

Abstract
Silanediol peptidomimetics are demonstrated to inhibit a serine protease. Asymmetric synthesis of the inhibitor was accomplished using Brown hydroboration and CBS reduction of an acylsilane intermediate. The silanediol product was found to inhibit the serine protease chymotrypsin with a Ki of 107 nM. Inhibition of the enzyme may involve exchange of a silane hydroxyl with the active site serine nucleophile, contrasting with previous silanediol protease inhibitors.
Design of protease inhibitors is a focus of many pharmaceutical research programs because of the myriad of biological processes that these enzymes mediate.1 More than 30 protease inhibitors have been approved for clinical use and many of these have reached the marketplace.2 Protease inhibitors are used to control hypertension,3 AIDS, thrombosis4 and cancer,5 but have the potential for many additional applications. Each inhibitor is tailored to the protease class, based on the active site mechanism, and employ the recognition elements that flank the active site to give the inhibitors specificity and potency.
Within each protease class, certain functionally relevant replacements for the tetrahedral intermediate have found broad utility. 6 Typical inhibitors of metalloproteases utilize zinc ion coordinating groups. 7 For aspartic proteases, a carbinol is used to hydrogen bond with the active site carboxylic acids. 8 Cysteine protease inhibitors often employ electrophilic functional groups to capture the cysteine nucleophile.9 Serine proteases are inhibited by electrophilic groups that interact with the active site serine alcohol nucleophile. Useful electrophilies include alpha-fluoro ketones and 1,2-dicarbonyls.10 A similar effect is found with the recently introduced anticancer agent bortezomib, that inhibits the closely related threonine protease found in the proteasome. This inhibition is effected by coordination of the active site hydroxyl by a Lewis acidic boronic acid.11 Each of these functional groups have steric, electronic and metabolic consequences that present challenges and opportunities in pharmaceutical design.
Serine proteases are contemporary targets for treatment of Alzheimer’s disease, cancer, obesity, diabetes, thromobosis and more.12 Novel functionality for serine protease inhibitor design may therefore have broad utility.
Silanols have potential as serine protease inhibitors because a silanol hydroxyl will rapidly exchange with water and with alcohols (Scheme 1).13 A covalent interaction between the silicon and the enzyme would contrast with previously described silanediol inhibitors that have been inhibitors of aspartic and metallo proteases where water is the nucleophilic group.14,15
Scheme 1.
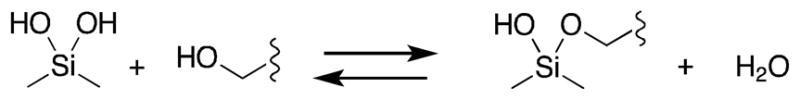
Silanols readily exchange with alcohols
Chymotrypsin is an archetype serine protease, characterized by a requirement for an aromatic sidechain at P1 (Figure 1).16 The use of the phenylalanine isostere 1 by Imperiali and Abeles underscored the importance of this aromatic substituent in chymotrypsin inhibitor design.17 To evaluate a silanediol as an inhibitor of this enzyme, we elected to utilize 2, an Ala–[Si]–Phe dipeptide analog. The choice of alanine substitution for P1′ in 2 was based on its effective earlier use as a steric shield for the silanediol to minimize oligomerization.18 Chymotrypsin is relatively insensitive toward substitution at P1′ in substrates19 and methyl substitution was effective in phosphoramide inhibitors developed by Bartlett, et al.20 Evaluation of 2 as an inhibitor used the commercially available chymotrypsin substrate 3 (vide infra). Termination with a primary amide ensures that the polarity and hydrogen bonding capabilities of the substrate are retained.
Figure 1.

Inhibition of the serine protease chymotrypsin requires an aromatic substitutent at P1
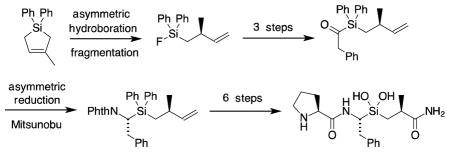
The central Ala–[Si]–Phe dipeptide of 2 had been previously prepared as part of a study of inhibitors of angiotensin-converting enzyme, but this synthesis employed a lengthy sequence.21 In this original preparation, the P1′ stereogenic center of 2 was derived from the commercially available, enantiomerically pure Roche ester.22 Subsequent separation of diastereomers led to the correct stereochemistry at P1. We have recently reported an asymmetric method for constructing the P1′ methyl stereogenic center and we employed this chemistry to prepare 2 (Scheme 2).23 Magnesium-mediated cycloaddition of isoprene with dichlorodiphenylsilane can be performed on >100 g scale.24 Hydroboration of 5 with monoisopinocampheylborane at −20 °C leads to silacyclopentane 6 in 93% ee. Warming this β-hydroxysilane with a 3:1 mixture of 48% HF in ethanol leads to Peterson fragmentation and formation of fluorosilane 7 in 90% distilled yield.19
Scheme 2.

