

Abstract
Brown–Vialetto–Van Laere syndrome was first described in 1894 as a rare neurodegenerative disorder characterized by progressive sensorineural deafness in combination with childhood amyotrophic lateral sclerosis. Mutations in the gene, SLC52A3 (formerly C20orf54), one of three known riboflavin transporter genes, have recently been shown to underlie a number of severe cases of Brown–Vialetto–Van Laere syndrome; however, cases and families with this disease exist that do not appear to be caused by SLC52A3 mutations. We used a combination of linkage and exome sequencing to identify the disease causing mutation in an extended Lebanese Brown–Vialetto–Van Laere kindred, whose affected members were negative for SLC52A3 mutations. We identified a novel mutation in a second member of the riboflavin transporter gene family (gene symbol: SLC52A2) as the cause of disease in this family. The same mutation was identified in one additional subject, from 44 screened. Within this group of 44 patients, we also identified two additional cases with SLC52A3 mutations, but none with mutations in the remaining member of this gene family, SLC52A1. We believe this strongly supports the notion that defective riboflavin transport plays an important role in Brown–Vialetto–Van Laere syndrome. Initial work has indicated that patients with SLC52A3 defects respond to riboflavin treatment clinically and biochemically. Clearly, this makes an excellent candidate therapy for the SLC52A2 mutation-positive patients identified here. Initial riboflavin treatment of one of these patients shows promising results.
Keywords: motor neuron disease, ALS, riboflavin, BVVL, SLC52A2
Introduction
Brown–Vialetto–Van Laere syndrome is an extremely rare neurological disorder characterized by progressive pontobulbar palsy associated with sensorineural deafness. This syndrome was first documented by Dr Charles Henry Brown in 1894 with his description of a 15-year-old German male at a meeting of the New York Neurological Society (Brown, 1894) and then further characterized by Vialetto (1936) and Laere (1977). Since the first documentation only ∼60 cases have been described in the literature. Approximately one-half of these cases are apparently sporadic in nature, a presentation consistent with the autosomal recessive mode of inheritance noted in several families (Sathasivam, 2008). On reviewing the clinical features of the reported families, the clinical course typically begins with sensorineural deafness, followed by involvement of the motor cranial nerves—usually the 7th and 9th to the 12th (Gallai et al., 1981). Patients with Brown–Vialetto–Van Laere syndrome often have other clinical features, the most common of which is not only respiratory compromise but also upper and lower limb weakness, wasting and even sensory symptoms in some patients. Brown–Vialetto–Van Laere syndrome also shares diagnostic overlap with other motor neuron disorders such as Madras motor neuron disease, Boltshauser syndrome, Nathalie syndrome, Fazio–Londe syndrome (possibly an allelic condition to Brown–Vialetto–Van Laere syndrome) and other cases with very early onset childhood amyotrophic lateral sclerosis. The childhood amyotrophic lateral sclerosis group have a broadly similar severe phenotype that can be divided into those with more severe forms of Brown–Vialetto–Van Laere syndrome, where children die in early infancy from respiratory problems, to the later onset forms and milder types of amyotrophic lateral sclerosis such as Madras motor neuron disease. Until now, there have been no disease modifying treatments for these disorders (Sathasivam, 2008).
Recently, mutations in the gene SLC52A3 (solute carrier family 52, riboflavin transporter, member 3; formerly C20orf54) were identified as a cause of autosomal recessive Brown–Vialetto–Van Laere syndrome in a consanguineous family with multiple affected individuals (Green et al., 2010). Mutations were subsequently found in six additional families. In humans, there are three members of the riboflavin transporter family, SLC52A1, SLC52A2 and SLC52A3. A mutation in SLC52A1 has recently been associated with maternal riboflavin deficiency leading to a profile consistent with multiple acyl-coA dehydrogenation deficiency (MADD) in a newborn infant (Ho et al., 2011). Notably, the symptoms and metabolic profile were completely resolved with riboflavin supplementation.
