

Abstract
Early secreted antigenic target of 6 kD (ESAT-6) of Mycobacterium tuberculosis is a T-cell antigen that is a potential vaccine candidate, but it is also a virulence factor that mediates pathogenicity. To better understand the effects of ESAT-6 on the immune response, we studied the effect of ESAT-6 on human dendritic cells (DCs). Peripheral blood monocytes were treated with GM-CSF and IL-4 to yield immature DCs, which were matured by addition of lipolysaccharide (LPS) and CD40 ligand (CD40L), with or without ESAT-6. ESAT-6 inhibited LPS/CD40L-induced DC expression of co-stimulatory molecules, reduced DC-stimulated allogeneic T cell proliferation and IL-2 and IFN-γ production, and enhanced IL-17 production. ESAT-6-treated DCs also increased IL-17 and reduced IFN-γ production by M. tuberculosis-specific autologous T cells. ESAT-6 inhibited LPS/CD40L-induced DC production of IL-12 and enhanced that of IL-23 and IL-1β, without affecting secretion of TNF-α, IL-6 or IL-8 through specific interaction with iDCs. The effects of ESAT-6 were not mediated through cAMP or p38 mitogen-activated protein kinase. Medium from ESAT-6-conditioned DCs increased IL-17 and reduced IFN-γ production by T cells stimulated with anti-CD3 plus anti-CD28, and ESAT-6-induced IL-17 production was blocked by neutralizing both IL-23 and IL-1β. ESAT-6 reduced LPS/CD40L-stimulated transcription of IL-12p35 and enhanced that of IL-23p19 through inhibition of interferon regulatory factor-1 and upregulation of activating transcription factor-2 and c-Jun, transcriptional regulators of IL-12p35 and IL-23p19, respectively. We conclude that ESAT-6 increases DC production of IL-23 and IL-1β, while inhibiting that of IL-12, thus enhancing Th17 at the expense of protective Th1 responses.
Keywords: Human, tuberculosis, dendritic cells, T cells, cytokines, ESAT-6
Introduction
Mycobacterium tuberculosis infects more than one-third of the world’s population, causing an estimated 1.8 million deaths in 2009 worldwide (1), accompanied by a staggering economic burden, especially in developing countries. Vaccination is the most cost-effective strategy for control and eventual elimination of tuberculosis. However, the most widely used tuberculosis vaccine, attenuated M. bovis Bacille Calmette Guerin (BCG), provides some protection against the most severe forms of childhood tuberculosis but does not prevent disease in adults, who comprise the majority of tuberculosis cases (2, 3). Therefore, development of an effective vaccine is essential for tuberculosis control, and depends in part on a better understanding of host-pathogen interactions.
Early secreted antigenic target of 6 kDa (ESAT-6) is a potent T-cell antigen identified in the short-term culture filtrate of M. tuberculosis (4, 5). ESAT-6-based vaccines confer protection against tuberculosis in animal models (6–9), and several such vaccines are either in clinical trials or undergoing preclinical development (10, 11). However, substantial evidence also indicates that ESAT-6 is a virulence factor. The gene encoding ESAT-6, Rv3875 (12), is in the region of difference (RD)1, which is present in many pathogenic mycobacteria, including M. tuberculosis and M. bovis, but not in attenuated BCG (13). ESAT-6 lyses alveolar epithelial cells and macrophages (14, 15), favoring intercellular spread of M. tuberculosis (15, 16), and can destabilize phagolysosomes, perhaps allowing M. tuberculosis and its products to escape the phagosome (17). Therefore, delineating the role of ESAT-6 in the immunopathogenesis of tuberculosis is important for optimizing ESAT-6 based-vaccines.
Previously, we demonstrated that ESAT-6 directly inhibits human T cell IFN-γ production (18) through a process that requires activation of p38 mitogen-activated protein kinase (MAPK) (19). However, the effect of ESAT-6 on human dendritic cells (DCs) has not been investigated. DCs are crucial in bridging innate and adaptive immunity, and play an essential role in initiation and maintenance of balanced T cell responses to infection (20). Upon encounter with pathogens, immature DCs (iDCs) in the local tissue take up the pathogen and mature after recognizing pathogen associated molecular patterns through their pattern recognition receptors, such as Toll-like receptors (TLRs), which induce increased expression of costimulatory molecules, such as CD80 and CD86, and production of cytokines, including interleukin (IL)-12, IL-23 and IL-β. Pathogen-experienced mature DCs then migrate to the local draining lymph nodes and initiate T cell responses by presenting microbial antigens in the context of costimulatory molecules and cytokines. IL-12 favors expansion of Th1 cells that produce interferon (IFN)-γ, and IL-23 and IL-1β induce development of Th17 cells that produce IL-17 (21). Studies in gene-deleted mice and in humans have clearly demonstrated that the IL-12/Th1 pathway is essential for immunity against tuberculosis (22–26). In contrast, the role of IL-23 and Th17 cells in protection against tuberculosis is more complex and controversial. One study found that the absence of IL-17 did not increase susceptibility to tuberculosis (27), whereas another showed that a gene deletion of IL-17A markedly increased bacillary burdens and impaired granuloma formation (28). IL-17 contributed significantly to vaccine-induced protection against challenge with M. tuberculosis (29), but also mediated tissue damage after repeated BCG vaccination in M. tuberculosis-infected mice (30) and may provide minimal protection during chronic infection (31).
M. tuberculosis infection can alter the normal process of DC maturation (32, 33), which is crucial for priming antigen-specific T cells. In this report, we studied the immune regulatory effects of ESAT-6 on human DCs. We found that ESAT-6 inhibits production of IL-12 but promotes production of IL-23 and IL-1β through inhibition of interferon regulatory factor (IRF)-1 and enhancement of activator protein (AP)-1 transcription factors. ESAT-6-treated DCs favor T-cell production of IL-17 over IFN-γ, providing a potentially novel mechanism for modulation of host immune responses by M. tuberculosis through its secreted proteins.
Materials and Methods
Human Subjects
Blood samples were obtained from 21 healthy donors without prior M. tuberculosis infection and 8 donors with latent tuberculosis infection, based on QuantiFERON-TB GOLD test results. All studies were approved by the Institutional Review Board of the University of Texas Health Science Center at Tyler and signed consent forms from all study subjects were obtained before collection of blood samples.
M. tuberculosis culture and cell stimulation
Heat-killed M. tuberculosis Erdman (provided by Dr. Patrick Brennan, Colorado State University, Fort Collins, CO) and live M. tuberculosis H37Rv and its esat-6 (Rv3875) deletion mutant H37RvΔ3875 (provided by Dr. David Sherman, University of Washington, Seattle, WA) were used. H37Rv and H37RvΔ3875 were grown in Middlebrook 7H9 medium supplemented with 0.2% glycerol and 10% ADC enrichment (Remel). Logarithmically growing cultures were sonicated briefly and centrifuged at 800 rpm for 10 min to eliminate clumped mycobacteria. The upper part of the culture was collected and the bacterial concentration was determined by measuring optical density (OD600), using the formula: 1 OD600=3 × 108 colony-forming units (CFU)/ml, which was confirmed by plating serially diluted bacterial suspensions on 7H10 agar and counting CFU after 3 wks.
Preparation of recombinant ESAT-6
The recombinant plasmid containing Rv3875, encoding ESAT-6, was obtained through the TB Vaccine Testing and Research Materials Contract (Colorado State University), and recombinant ESAT-6 was prepared, as described previously (18). Analysis of recombinant ESAT-6 preparations with gel filtration chromatography by FPLC showed no protein aggregates, and > 95% of the protein formed a peak of approximately 24 kDa, probably representing ESAT-6 homodimers (results not shown).
