1/PU.1. Mol Cell Biol. 1995;15:58–68. [PMC free article] [PubMed] [Google Scholar]
- 34.Shin MK, Koshland ME. Ets-related protein PU.1 regulates expression of the immunoglobulin J-chain gene through a novel Ets-binding element. Genes Dev. 1993;7:2006–2015. doi: 10.1101/gad.7.10.2006. [DOI] [PubMed] [Google Scholar]
- 35.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 36.Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- 37.Erman B, Sen R. Context dependent transactivation domains activate the immunoglobulin mu heavy chain gene enhancer. Embo J. 1996;15:4665–4675. [PMC free article] [PubMed] [Google Scholar]
- 38.Nishiyama C, Nishiyama M, Ito T, Masaki S, Masuoka N, Yamane H, Kitamura T, Ogawa H, Okumura K. Functional analysis of PU.1 domains in monocyte-specific gene regulation. FEBS Lett. 2004;561:63–68. doi: 10.1016/S0014-5793(04)00116-4. [DOI] [PubMed] [Google Scholar]
- 39.Karim FD, Urness LD, Thummel CS, Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA, Gunther CV, Nye JA, et al. The ETS-domain: a new DNA-binding motif that recognizes a purine-rich core DNA sequence. Genes Dev. 1990;4:1451–1453. doi: 10.1101/gad.4.9.1451. [DOI] [PubMed] [Google Scholar]
- 40.Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA. The macrophage and B cell-specific transcription factor PU.1 is related to the ets oncogene. Cell. 1990;61:113–124. doi: 10.1016/0092-8674(90)90219-5. [DOI] [PubMed] [Google Scholar]
- 41.Nagulapalli S, Pongubala JM, Atchison ML. Multiple proteins physically interact with PU.1. Transcriptional synergy with NF-IL6 beta (C/EBP delta, CRP3) J Immunol. 1995;155:4330–4338. [PubMed] [Google Scholar]
- 42.Hlubek F, Lohberg C, Meiler J, Jung A, Kirchner T, Brabletz T. Tip60 is a cell-type-specific transcriptional regulator. J Biochem. 2001;129:635–641. doi: 10.1093/oxfordjournals.jbchem.a002901. [DOI] [PubMed] [Google Scholar]
- 43.Katsumoto T, Aikawa Y, Iwama A, Ueda S, Ichikawa H, Ochiya T, Kitabayashi I. MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 2006;20:1321–1330. doi: 10.1101/gad.1393106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stopka T, Amanatullah DF, Papetti M, Skoultchi AI. PU.1 inhibits the erythroid program by binding to GATA-1 on DNA and creating a repressive chromatin structure. Embo J. 2005;24:3712–3723. doi: 10.1038/sj.emboj.7600834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32:335–343. doi: 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, Zhang Y, Rembold M, Gaisberger M, Hartl A, Epstein MM, Matthias P, Seiser C, Ellmeier W. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol. 2010;185:3489–3497. doi: 10.4049/jimmunol.0903610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laribee RN, Klemsz MJ. Loss of PU.1 expression following inhibition of histone deacetylases. J Immunol. 2001;167:5160–5166. doi: 10.4049/jimmunol.167.9.5160. [DOI] [PubMed] [Google Scholar]
- 48.Ramming A, Druzd D, Leipe J, Schulze-Koops H, Skapenko A. Maturation-related histone modifications in the PU.1 promoter regulate Th9-cell development. Blood. 2012;119:4665–4674. doi: 10.1182/blood-2011-11-392589. [DOI] [PubMed] [Google Scholar]
- 49.Sanso M, Vargas-Perez I, Quintales L, Antequera F, Ayte J, Hidalgo E. Gcn5 facilitates Pol II progression, rather than recruitment to nucleosome-depleted stress promoters, in Schizosaccharomyces pombe. Nucleic Acids Res. 2011;39:6369–6379. doi: 10.1093/nar/gkr255. [DOI] [PMC free article] [PubMed] [Google Scholar]