Setting the P1′ methyl stereochemistry23
Construction of the α-aminosilane component utilized a Brook-Corey acylsilane approach (Scheme 3).25 Addition of 2-lithio-1,3-dithiane to fluorosilane 7, followed by deprotonation and alkylation with benzyl bromide, gave 8. Hydrolysis of the dithiane then gave the silyl ketone 9. Reduction of this ketone using the (S)-B-Me-CBS reagent gave a clean reduction with a diastereoselectivity of > 90% dr (NMR).26 The resulting alcohol 10 underwent Mitsunobu inversion with phthalimide to produce 11.
Scheme 3.

Asymmetric construction of the α-amino silane
Completion of the inhibitor (Scheme 4) began with ozonolysis of the terminal alkene, followed by Pinnick oxidation of the resulting aldehyde. Acid 12 was then converted to the primary amide through the mixed anhydride using ethyl chloroformate. Hydrolysis of the phthalimide with hydrazine gave the corresponding amine, which was then coupled with Boc-protected proline using HATU to give tripeptide analog 14.
Scheme 4.

Completion of the inhibitor synthesis
Hydrolysis of diphenylsilane 14 to silanediol 15 used our protocol developed in earlier work.27 Treatment of 14 with triflic acid in methylene chloride at 0 °C cleaved the Si–phenyl bonds (as well as the Boc group). This was followed by dilution with ammonium hydroxide. The resulting product was then treated with 48% aqueous HF, to form the difluorosilane. This monomeric product is readily hydrolyzed to the corresponding silanediol under mildly basic aqueous conditions.
Evaluation of 15 as an inhibitor of chymotrypsin employed the method of Bartlett et al. in which hydrolysis of substrate 3 was followed by the increase in UV absorbance at 410 nm.28,29 Chymotrypsin is inhibited by 15 in a concentration-dependent manner. The results are shown in the Dixon plot (Figure 2). These experiments found the Ki for this inhibition to be 107 nM.
Figure 2.