Generation and culture of DCs, and detection of cytokines
PBMC were isolated by differential centrifugation of heparinized blood over Ficoll-Paque (GE Healthcare Life Science). CD14+ and CD4+ cells were purified from PBMC by positive immunomagnetic selection, and CD3+ cells were purified by negative selection (Human Pan T cell isolation kit) (all from Miltenyi Biotec). Cell purity was > 98%, as measured by immunolabeling and flow cytometry analysis with a FACSCalibur (BD Biosciences). To generate immature DCs, CD14+ cells were cultured at 106 cells/ml in RPMI 1640 (Invitrogen), supplemented with 10% heat-inactivated pooled human serum (Atlanta Biologicals), 100 units or 100 µg/ml penicillin and streptomycin, 1 mM sodium pyruvate, 0.1 mM MEM nonessential amino acids (all from Invitrogen), and 25 ng/ml human granulocyte-macrophage-colony-stimulating factor and IL-4 (both from R&D systems) for 3–5 d. The immature DCs were further stimulated with human CD40LT (Immunex, Seattle, Washington) at 2.5 µg/ml, plus LPS (Sigma) at 1 µg/ml, with or without ESAT-6, for 24 h. In some experiments, CD3+ cells were cultured at 2 × 106/ml with autologous or allogeneic DCs in 96-well flat-bottom plates for 3–5 d. Cell-free supernatants were collected and cytokine concentrations were measured by ELISA, using capture and detection antibodies for IFN-γ and TNF-α (BD Biosciences), and ELISA kits for IL-2, IL-12p70 (both from BD Biosciences), IL-17, IL-8 (both from R&D systems), IL-1β, IL-23 (both from Mabtech), and IL-6 (BioLegend). The detection limits of these kits are 15–25 pg/ml.
In some experiments, iDC were infected with live H37Rv and H37RvΔ3875 at a multiplicity of infection of 10 for 4 h, and free bacteria were washed off with prewarmed culture media, before stimulation with LPS plus CD40LT. The concentrations of IL-12p70, IL-23 and IL-1β in 24 h culture supernatants were measured by ELISA, as described above.
Flow cytometry
Expression of CD80, CD86, HLA-DR and CD83 was measured by incubating DCs with FITC-anti-CD80 (clone 2D10), PE-anti-CD86 (clone IT2.2), FITC-anti-HLA-DR (clone L243) (all from Biolegend) or FITC-anti-CD83 (clone HB15) (eBioscience) on ice for 30 min. Interaction of ESAT-6 with DCs were examined by incubation of iDCs with Alexa Fluor 488-labeled ESAT-6 after confirming that labeled ESAT-6 inhibits T cell IFN-γ production with same potency as unlabeled ESAT-6. The specificity of interaction was examined by blocking experiments by incubating iDCs with increasing concentrations of unlabeled ESAT-6 or BSA as control prior to incubation with 20 µg/ml labeled ESAT-6. Flow cytometry analysis was performed with a FACSCalibur, using Flowjo software (Tree star).
Cell viability and proliferation assays
Cell viability was measured by the MTT cleavage assay (USB Corporation). Proliferation of CD4+ cells in response to allogeneic stimulation was measured by the CFSE dilution assay. Briefly, purified cells were labeled with CFSE (Invitrogen) as described (18) and incubated with allogeneic DCs in a 96-well flat-bottom plate. After 4 d, the cells were collected and proliferation was analyzed by flow cytometry.
Cytokine mRNA quantification by real-time PCR
Total RNA was extracted from 2.5 × 105 DCs with TRIzol reagent (Invitrogen), cDNA was synthesized and IL-12p35 (p35), IL-23p19 (p19) and IL-12/23p40 mRNA were quantified by minor modifications of our published methods (18). The relative quantity of each mRNA was calculated by the ΔΔCt method (34).
Depletion of ESAT-6
ESAT-6 was removed from the recombinant protein preparations, as described previously (18). Briefly, 500 µg of rESAT-6 was incubated with 250 µl of activated nickel resin in a 1.5 ml Eppendorf tube at room temperature. For sham depletion, ESAT-6 was incubated with unactivated nickel resin. After 30 min incubation, samples were centrifuged to pellet the resin, and the resin-free supernatants were collected as ESAT-6-depleted samples. The protein content of the samples was measured by the bicinchoninic acid assay. Samples were also subjected to SDS-PAGE, followed by Coomassie blue staining and Western blotting with anti-ESAT-6 (HYB 76-8, provided by Dr. Peter Andersen, Statens Seruminstitut, Copenhagen, Denmark).
The effect of ESAT-6-conditioned DC medium on T cell cytokine production
iDCs were stimulated with LPS plus CD40LT, with or without ESAT-6, for maturation. Twenty-four hrs later, the cell-free supernatants were harvested and stored at −20°C. Negatively selected CD3+ T cells from PBMC of healthy donors were resuspended in RPMI-1640 with 10% heat-inactivated pooled human AB serum and plated at 4 × 105 cells in 100 µl/well in a 96-well flat bottom culture plate precoated with anti-CD3 and anti-CD28. Another 100 µl of conditioned media was added to each well. The cells were incubated at 37°C and 5% CO2. Forty-eight hrs later, cell-free supernatants were harvested and IFN-γ and IL-17 levels were measured by ELISA. For cytokine neutralization experiments, the CD3+ cells with conditioned medium were incubated with anti-IL-23 or anti-IL-1β at 10 μg/ml for one hour before cultured in a 96-well plate precoated with anti-CD3 plus anti-CD28.
Preparation of DC nuclear protein extracts
DCs were incubated in a 12-well plate at 1 × 106 cells/ml, with or without 20 µg/ml ESAT-6 for 1 h, followed by stimulation with LPS plus CD40LT for 4 h. DCs were then collected by scraping with rubber policemen, washed twice with pre-chilled PBS, and cytosolic and nuclear protein extracts were prepared, as described (35). The protein concentration was measured by the bicinchoninic acid assay, and the extracts were kept at −70°C in aliquots until use.
Detection of transcription factors by Western blotting
Twenty-five µg of nuclear and cytosolic protein extracts of DCs were resolved by 10% SDS-PAGE in reducing conditions, electroblotted to a nitrocellulose membrane, and expression and phosphorylation of the transcription factors, IRF-1, activating transcription factor (ATF)-2 and c-Jun AP-1 were evaluated by Western blotting, as described previously (18), using antibodies against IRF-1 (H-205), ATF-2 (c-19), and c-Jun AP-1 (H79), all from Santa Cruz Biotechnology. To control for protein loading, the blot was stripped, and expression of GAPDH was evaluated by immunoblotting (FL-335, Santa Cruz Biotechnology).
Detection of DNA bindng activities of AP-1 transcription factors by electrophoretic mobility shift assay
The promoter binding activity of AP-1 transcription factors in DC was evaluated by electrophoretic mobility shift assays, using our published methods (35). DC nuclear protein extracts were used as transcription factor sources and the probe was a radio-labeled AP-1 binding site of the human p19 promoter, which corresponds to the nucleotides from −228 to −198, relative to the p19 transcription start site. To characterize the binding specificity, we used unlabeled oligo DNAs: consensus AP-1 binding site, human p19 AP-1 binding site and NF-κB consensus binding site for competition assays. To identify proteins in DNA binding complexes, we used Abs against ATF-2 (polyclonal C-19 and monoclonal F2BR-1), anti-c-Jun (H79), anti-cyclic AMP response element binding protein1 (CREB) mAb, and control IgG, all from Santa Cruz Biotechnology.
Detection of recruitment of IRF-1 to the IL-12p35 promoter in live DC by chromatin immunoprecipitation
Immature DCs were stimulated with LPS plus CD40LT, with or without ESAT-6, for 6 h, and then treated with 1% formaldehyde at room temperature with shaking. After 10 min, the cells were treated with 2 M glycine to neutralize free formalydehyde. The preparation of DC chromatin and the chromatin immunoprecipitation assay was performed as described previously (36), using anti-IRF-1 (H-205) for immunoprecipitation and a Chip Assay Kit (Millipore). The amount of IRF-1-bound IL-12p35 promoter in the samples was determined by PCR, using specific primer sets: forward, 5’GAACATTTCGCTTTCATTTTGGG3’; reverse, 5’ACTCTGGTCTCTTGCTTTCT3’, which yields a 187 bp fragment of the p35 promoter containing the IRF-1 binding site. To control for equal sample loading, the same DNA fragment of p35 was amplified by PCR from an equal amount of chromatin supernatants, prior to immunoprecipitation.
Statistical analysis
Student’s t test was used to evaluate differences between groups. p values < 0.05 were considered to be statistically significant.
Results
ESAT-6 inhibits DC maturation
Because DCs are essential for eliciting protective immunity against M. tuberculosis (37), and this bacterium targets DC functions (38, 39), we hypothesized that ESAT-6 may play a role in this process. To test this hypothesis, we first generated iDCs by culturing CD14+ monocytes with granulocyte-macrophage-colony-stimulating factor and IL-4 for 5 days (40). We stimulated iDC through TLR4 and CD40 with their respective ligands, lipopolysaccharide (LPS) and CD40ligand trimer (CD40LT), which induce DC maturation (41). LPS/CD40LT treatment yielded mature DCs (mDCs), which showed upregulation of the co-stimulatory molecules, CD86 and CD80, MHC class II molecule HLA-DR, and the DC maturation marker, CD83 (Fig. 1). The presence of ESAT-6 during maturation diminished expression of these surface markers (Fig. 1). When human DCs were purified directly from PBMC by positive selection, using magnetic beads conjugated with anti-CD304 (BDCA-4/neuropilin) (Miltenyi Biotec) and treated with LPS/CD40LT, with or without ESAT-6, ESAT-6 inhibited upregulation of CD86, CD80 and HLA-DR (data not shown). Because of the extremely low numbers of primary DCs, further experiments were performed with monocyte-derived DCs.
Figure 1. ESAT-6 inhibits DC maturation.

iDCs from four donors were stimulated with LPS/CD40LT for maturation, with or without 20 µg/ml of ESAT-6. Twenty-four hrs post-stimulation, cells were stained with different antibodies to costimulatory molecules and maturation markers, and analyzed by flow cytometry.
ESAT-6 inhibits Th1 and enhances Th17 responses induced by DCs
To evaluate the functional effects of ESAT-6 on DC maturation, we examined the allostimulatory capacity of DCs matured in the presence of ESAT-6 by co-culturing with allogeneic T cells. iDCs from four donors were stimulated with LPS/CD40LT, with or without ESAT-6, and cultured with CFSE-labeled allogeneic CD4+ T cells. ESAT-6 reduced the capacity of DCs to stimulate proliferation by allogeneic T cells, particularly at low DC:T cell ratios (Figs. 2A and B). ESAT-6 also markedly inhibited T cell production of IFN-γ by five donors (Fig. 2C), and had similar effects on IL-2 production (Fig. 2D). DCs matured in the presence of ESAT-6 increased T-cell IL-17 production, with marked differences at higher DC:T cell ratios (Fig. 2E). Thus, iDC matured in the presence of ESAT-6 reduced proliferation and Th1 cytokine production by allogeneic T cells, but enhanced production of IL-17. To determine if ESAT-6 had similar effects on T cells stimulated with mycobacterial antigens, we prepared iDCs and autologous CD4+ T cells from four donors with latent tuberculosis infection. ESAT-6-treated DCs induced less IFN-γ and more IL-17 production by T cells, stimulated with heat-killed M. tuberculosis or infected with live bacilli (Fig. 3). These results together suggested that DCs matured with ESAT-6 support Th17 cells at the expense of Th1 cells.
Figure 2. DC matured in the presence of ESAT-6 inhibits production of Th1 cytokines and enhances production of IL-17 by allogeneic T cells.

iDCs generated from four healthy donors were matured with LPS/CD40LT, with or without 20 µg/ml of ESAT-6. After extensive washing to remove free ESAT-6 and LPS/CD40LT, varying number of mDC ranging from 4 × 103 to 4 × 104 were incubated with 4 × 105 CFSE-labeled, allogeneic CD4+ T cells at indicated ratios. A. After 4 days, the percentages of proliferating cells were determined by flow cytometry. A representative result is shown. B. The means and SEMs of the percentage of proliferated CD4+ cells are shown from four experiments. C–E. The supernatants from cocultures of mDCs and allogeneic CD4+ T cells from five donors were collected after 24 h for measurement of IL-2 (D) and after 96 h for measurement of IFN-γ (C) and IL-17 (E) by ELISA. Means and SEMs are shown. *, p < 0.05 (B–E), compared to cells stimulated with LPS/CD40LT at the same DC:T cell ratio.
Figure 3. DCs matured with ESAT-6 stimulate IL-17 and inhibit IFN-γ production by M. tuberculosis-responsive T cells.

iDCs from four donors with latent tuberculosis infection were matured with LPS/CD40LT, with or without ESAT-6, for 24 h. The cells were then washed and incubated with autologous CD3+ cells at different ratios, and stimulated with heat-killed Mtb at 2.5 µg/ml (left panels) or infected with H37Rv at a MOI of 20 (right panels). Forty-eight hrs later, the supernatants were harvested and IFN-γ and IL-17 levels were measured by ELISA. Means and SEMs are shown. *, p < 0.05, compared to cells matured with LPS/CD40LT at the same DC:T cell ratio.
ESAT-6 differentially regulates DC cytokine production
Because DCs matured in the presence of ESAT-6 increased IL-17 and reduced IFN-γ production by T cells, and cytokines produced by DCs strongly affect T cell differentiation (42), we examined the effect of ESAT-6 on DC cytokine production. Stimulation of iDC with LPS/CD40LT induced robust production of IL-12, IL-23, TNF-α, IL-6 and IL-8 but minimal amounts of IL-1β (Figs. 4A–F). ESAT-6 inhibited DC maturation-induced IL-12 production and induced production of IL-1β in a dose-dependent manner (Figs. 4A and C), and increased IL-23 production (Fig. 4B). However, ESAT-6 did not affect secretion of TNF-α, IL-6 or IL-8 (Figs. 4D–F). ESAT-6 alone, in the absence of LPS/CD40LT, did not stimulate production of any cytokines except for minimal levels of IL-6 and IL-8. Because ESAT-6 activates the inflammasome and caspase-1-dependent production of IL-1β in monocytes (43), we incubated iDC from 4 donors with a caspase-1 inhibitor before treatment with LPS/CD40LT and ESAT-6. ESAT-6 increased IL-1β production in a dose-dependent manner, but this was markedly reduced by caspase-1 inhibition (Fig. 4G), demonstrating that this effect requires caspase-1.
Figure 4. DCs matured in the presence of ESAT-6 produce less IL-12 and more IL-23 and IL-1β.

iDCs were stimulated with LPS/CD40LT, with or without ESAT-6, and some cells were incubated with ESAT-6 only. Twenty-four hrs post stimulation, the supernatants were collected and levels of IL-12p70 (A), IL-23 (B), IL-1β (C), TNFα (D), IL-6 (E) and IL-8 (F) were measured by ELISA. G. Some cells were treated with a caspase 1 inhibitor (20 µM) prior to stimulation with LPS/CD40LT and ESAT-6. Twenty-four h later, supernatants were collected and IL-1β levels were measured. The far right grey bar represents cells treated with DMSO only as a vehicle control. For all panels, means and SEMs are shown. *, p < 0.05, compared to cells stimulated with LPS/CD40LT only.
The effects of ESAT-6 on DC cytokine production are not mimicked by other M. tuberculosis antigens and are not due to contaminants in recombinant ESAT-6
To determine if the effects of ESAT-6 on DC cytokine production were specific for this mycobacterial protein, we tested antigen 85A (Ag85A), an immunogenic secreted protein of M. tuberculosis (44). Using iDCs from four donors, ESAT-6 reduced IL-12 secretion and increased IL-23 and IL-1β production, but Ag85 did not (Fig. 5A). To further test if the effect of ESAT-6 on DC cytokine production is not due to nonspecific effect of ESAT-6 aggregates, we dissolved both recombinant ESAT-6 and Ag85A with organic solvent DMSO, and tested the effects of solubilized proteins on cytokine production by iDCs from four donors. DMSO treated ESAT-6 but not Ag85A inhibited IL-12 production by iDCs in response to maturation stimulation in the same manner as non DMSO treated ESAT-6, further suggesting the specific effect of ESAT-6 on DC cytokine production (data not shown).
Figure 5. The effects of ESAT-6 on DC cytokine production are not mimicked by Ag85A and are not due to contaminants in recombinant ESAT-6 or cell death.

A. iDCs were matured by stimulation with LPS/CD40LT, with or without ESAT-6 or Ag85A at different concentrations as indicated. Twenty-four h later, culture supernatants were collected and IL-12p70, IL-23 and IL-1β levels were measured by ELISA. B. iDCs were stimulated with LPS/CD40LT, in the presence of ESAT-6 preparations, depleted or sham-depleted of ESAT-6 with a nickel column, or with a depletion buffer without ESAT-6. Twenty four h later, supernatants were harvested and IL-12 levels were measured. C. iDCs were stimulated with LPS/CD40LT, with or without ESAT-6, for 24 h, and cell viability was evaluated with a MTT assay. For all panels, means and SEMs are shown. *, p < 0.05, compared to cells stimulated with LPS/CD40LT only.