Dixon Plot showing inhibition of chymotrypsin by 15, with concentration of substrate 3 at 50 μM (circles) and 45 μM (triangles)
Inhibition of chymotrypsin by 15 is consistent with exchange of a silanediol hydroxyl group with the nucleophilic active site serine hydroxyl of the enzyme. It is likely that this hydroxyl exchange occurs through a pentavalent intermediate.30
Synthesis of this inhibitor took advantage of an asymmetric hydroboration and an asymmetric borane reduction to control the two stereogenic centers flanking the silicon. The inhibition of chymotrypsin illustrates the use of silanediols as inhibitors of a third family of proteases, adding to metallo and aspartic examples.15 Additional studies are in progress.
Supplementary Material
Acknowledgments
The authors thank Yingmei Qi for preliminary studies, and generous support for this program by the National Institutes of Health (R01 GM076471).
Footnotes
Supporting Information Available. Experimental details and characterization data for new compounds.
References
- 1.Babine RE, Bender SL. Chem Rev. 1997;97:1359–1472. doi: 10.1021/cr960370z. [DOI] [PubMed] [Google Scholar]; Leung D, Abbenante G, Fairlie DP. J Med Chem. 2000;43:305–341. doi: 10.1021/jm990412m. [DOI] [PubMed] [Google Scholar]
- 2.Turk B. Nat Rev Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 3.Matchar DB, McCrory DC, Orlando LA, Patel MR, Patel UD, Patwardhan MB, Powers B, Samsa GP, Gray RN. Ann Intern Med. 2008;148:16–29. doi: 10.7326/0003-4819-148-1-200801010-00189. [DOI] [PubMed] [Google Scholar]
- 4.Mackman N. Nature. 2008;451:914–918. doi: 10.1038/nature06797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kessenbrock K, Plaks V, Werb Z. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; Page-McCaw A, Ewald AJ, Werb Z. Nature Rev Molec Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meanwell NA. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 7.Lead references: Thiols: Cushman DW, Cheung HS, Sabo EF, Ondetti MA. Biochemistry. 1977;16:5484–5491. doi: 10.1021/bi00644a014.Carboxylates: DePriest SA, Mayer D, Naylor CB, Marshall GR. J Am Chem Soc. 1993;115:5372–5384.Phosphinates: Akif M, Schwager SL, Anthony CS, Czarny B, Beau F, Dive V, Sturrock ED, Acharya KR. Biochemical Journal. 2011;436:53–59. doi: 10.1042/BJ20102123.Collinsova M, Jiracek J. Curr Med Chem. 2000;7:629–647. doi: 10.2174/0929867003374831.Hydroxamic acids: Jacobsen JA, Major Jourden JL, Miller MT, Cohen SM. Biochim Biophys Acta, Molec Cell Res. 2010;1803:72–94. doi: 10.1016/j.bbamcr.2009.08.006.
- 8.Rich DH, Bernatowicz MS, Agarwal NS, Kawai M, Salituro FG, Schmidt PG. Biochemistry. 1985;24:3165–3173. doi: 10.1021/bi00334a014. [DOI] [PubMed] [Google Scholar]; Dunn BM. Chem Rev. 2002;102:4431–4458. doi: 10.1021/cr010167q. [DOI] [PubMed] [Google Scholar]; Eder J, Hommel U, Cumin F, Martoglio B, Gerhartz B. Curr Pharm Des. 2007;13:271–285. doi: 10.2174/138161207779313560. [DOI] [PubMed] [Google Scholar]
- 9.Powers JC, Asgian JL, Ekici D, James KE. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 10.Zhong J, Groutas WC. Curr Top Med Chem. 2004;4:1203–1216. doi: 10.2174/1568026043387971. [DOI] [PubMed] [Google Scholar]; Bachovchin DA, Cravatt BF. Nat Rev Drug Discov. 2012;11:52–68. doi: 10.1038/nrd3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groll M, Berkers CR, Ploegh HL, Ovaa H. Structure. 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 12.Bachovchin DA, Cravatt BF. Nat Rev Drug Discov. 2012;11:52–68. doi: 10.1038/nrd3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tacke R, Linoh H, Ernst L, Moser U, Mutschler E, Sarge S, Cammenga HK, Lambrecht G. Chem Ber. 1987;120:1229–1237. [Google Scholar]
- 14.A silanediol inhibitor of the serine protease elastase was reported: Showell GA, Montana JG, Chadwick JA, Higgs C, Hunt HJ, MacKenzie RE, Price S, Wilkinson TJ. Silicon diols, effective inhibitors of human leukocyte elastase. In: Auner N, Weis J, editors. Organosilicon Chem VI, [Eur Silicon Days] 2. Wiley-VCH Verlag GmbH & Co. KGaA; 2005. pp. 569–574.. This silanediol has also been prepared by Skrydstrup et al.: Hernández D, Lindsay KB, Nielsen L, Mittag T, Bjerglund K, Friis S, Mose R, Skrydstrup T. J Org Chem. 2010;75:3283–3293. doi: 10.1021/jo100301n.
- 15.Sieburth SMcN, Chen C-A. Eur J Org Chem. 2006:311–322. [Google Scholar]
- 16.Schecter I, Berger A. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 17.Imperiali B, Abeles RH. Biochemistry. 1986;25:3760–3767. doi: 10.1021/bi00361a005. [DOI] [PubMed] [Google Scholar]
- 18.Mutahi Mwa, Nittoli T, Guo L, Sieburth SMcN. J Am Chem Soc. 2002;124:7363–7375. doi: 10.1021/ja026158w. [DOI] [PubMed] [Google Scholar]
- 19.Schellenberger V, Braune K, Hofmann H, Jaubke H. Eur J Biochem. 1991;199:623–636. doi: 10.1111/j.1432-1033.1991.tb16163.x. [DOI] [PubMed] [Google Scholar]
- 20.Bartlett PA, Lamden LA. Bioorg Chem. 1986;14:356–377. [Google Scholar]
- 21.Kim J, Hewitt G, Carroll P, Sieburth SMcN. J Org Chem. 2005;70:5781–5789. doi: 10.1021/jo048121v. [DOI] [PubMed] [Google Scholar]
- 22.Cohen N, Eichel WF, Lopresti RJ, Neukom C, Saucy G. J Org Chem. 1976;41:3505–3511. doi: 10.1021/jo00884a002. [DOI] [PubMed] [Google Scholar]
- 23.Sen S, Purushotham M, Qi Y, Sieburth SMcN. Org Lett. 2007;9:4963–4965. doi: 10.1021/ol7021559. [DOI] [PubMed] [Google Scholar]
- 24.Manuel G, Mazerolles P, Lesbre M, Pradel J-P. J Organomet Chem. 1973;61:147–165. [Google Scholar]
- 25.Brook AG, Duff JM, Jones PF, Davis NR. J Am Chem Soc. 1967;89:431–434. [Google Scholar]; Corey EJ, Seebach D, Freedman R. J Am Chem Soc. 1967;89:434–436. [Google Scholar]
- 26.Corey EJ, Helal CJ. Angew Chem Int Ed Engl. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, Sieburth SMcN. J Org Chem. 2004;69:3008–3014. doi: 10.1021/jo049929i. [DOI] [PubMed] [Google Scholar]
- 28.Delmar EG, Largman C, Brodrick JW, Geokas MC. Anal Biochem. 1979;99:316–320. doi: 10.1016/s0003-2697(79)80013-5. [DOI] [PubMed] [Google Scholar]
- 29.Hansen KB, Grosch B, Greiveldinger-Poenaru S, Bartlett PA. J Org Chem. 2003;68:8465–8470. doi: 10.1021/jo034837z. [DOI] [PubMed] [Google Scholar]
- 30.Lickiss PD, Rappoport Z, Apeloig Y. The Chemistry of Organic Silicon Compounds. 3. John Wiley & Sons; New York: 2001. Polysilanols; pp. 695–744.Trivalent species have also been postulated: Oostendorp DJ, Bertrand GL, Stoffer JO. J Adhes Sci Technol. 1992;6:171–191.It is unlikely however that a pentavalent silanol is the inhibitor: Karsch HH, Bienlein F, Sladek A, Heckel M, Burger K. J Am Chem Soc. 1995;117:5160–5161.Bassindale AR, Parker DJ, Taylor PG, Auner N, Herrschaft B. Chem Commun. 2000;7:565–566.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.