To confirm that the effects of ESAT-6 were not due to contaminants, we depleted ESAT-6 from recombinant protein preparations, using a nickel resin, as outlined in the methods. The depletion process removed 99% of total protein, as measured by the bicinchoninic acid protein assay, and confirmed by Western blot for ESAT-6 (data not shown). Depletion of ESAT-6 restored IL-12 production by iDC, but sham depletion had no effect (Fig. 5B), indicating that IL-12 inhibition was not due to contaminants. Because ESAT-6 lyses human lung epithelial cells and monocytic cells, probably through apoptosis (14, 45), and apoptotic cells inhibit DC IL-12 production (46), we determined whether ESAT-6 affects DC viability. The highest concentration of ESAT-6 used in our experiments (20 µg/ml), either alone or with LPS/CD40LT, did not reduce viability of iDCs after 24–96 h (Fig. 5C and data not shown), based on the MTT assay, indicating that ESAT-6 was not cytotoxic in our experimental system.
ESAT-6 does not affect DC cytokine production through cAMP or p38 MAPK
We considered the possibility that ESAT-6 reduced DC IL-12 production through increasing intracellular cyclic AMP (cAMP), as other bacterial toxins, such as adenylate cyclase toxin of Bordetella pertusis act through this mechanism (47, 48). However, blocking cAMP with the cAMP-specific chemical inhibitor, Rp-cAMP, did not affect ESAT-6-induced differential regulation of DC cytokine production (Supplementary Fig. 1), and treatment with ESAT-6 did not affect cAMP levels in iDC (data not shown), indicating that cAMP does not mediate the effects of ESAT-6 on DC cytokine production.
Next, we determined if p38 MAPK mediates the effects of ESAT-6 on DC cytokine production, because ESAT-6 inhibits T cell IFN-γ production through this signaling pathway (19). Consistent with previous reports (49, 50), SB203580, a specific p38 MAPK inhibitor, reduced LPS/CD40LT-stimulated DC IL-12 production. SB203580 further reduced ESAT-6 inhibition of IL-12 production (Supplementary Fig. 2), making it unlikely that ESAT-6 acted through p38 MAPK. The results were more definitive for IL-23 and IL-1β. In both cases, SB203580 did not affect cytokine production and did not abrogate the effects of ESAT-6 (Supplementary Fig. 2). Thus, p38 MAPK does not contribute to ESAT-6-mediated differential regulation of DC cytokine production.
ESAT-6 binds to DCs with specificity
Although DCs are known to take up non-self components, such as bacterial proteins, for antigen presentation, we tested if ESAT-6 interacts with DCs specifically by incubating iDCs from three different donors with Alexa Fluor 488-labeled ESAT-6. The results demonstrated that ESAT-6 binds to iDCs (Fig. 6A), and this binding is saturable (Fig. 6B), as successive doublings of ESAT-6 concentrations led to proportionately smaller increases in mean fluorescence intensities (MFI) of DC-bound labeled ESAT-6. Prior incubation of iDCs with increased concentrations of unlabeled ESAT-6 (Fig. 6C) but not BSA (Fig. 6D) progressively reduced binding of labeled ESAT-6 to iDCs with significant reduction at 160 µg/ml (from MFI of 55 to 22), suggesting that interaction of ESAT-6 with iDCs is specific and reversible. We could not achieve full blocking in binding since ESAT-6 at 320 µg/ml or above was toxic. In conclusion, ESAT-6 interacts with human iDCs specifically, providing mechanistic clues for specific effects of ESAT-6 on DC maturation and cytokine production.
Figure 6. ESAT-6 interacts DCs with specificity.
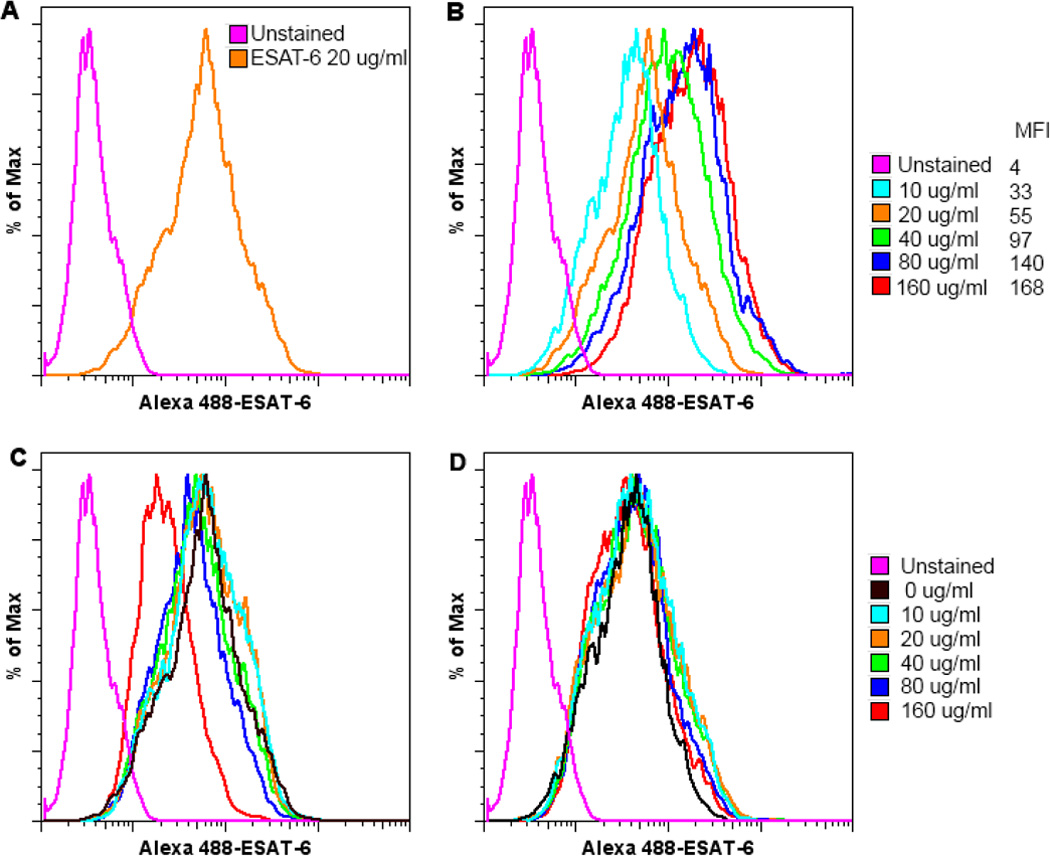
iDCs from three healthy donors were incubated with 20 µg/ml (A) or different concentrations of Alexa Fluor 488-labeled ESAT-6, as indicated (B). One hour later, the cells were washed and examined by flow cytometry. MFI indicates mean fluorescence intensities for corresponding concentrations of labeled ESAT-6. For blocking experiments, iDCs were incubated with increasing concentrations of unlabeled ESAT-6 (C) or BSA (D) as indicated for 30 min prior to incubation with 20 µg/ml labeled ESAT-6. The cells were washed and analyzed by flow cytometry. One representative result from three different experiments is shown for all panels.
ESAT-6-treated DCs favor T cell production of IL-17 through IL-23 and IL-1β
The data above (Figs. 2–4) show that ESAT-6-treated DCs enhanced T cell IL-17 and reduced IFN-γ production, and that ESAT-6 increased DC production of IL-23 and IL-1β, and reduced that of IL-12. To link these findings, we added media from LPS/CD40LT-treated iDC, with or without ESAT-6, to CD3+ cells from six donors, stimulated with anti-CD3 plus anti-CD28. T cells treated with conditioned media from unstimulated DCs induced low IL-17 levels (132 ± 22 pg/ml), which were increased slightly by medium from DCs stimulated with LPS/CD40LT or ESAT-6 alone (Fig. 7A). However, medium from DCs treated with LPS/CD40LT in the presence of ESAT-6 increased IL-17 levels 6-fold (739 ± 80 pg/ml). In contrast to the effects on IL-17, medium from LPS/CD40LT-treated DCs almost doubled IFN-γ levels compared to that with DC medium without stimulation, but medium from DCs treated with LPS/CD40LT in the presence of ESAT-6 significantly reduced IFN-γ concentrations close to the level of T cells with conditioned medium from unstimulated DC (Fig. 7B). Addition of neutralizing antibodies to IL-23 or IL-1β to conditioned media from DCs treated with ESAT-6 and LPS/CD40LT modestly reduced IL-17 production by activated CD3+ cells, and addition of both antibodies together decreased IL-17 concentrations by 70–80% (Fig. 7C), suggesting that IL-23 and IL-1β contributed significantly to the capacity of ESAT-6-treated DCs to elicit IL-17 secretion by T cells. These results together indicated that DCs matured in the presence of ESAT-6 generate a condition that favors expansion of Th17 over Th1 cells.
Figure 7. ESAT-6-treated DCs stimulate T cell IL-17 production through IL-23 and IL-1β.

Purified CD3+ cells from healthy donors were stimulated with plate-bound anti-CD3 plus anti-CD28 in a 96-well plate, with each well containing 100 µl of RPMI-1640 and 10% human serum and 100 µl of conditioned medium from DCs, matured under the conditions as indicated. Forty-eight h later, supernatants were harvested and IL-17 (A) and IFN-γ (B) levels were measured by ELISA. C, Purified CD3+ cells were cultured in 180 µl of RPMI-1640 with 10% human serum and 20 µl of conditioned medium from DCs, matured by LPS/CD40LT in the presence of ESAT-6. Some cells were treated with neutralizing Abs or isotype control IgG for 1 h, prior to stimulation with anti-CD3 plus anti-CD28 in a 96-well plate. Forty-eight h later, supernatants were harvested and IL-17 levels were measured by ELISA. Means and SEMs are shown for all three panels. *, p < 0.05, compared to the cells stimulated in the presence of conditioned medium from DC matured with LPS/CD40LT (A and B), or compared to the cells stimulated in the presence of control IgG (C).
ESAT-6 inhibits IL-12 production by DC stimulated through CD40 and multiple TLR
To determine if ESAT-6 affects DC cytokine production in response to TLR ligands other than TLR4, we stimulated iDC from three donors with nine different TLR ligands, together with CD40LT. Agonists for TLR1/2 (Pam3CSK4), TLR2 (heat-killed Listeria monocytogene), TLR5 (flagellin), TLR6/2 (FSL-1, or Pam2CGDPKHPKSF), and TLR8 (single-stranded RNA), induced significant levels of both IL-12p70 (Supplementary Fig. 3A) and IL-23 (data not shown). ESAT-6 significantly reduced IL-12 production by DC from six different donors in response to all five TLR ligands (Supplementary Fig. 3B), but did not clearly affect IL-23 production (data not shown). Therefore, ESAT-6 may inhibit IL-12 production by DC in response to multiple TLR agonists by targeting a common signaling pathway.
ESAT-6 inhibits transcription of p35 and enhances that of p19
IL-12 and IL-23 are heterodimeric cytokines, composed of the p35 and p19 polypeptides, respectively, and a shared p40 chain (51, 52). To determine if ESAT-6 regulated DC production of IL-12 and IL-23 through transcriptional regulation, we measured mRNA expression by real-time PCR. Consistent with previous reports (52, 53), LPS/CD40LT stimulation of iDC induced robust transcription of all three polypeptide genes. ESAT-6 markedly inhibited p35 transcripts (Fig. 8A) in a dose-dependent manner, greatly enhanced p19 mRNA expression (Fig. 8C), and did not affect that of p40 (Fig. 8B). These results support previous findings that expression of p35 and p19 are the rate limiting factors for antigen-presenting cell production of functional IL-12 and IL-23, respectively (54, 55), and that ESAT-6 reduced IL-12 and increased IL-23 production by DC through differentially regulating expression of these peptides.
Figure 8. ESAT-6 inhibits DC transcription of p35 and enhances that of p19.

iDCs were matured with LPS/CD40LT, with or without ESAT-6 at 20 µg/ml. Sixteen h later, the cells were collected and mRNA was quantified by real-time PCR for p35 (A), p40 (B), and p19 (C), after normalization for 18S rRNA content. Results are shown as the fold-change, relative to that of unstimulated DCs. Means and SEMs for four donors are shown. *, p < 0.05, compared to cells stimulated with LPS/CD40LT only.
ESAT-6 affects transcription factors that control p35 and p19 mRNA expression
To understand the mechanisms by which ESAT-6 affects mRNA expression of p35 and p19, we evaluated the effect of ESAT-6 on transcription factors that are downstream from the TLR signaling pathways and control transcription of these polypeptides. Interferon regulatory factor (IRF)-1 induces transcription of p35 by binding to the p35 promoter (56), but also inhibits p19 transcription by blocking RelA binding to the p19 κ-site (57). In contrast, STAT-3 enhances transcription of p19 through its promoter but inhibits that of p35 by preventing binding of c-Rel to the p35 promoter (58). The AP-1 transcription factors, activating transcription factor (ATF)-2 and c-Jun, positively regulate transcription of p19 (59).
IRF-1 expression in DCs was increased by stimulation with LPS/CD40LT, but reduced by addition of ESAT-6 during DC maturation (Fig. 9A). Furthermore, binding of IRF-1 to the p35 promoter in live DCs was reduced, based on chromatin immunoprecipitation (Fig. 9D). LPS/CD40LT increased expression of phospho-STAT3 in DCs, but ESAT-6 inhibited STAT3 phosphorylation (data not shown), indicating that STAT3 does not mediate the effects of ESAT-6 on transcription of p35 and p19. Phosphorylation of ATF-2 and c-Jun was increased by stimulation with LPS/CD40LT, and further enhanced by ESAT-6 (Fig. 9A). To determine if phosphorylation of these transcription factors correlates with promoter binding activity, we performed an electrophoretic mobility shift assay, using the AP-1 binding site of p19 as a probe. Stimulation of iDC with LPS/CD40LT increased AP-1 binding activity, and this was further enhanced by ESAT-6 (Fig. 9B). DNA-binding was specific, as excess unlabeled oligonucleotides with the consensus AP-1 sequence or the p19 AP-1 site blocked the formation of DNA-protein complexes, whereas the NF-κB consensus oligonucleotide did not (Fig. 9C). Anti-c-Jun super-shifted and anti-ATF-2 blocked the formation of this complex (Fig. 9C), suggesting that ATF-2 and c-Jun contribute to the increased DNA binding activity that accompanies iDC maturation. In summary, these results suggest that ESAT-6 induces differential transcription of p35 and p19 through inhibition of IRF-1 expression and upregulation of ATF-2 and c-Jun activation.
Figure 9. ESAT-6 differentially regulates expression and DNA-binding activities of IRF-1 and AP-1 transcription factors.

A. iDCs from four donors were stimulated with LPS/CD40LT, with or without ESAT-6. Four h later, total cell protein extracts were prepared, and Western blotting was performed to determine expression of IRF-1 and phosphorylation of the AP-1 transcription factors, ATF-2 and c-Jun. The blots were stripped and reblotted for expression of GAPDH as a protein loading control. A representative result is shown. B. iDCs from four donors were matured as in A, and DNA binding activity of the nuclear protein extracts was evaluated by electrophoretic mobility shift assays, using the radiolabeled AP-1 binding site of the p19 promoter as a probe. The DNA:protein complexes were resolved by 5% non-denaturing polyacrilamide gel electrophoresis and visualized by autoradiography. A representative result is shown. C. Nuclear extracts of DCs from four donors, matured with LPS/CD40LT and ESAT-6 were incubated with unlabeled double-stranded DNA or different IgGs, as indicated, for 30 min, followed by incubation with the radiolabeled AP-1 site of p19, and visualized as in B. A representative result is shown. (ATF-21, pAb and ATF-22, mAb) D. Chromatin immunoprecipitation. iDCs from four donors were treated with LPS/CD40LT, with or without ESAT-6, as described in A. Six hrs later, the cells were incubated with 1% formaldehyde, followed by preparation of nuclear chromatin supernatants and immunoprecipitation with anti-IRF-1 or isotype-matched control IgG (c.Ig). The precipitated DNA samples were amplified with primers for a 187 bp DNA fragment that contains the IRF-1 binding site of the p35 promoter. The PCR product was visualized by 1% agarose gel analysis. A representative result is shown.
M. tuberculosis lacking ESAT-6 elicit differential cytokine production by iDCs
The studies above were performed with recombinant ESAT-6. To determine if ESAT-6 produced by live M. tuberculosis had similar effects on DCs, we infected iDCs with M. tuberculosis H37Rv and its esat-6 deletion mutant, H37RvΔ3875. Both H37Rv and H37RvΔ3875 inhibited LPS/CD40LT-induced IL-12 production, but inhibition was significantly greater with H37Rv (Fig. 10A), confirming our findings with recombinant ESAT-6. Similarly, H37Rv induced 3–4-fold more IL-1β than H37RvΔ3875 (Fig. 10C), mimicking the effect of ESAT-6. In contrast, IL-23 levels produced by uninfected DCs and DCs infected with either H37Rv or H37RvΔ3875 were similar (Fig.10B), suggesting that the effects of other M. tuberculosis components mask those of ESAT-6, or compensate for its deletion, in eliciting IL-23 production.
Figure 10. Differential effects of M. tuberculosis H37Rv and its esat-6 deletion mutant on DC cytokine production.

iDCs from four donors were infected with live H37Rv and its esat-6 deletion mutant, H37RvΔ3875, at a multiplicity of infection of 10. Four h later, the cells were stimulated with LPS plus CD40LT for 24 h, culture supernatants were collected and levels of IL-12p70 (A), IL-23 (B), and IL-1β (C) were measured by ELISA. Means and SEMs of results from four donors are shown. *, p < 0.05 compared to H37Rv.
Discussion
DCs are pivotal to initiate, shape and maintain protective T cell responses against microbial pathogens. During M. tuberculosis infection, DCs present mycobacterial peptides to T cells, activating them in lymph nodes that drain the lung (37). However, M. tuberculosis also evades immunity by interfering with these DC:T cell interactions (38). Previously we showed that ESAT-6 directly inhibits T cell IFN-γ production in a p38 MAPK-dependent manner (18, 19). In this study, we demonstrate that ESAT-6 programs human DCs to favor stimulation of Th17 cells at the expense of Th1 cells, by increasing DC production of IL-23 and IL-1β, while reducing production of IL-12. These effects of ESAT-6 on DCs are probably mediated through specific interaction of ESAT-6 with DCs (Fig. 6), followed by altered expression of the transcription factors, IRF-1, ATF-2 and c-Jun. Our findings uncover a novel role for ESAT-6 in altering DC function to suppress protective immunity and elicit potentially immunopathologic responses.
Treatment with ESAT-6 during iDC maturation enhanced T cell production of IL-17, in response to allostimulation (Fig. 2E) or M. tuberculosis (Fig. 3), and conditioned medium from ESAT-6-treated DC markedly increased IL-17 production by activated T cells, compared to medium from untreated DC (Fig. 7A). This was not due to direct effects of free ESAT-6 in the DC conditioned medium on T cells, as medium from DC treated with ESAT-6 but not LPS/CD40LT did not increase IL-17 production (Fig. 7A), and addition of ESAT-6 to T cells inhibits IL-17 production (18).
ESAT-6 enhanced DC secretion of IL-23 and IL-1β (Figs. 4B and C), and neutralization of these cytokines strongly inhibited T cell IL-17 production (Figs. 7A and C). These results are consistent with the important roles of IL-23 and IL-1β in expanding human Th17 cells (60, 61). IL-6 and transforming growth factor-β favor development of murine Th17 cells from naïve T cells (62, 63), but their roles in human Th17 cells are more controversial (60, 61), perhaps because it is difficult to identify truly naïve T cells in adult humans, and because serum used in cell culture contains variable levels of transforming growth factor-β (64). In our experimental system, ESAT-6 did not increase DC production of IL-6 (Fig. 4E) and anti-transforming growth factor-β did not reduce the capacity of ESAT-6-treated DCs to elicit IL-17 production (Xisheng Wang, unpublished data), suggesting that these cytokines did not stimulate IL-17 production. The intracellular signaling molecules through which ESAT-6 increases IL-17 production remain uncertain. Inhibition of p38 MAPK did not reduce ESAT-6-induced IL-23 and IL-1β production by DC (Supplementary Fig. 2), unlike the case in T cells, where ESAT-6 inhibits IFN-γ production through activation of p38 MAPK (19), suggesting that ESAT-6 may target different signaling molecules in different cells.
IL-12 is central to defense against many pathogens, which have developed several strategies to reduce its production, such as increased cAMP levels elicited by the adenylate cyclase toxin of B. pertussis (47). However, blocking cAMP did not alter the effects of ESAT-6 on DC cytokine production (Supplementary Fig. 1). ESAT-6 also did not reduce DC IL-12 transcription by increasing IL-10 production (65, 66), as IL-10 levels of ESAT-6-treated DCs were not elevated (Xisheng Wang, unpublished data). ESAT-6 inhibits LPS-stimulated production of IL-12p40 by the murine RAW macrophage cell line by binding to macrophage TLR2 through its C-terminus 20-amino acid domain. (67). However, deletion of the C-terminal 20 amino acids of ESAT-6 or neutralizing antibodies to TLR2 and TLR1 did not abrogate the effects of ESAT-6 on LPS/CD40LT stimulated DC IL-12 production (data not shown), suggesting that the mechanism for ESAT-6 inhibition of IL-12 production by human DCs differs from that described in murine macrophages.
Transcription factors of the NF-κB (68) and IRF families (69), are critical for development, differentiation and activation of DCs. Production of IL-12 and IL-23 are regulated through transcription of p35 and p19, which are the rate-limiting factors in generating the mature IL-12 and IL-23 polypeptides, respectively. The NF-κB components, c-Rel and Rel A, enhance transcription of p35 and p19, respectively. ESAT-6 did not affect the DNA-binding activity of NF-κB in DC (Xisheng Wang, unpublished data), but reduced the expression and DNA-binding activity of IRF-1 in live DCs (Figs. 9A and D). IRF-1 and IRF-8 regulate expression of IL-12 in DCs in response to TLR signaling through pathogen-associated molecular patterns, such as LPS (69). IRF-1 also enhances p35 transcription, but inhibits that of p19 by interfering with binding of Rel A (56, 57) to the promoter. Mice with a deleted IRF-1 gene are highly susceptible to tuberculosis (70–73), indicating that it regulates genes that are central for resistance to mycobacterial infection. Our results indicate that ESAT-6 can differentially regulate both IL-12 and IL-23 by targeting a single transcription factor, illustrating a novel and efficient means of manipulating the immune response to reduce protective immunity while enhancing immunopathology and inflammation. ESAT-6 also increased activation and DNA binding capacities of ATF-2 and c-Jun (Fig. 9A, B and C), which are known to facilitate p19 transcription by binding the p19 promoter (59).
Although most of our studies were performed with human DCs in vitro, experiments with DCs infected with live M. tuberculosis with a deleted esat-6 gene showed that ESAT-6 expression inhibited DC production of IL-12 and enhanced that of IL-1β, mimicking the effects of recombinant ESAT-6. H37Rv and the esat-6 deletion mutant did not increase IL-23 production by DCs (Fig. 10), perhaps because other mycobacterial components also affect IL-23 production and mask the effects of ESAT-6 deletion. Nevertheless, ESAT-6 is likely to affect IL-17 production in vivo, as this secreted protein is present in the lungs during mycobacterial infection (74), and aerosol infection of mice with M. tuberculosis H37Rv generated higher IL-17 levels in the lungs than infection with an RD1 deletion mutant or with BCG, both of which lack ESAT-6 (75).
In summary, we found that ESAT-6, a virulence determinant of M. tuberculosis that is also a candidate vaccine antigen, favors Th17 responses at the expense of protective Th1 responses by differentially regulating production of DC cytokines and expression of transcription factors that bind to the promoters of p35 and p19. These findings provide a novel mechanism through which M. tuberculosis modulates host immune responses.
Supplementary Material
Acknowledgements
We thank Dr. Patrick Brennan for provision of heat-killed M. tuberculosis Erdman, Dr. Karen M. Dobos-Elder for providing the ESAT-6 expression construct, and Dr. Michael K. Pangburn for helpful discussions.
Abbreviations used in this aricle
- ESAT-6
early secreted antigenic target of 6 kDa
- MAPK
mitogen activated protein kinase
- BDG
bacillus Calmette-Guerin
- RD
region of difference
- IRF
interferon regulatory factor
- ATF
activating transcription factor
Footnotes
Disclosure: The authors have no financial conflict of interest.
This work was supported in part by grants from the National Institutes of Health (AI063514, AI082335, and AI099345), The Potts Memorial Foundation (to X.W.), the James Byers Cain Research Endowment and the Center for Pulmonary and Infectious Disease Control. Peter F. Barnes holds the Margaret E. Byers Cain Chair for Tuberculosis Research. Ivana B Alvarez was supported by a Fulbright-Bunge & Born Scholarship.
References
- 1.WHO. Multidrug and extensively drug-resistant TB (M/XDR-TB):2010 global report on surveillance and response. 2010:71.
- 2.Colditz GA, Brewer TF, Berkey CS, Wilson ME, Burdick E, Fineberg HV, Mosteller F. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. Jama. 1994;271:698–702. [PubMed] [Google Scholar]
- 3.Sterne JA, Rodrigues LC, Guedes IN. Does the efficacy of BCG decline with time since vaccination? Int J Tuberc Lung Dis. 1998;2:200–207. [PubMed] [Google Scholar]
- 4.Andersen P, Andersen AB, Sorensen AL, Nagai S. Recall of long-lived immunity to Mycobacterium tuberculosis infection in mice. J Immunol. 1995;154:3359–3372. [PubMed] [Google Scholar]
- 5.Sorensen AL, Nagai S, Houen G, Andersen P, Andersen AB. Purification and characterization of a low-molecular-mass T-cell antigen secreted by Mycobacterium tuberculosis. Infection and immunity. 1995;63:1710–1717. doi: 10.1128/iai.63.5.1710-1717.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langermans JA, Doherty TM, Vervenne RA, van der Laan T, Lyashchenko K, Greenwald R, Agger EM, Aagaard C, Weiler H, van Soolingen D, Dalemans W, Thomas AW, Andersen P. Protection of macaques against Mycobacterium tuberculosis infection by a subunit vaccine based on a fusion protein of antigen 85B and ESAT-6. Vaccine. 2005;23:2740–2750. doi: 10.1016/j.vaccine.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 7.Dietrich J, Billeskov R, Doherty TM, Andersen P. Synergistic effect of bacillus calmette guerin and a tuberculosis subunit vaccine in cationic liposomes: increased immunogenicity and protection. J Immunol. 2007;178:3721–3730. doi: 10.4049/jimmunol.178.6.3721. [DOI] [PubMed] [Google Scholar]
- 8.Pym AS, Brodin P, Majlessi L, Brosch R, Demangel C, Williams A, Griffiths KE, Marchal G, Leclerc C, Cole ST. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nature medicine. 2003;9:533–539. doi: 10.1038/nm859. [DOI] [PubMed] [Google Scholar]
- 9.Olsen AW, Williams A, Okkels LM, Hatch G, Andersen P. Protective effect of a tuberculosis subunit vaccine based on a fusion of antigen 85B and ESAT-6 in the aerosol guinea pig model. Infection and immunity. 2004;72:6148–6150. doi: 10.1128/IAI.72.10.6148-6150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aagaard C, Hoang T, Dietrich J, Cardona PJ, Izzo A, Dolganov G, Schoolnik GK, Cassidy JP, Billeskov R, Andersen P. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nature medicine. 2011;17:189–194. doi: 10.1038/nm.2285. [DOI] [PubMed] [Google Scholar]
- 11.Andersen P. Tuberculosis vaccines - an update. Nat Rev Microbiol. 2007;5:484–487. doi: 10.1038/nrmicro1703. [DOI] [PubMed] [Google Scholar]
- 12.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 13.Harboe M, Oettinger T, Wiker HG, Rosenkrands I, Andersen P. Evidence for occurrence of the ESAT-6 protein in Mycobacterium tuberculosis and virulent Mycobacterium bovis and for its absence in Mycobacterium bovis BCG. Infection and immunity. 1996;64:16–22. doi: 10.1128/iai.64.1.16-22.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR., Jr The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao LY, Guo S, McLaughlin B, Morisaki H, Engel JN, Brown EJ. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Molecular microbiology. 2004;53:1677–1693. doi: 10.1111/j.1365-2958.2004.04261.x. [DOI] [PubMed] [Google Scholar]
- 16.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. 2007;129:1287–1298. doi: 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 17.de Jonge MI, Pehau-Arnaudet G, Fretz MM, Romain F, Bottai D, Brodin P, Honore N, Marchal G, Jiskoot W, England P, Cole ST, Brosch R. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. Journal of bacteriology. 2007;189:6028–6034. doi: 10.1128/JB.00469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Barnes PF, Dobos-Elder KM, Townsend JC, Chung YT, Shams H, Weis SE, Samten B. ESAT-6 inhibits production of IFN-gamma by Mycobacterium tuberculosis-responsive human T cells. J Immunol. 2009;182:3668–3677. doi: 10.4049/jimmunol.0803579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng H, Wang X, Barnes PF, Tang H, Townsend JC, Samten B. The Mycobacterium tuberculosis early secreted antigenic target of 6 kDa inhibits T cell interferon-gamma production through the p38 mitogen-activated protein kinase pathway1. The Journal of biological chemistry. 2011;286:24508–24518. doi: 10.1074/jbc.M111.234062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCullough KC, Ruggli N, Summerfield A. Dendritic cells--at the front-line of pathogen attack. Veterinary immunology and immunopathology. 2009;128:7–15. doi: 10.1016/j.vetimm.2008.10.290. [DOI] [PubMed] [Google Scholar]
- 21.Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunological reviews. 2008;226:112–131. doi: 10.1111/j.1600-065X.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. The Journal of experimental medicine. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. The Journal of experimental medicine. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ottenhoff TH, Kumararatne D, Casanova JL. Novel human immunodeficiencies reveal the essential role of type-I cytokines in immunity to intracellular bacteria. Immunology today. 1998;19:491–494. doi: 10.1016/s0167-5699(98)01321-8. [DOI] [PubMed] [Google Scholar]
- 25.Fortin A, Abel L, Casanova JL, Gros P. Host genetics of mycobacterial diseases in mice and men: forward genetic studies of BCG-osis and tuberculosis. Annual review of genomics and human genetics. 2007;8:163–192. doi: 10.1146/annurev.genom.8.080706.092315. [DOI] [PubMed] [Google Scholar]
- 26.van de Vosse E, van Dissel JT, Ottenhoff TH. Genetic deficiencies of innate immune signalling in human infectious disease. The Lancet infectious diseases. 2009;9:688–698. doi: 10.1016/S1473-3099(09)70255-5. [DOI] [PubMed] [Google Scholar]
- 27.Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, Ghilardi N, deSauvage F, Cooper AM. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 28.Okamoto Yoshida Y, Umemura M, Yahagi A, O'Brien RL, Ikuta K, Kishihara K, Hara H, Nakae S, Iwakura Y, Matsuzaki G. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J Immunol. 2010;184:4414–4422. doi: 10.4049/jimmunol.0903332. [DOI] [PubMed] [Google Scholar]
- 29.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nature immunology. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 30.Cruz A, Fraga AG, Fountain JJ, Rangel-Moreno J, Torrado E, Saraiva M, Pereira DR, Randall TD, Pedrosa J, Cooper AM, Castro AG. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. The Journal of experimental medicine. 2010;207:1609–1616. doi: 10.1084/jem.20100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Junecko BA, Fountain JJ, Martino C, Pearl JE, Tighe M, Lin YY, Slight S, Kolls JK, Reinhart TA, Randall TD, Cooper AM. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol. 2011;187:5402–5407. doi: 10.4049/jimmunol.1101377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henderson RA, Watkins SC, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J Immunol. 1997;159:635–643. [PubMed] [Google Scholar]
- 33.Hanekom WA, Mendillo M, Manca C, Haslett PA, Siddiqui MR, Barry C, 3rd, Kaplan G. Mycobacterium tuberculosis inhibits maturation of human monocyte-derived dendritic cells in vitro. The Journal of infectious diseases. 2003;188:257–266. doi: 10.1086/376451. [DOI] [PubMed] [Google Scholar]
- 34.Samten B, Townsend JC, Sever-Chroneos Z, Pasquinelli V, Barnes PF, Chroneos ZC. An antibody against the surfactant protein A (SP-A)-binding domain of the SP-A receptor inhibits T cell-mediated immune responses to Mycobacterium tuberculosis. Journal of leukocyte biology. 2008;84:115–123. doi: 10.1189/jlb.1207835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samten B, Ghosh P, Yi AK, Weis SE, Lakey DL, Gonsky R, Pendurthi U, Wizel B, Zhang Y, Zhang M, Gong J, Fernandez M, Safi H, Vankayalapati R, Young HA, Barnes PF. Reduced expression of nuclear cyclic adenosine 5'-monophosphate response element-binding proteins and IFN-gamma promoter function in disease due to an intracellular pathogen. J Immunol. 2002;168:3520–3526. doi: 10.4049/jimmunol.168.7.3520. [DOI] [PubMed] [Google Scholar]
- 36.Samten B, Townsend JC, Weis SE, Bhoumik A, Klucar P, Shams H, Barnes PF. CREB, ATF, and AP-1 transcription factors regulate IFN-gamma secretion by human T cells in response to mycobacterial antigen. J Immunol. 2008;181:2056–2064. doi: 10.4049/jimmunol.181.3.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. The Journal of experimental medicine. 2008;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol. 2007;179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 39.Balboa L, Romero MM, Yokobori N, Schierloh P, Geffner L, Basile JI, Musella RM, Abbate E, de la Barrera S, Sasiain MC, Aleman M. Mycobacterium tuberculosis impairs dendritic cell response by altering CD1b, DC-SIGN and MR profile. Immunology and cell biology. 2010;88:716–726. doi: 10.1038/icb.2010.22. [DOI] [PubMed] [Google Scholar]
- 40.Tuyaerts S, Aerts JL, Corthals J, Neyns B, Heirman C, Breckpot K, Thielemans K, Bonehill A. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol Immunother. 2007;56:1513–1537. doi: 10.1007/s00262-007-0334-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Sullivan B, Thomas R. CD40 and dendritic cell function. Critical reviews in immunology. 2003;23:83–107. doi: 10.1615/critrevimmunol.v23.i12.50. [DOI] [PubMed] [Google Scholar]
- 42.Mortellaro A, Robinson L, Ricciardi-Castagnoli P. Spotlight on mycobacteria and dendritic cells: will novel targets to fight tuberculosis emerge? EMBO molecular medicine. 2009;1:19–29. doi: 10.1002/emmm.200900008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, Anes E. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cellular microbiology. 2010;12:1046–1063. doi: 10.1111/j.1462-5822.2010.01450.x. [DOI] [PubMed] [Google Scholar]
- 44.Launois P, DeLeys R, Niang MN, Drowart A, Andrien M, Dierckx P, Cartel JL, Sarthou JL, Van Vooren JP, Huygen K. T-cell-epitope mapping of the major secreted mycobacterial antigen Ag85A in tuberculosis and leprosy. Infection and immunity. 1994;62:3679–3687. doi: 10.1128/iai.62.9.3679-3687.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Derrick SC, Morris SL. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cellular microbiology. 2007;9:1547–1555. doi: 10.1111/j.1462-5822.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- 46.Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–653. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 47.Spensieri F, Fedele G, Fazio C, Nasso M, Stefanelli P, Mastrantonio P, Ausiello CM. Bordetella pertussis inhibition of interleukin-12 (IL-12) p70 in human monocyte-derived dendritic cells blocks IL-12 p35 through adenylate cyclase toxin-dependent cyclic AMP induction. Infection and immunity. 2006;74:2831–2838. doi: 10.1128/IAI.74.5.2831-2838.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hickey FB, Brereton CF, Mills KH. Adenylate cycalse toxin of Bordetella pertussis inhibits TLR-induced IRF-1 and IRF-8 activation and IL-12 production and enhances IL-10 through MAPK activation in dendritic cells. Journal of leukocyte biology. 2008;84:234–243. doi: 10.1189/jlb.0208113. [DOI] [PubMed] [Google Scholar]
- 49.Bohnenkamp HR, Papazisis KT, Burchell JM, Taylor-Papadimitriou J. Synergism of Toll-like receptor-induced interleukin-12p70 secretion by monocyte-derived dendritic cells is mediated through p38 MAPK and lowers the threshold of T-helper cell type 1 responses. Cellular immunology. 2007;247:72–84. doi: 10.1016/j.cellimm.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 50.Makela SM, Strengell M, Pietila TE, Osterlund P, Julkunen I. Multiple signaling pathways contribute to synergistic TLR ligand-dependent cytokine gene expression in human monocyte-derived macrophages and dendritic cells. Journal of leukocyte biology. 2009;85:664–672. doi: 10.1189/jlb.0808503. [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. The Journal of experimental medicine. 1989;170:827–845. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schulz O, Edwards AD, Schito M, Aliberti J, Manickasingham S, Sher A, Reise Sousa C. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 2000;13:453–462. doi: 10.1016/s1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 53.Sender LY, Gibbert K, Suezer Y, Radeke HH, Kalinke U, Waibler Z. CD40 ligand-triggered human dendritic cells mount interleukin-23 responses that are further enhanced by danger signals. Molecular immunology. 2010;47:1255–1261. doi: 10.1016/j.molimm.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 54.Snijders A, Hilkens CM, van der Pouw Kraan TC, Engel M, Aarden LA, Kapsenberg ML. Regulation of bioactive IL-12 production in lipopolysaccharide-stimulated human monocytes is determined by the expression of the p35 subunit. J Immunol. 1996;156:1207–1212. [PubMed] [Google Scholar]
- 55.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 56.Liu J, Cao S, Herman LM, Ma X. Differential regulation of interleukin (IL)-12 p35 and p40 gene expression and interferon (IFN)-gamma-primed IL-12 production by IFN regulatory factor 1. The Journal of experimental medicine. 2003;198:1265–1276. doi: 10.1084/jem.20030026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheikh SZ, Kobayashi T, Matsuoka K, Onyiah JC, Plevy SE. Characterization of an interferon-stimulated response element (ISRE) in the IL-23a promoter. The Journal of biological chemistry. 2011;286:1174–1180. doi: 10.1074/jbc.M110.147884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, Drake C, Pardoll D, Yu H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu W, Ouyang X, Yang J, Liu J, Li Q, Gu Y, Fukata M, Lin T, He JC, Abreu M, Unkeless JC, Mayer L, Xiong H. AP-1 activated by toll-like receptors regulates expression of IL-23 p19. The Journal of biological chemistry. 2009;284:24006–24016. doi: 10.1074/jbc.M109.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28:445–453. doi: 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 61.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nature immunology. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 62.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 63.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 64.O'Garra A, Stockinger B, Veldhoen M. Differentiation of human T(H)-17 cells does require TGF-beta! Nature immunology. 2008;9:588–590. doi: 10.1038/ni0608-588. [DOI] [PubMed] [Google Scholar]
- 65.Bhattacharyya S, Sen P, Wallet M, Long B, Baldwin AS, Jr, Tisch R. Immunoregulation of dendritic cells by IL-10 is mediated through suppression of the PI3K/Akt pathway and of IkappaB kinase activity. Blood. 2004;104:1100–1109. doi: 10.1182/blood-2003-12-4302. [DOI] [PubMed] [Google Scholar]
- 66.Loscher CE, Draper E, Leavy O, Kelleher D, Mills KH, Roche HM. Conjugated linoleic acid suppresses NF-kappa B activation and IL-12 production in dendritic cells through ERK-mediated IL-10 induction. J Immunol. 2005;175:4990–4998. doi: 10.4049/jimmunol.175.8.4990. [DOI] [PubMed] [Google Scholar]
- 67.Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A, Kaisho T, Kundu M, Basu J. Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nature immunology. 2007;8:610–618. doi: 10.1038/ni1468. [DOI] [PubMed] [Google Scholar]
- 68.Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 2002;16:257–270. doi: 10.1016/s1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 69.Gabriele L, Ozato K. The role of the interferon regulatory factor (IRF) family in dendritic cell development and function. Cytokine & growth factor reviews. 2007;18:503–510. doi: 10.1016/j.cytogfr.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 70.Pine R. IRF and tuberculosis. J Interferon Cytokine Res. 2002;22:15–25. doi: 10.1089/107999002753452629. [DOI] [PubMed] [Google Scholar]
- 71.Qiao Y, Prabhakar S, Canova A, Hoshino Y, Weiden M, Pine R. Posttranscriptional inhibition of gene expression by Mycobacterium tuberculosis offsets transcriptional synergism with IFN-gamma and posttranscriptional up-regulation by IFN-gamma. J Immunol. 2004;172:2935–2943. doi: 10.4049/jimmunol.172.5.2935. [DOI] [PubMed] [Google Scholar]
- 72.Yamada H, Mizuno S, Sugawara I. Interferon regulatory factor 1 in mycobacterial infection. Microbiology and immunology. 2002;46:751–760. doi: 10.1111/j.1348-0421.2002.tb02760.x. [DOI] [PubMed] [Google Scholar]
- 73.Cooper AM, Pearl JE, Brooks JV, Ehlers S, Orme IM. Expression of the nitric oxide synthase 2 gene is not essential for early control of Mycobacterium tuberculosis in the murine lung. Infection and immunity. 2000;68:6879–6882. doi: 10.1128/iai.68.12.6879-6882.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Majlessi L, Brodin P, Brosch R, Rojas MJ, Khun H, Huerre M, Cole ST, Leclerc C. Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J Immunol. 2005;174:3570–3579. doi: 10.4049/jimmunol.174.6.3570. [DOI] [PubMed] [Google Scholar]
- 75.Chatterjee S, Dwivedi VP, Singh Y, Siddiqui I, Sharma P, Van Kaer L, Chattopadhyay D, Das G. Early Secreted Antigen ESAT-6 of Mycobacterium tuberculosis promotes protective T helper 17 cell responses in a Toll-like receptor-2-dependent manner. PLoS pathogens. 2011;7 doi: 10.1371/journal.ppat.1002378. e1002378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.