Patients with Brown–Vialetto–Van Laere syndrome have been found negative for coding mutations in SLC52A3, suggesting that other responsible gene mutations exist (Green et al., 2010; Johnson et al., 2010). During the last few years, genotyping and DNA sequencing technologies have revolutionized Mendelian genetics (Gibbs and Singleton, 2006; Kuhlenbaumer et al., 2011). Here, we demonstrate the efficacy of array technology in combination with exome sequencing to discover the molecular basis of a rare monogenic disorder. We describe a series of experiments that aimed to identify the underlying genetic lesion in a family with a childhood motor neuron disease and Brown–Vialetto–Van Laere syndrome features from northern Lebanon. The family had similar features to reported Brown–Vialetto–Van Laere syndrome cases but had a later age of onset and slower clinical progression. We screened the identified gene in a series of European and Asian patients with Brown–Vialetto–Van Laere syndrome features and childhood motor neuron disease.
Materials and methods
Sample collection
Brown–Vialetto–Van Laere syndrome was previously described in four individuals of a consanguineous family from northern Lebanon (Fig. 1) (Mégarbané et al., 2000). DNA samples of this family were obtained from Dr A Mégarbané at the Unité de Génétique Médicale at the Université Saint Joseph (Beirut, Lebanon). Case 1 (proband) had a normal delivery and a normal neonatal and early childhood medical history up to the age of 2.5 years when his hearing and walking were noticed to be abnormal. His condition progressed and when he was examined at the age of 8 years, he was found to have evidence of profound deafness, tongue fasciculations, bulbar dysfunction and axial and limb hypotonia. There was severe weakness of neck flexion and extension resulting in an inability to hold his head up. He had weakness of the proximal and distal upper and lower limbs with distal atrophy and clawed hands. Deep tendon reflexes and plantar responses were absent. There was evidence of scoliosis. Investigations revealed a normal MRI of brain and spinal cord, normal blood screen that included full blood count, clotting, renal, liver, bone, endocrine profiles, white cell enzymes and very long chain fatty acids. Further analyses of intermediary metabolism including plasma acylcarnitine profile and urinary organic acids were not carried out in this family prior to the identification of the SLC52A3 mutation. Brainstem auditory-evoked responses were absent. The patient died at the age of 11 years from respiratory failure.
Figure 1.

Pedigree of Lebanese family with Brown–Vialetto–Van Laere syndrome. Males are represented by squares and females are represented by circles. A blackened symbol indicates an affected individual, and a slash through a symbol indicates the individual is deceased. A double horizontal line indicates a consanguineous marriage.
Case 2 was born 5 years after his male sibling (Case 1) and had a similar age of onset and presentation. When he was examined at the age of 7 years, he had evidence of multiple cranial nerve palsies with poor papillary responses and nystagmus, axial and limb hypotonia, upper and lower limb weakness, and wasting and kyphoscoliosis. Brainstem auditory-evoked responses revealed severe sensorineural hearing loss. Motor responses had normal velocities and EMG revealed evidence of fibrillations. He died suddenly at home 4 months after the time of examination. His female sibling (Case 3) presented in the same way but died at the age of 4 years after a severe episode of gastroenteritis.
Case 4, a male cousin to Cases 1–3, presented in a similar fashion to his cousins except that his muscle weakness was less severe. EMG and nerve conduction studies showed denervation with absent sensory response of the sural and median nerves but normal motor velocities. Affected individuals were screened and determined negative for mutations in SMN, which encodes survival motor neuron protein (Mégarbané et al., 2000).
The screening series consisted of 44 European and Asian patients with Brown–Vialetto–Van Laere syndrome features and childhood motor neuron disease. A number of these families were consanguineous with the core clinical features of childhood onset bulbar or breathing problems, multiple cranial nerve palsies and peripheral weakness and wasting. A number of these cases were ventilator dependent and several died due to respiratory complications. Subjects from this series have been previously screened and found to be negative for SMN deletions, the common mitochondrial mutations (8243, 3244 and mitochondrial deletions on Southern blotting) and SOD1 mutations.
Genome-wide single-nucleotide polymorphism genotyping
Genome-wide single-nucleotide polymorphism (SNP) genotyping was performed for Cases VI-1, VI-6 and VI-7 using Infinium II 550 K SNP chips (Illumina Inc.), which assay 555 352 unique SNPs derived from the International Human HapMap project. SNP genotyping was performed for Cases VI-1, VI-8, VI-10 and VI-11, and for Case VI-7 using 300 K chips, which assay 317 511 unique SNPs across the human genome. These assays were performed according to manufacturer’s instructions (Illumina Inc.). Chromosomes were assessed for structural changes using the Genome Viewer tool in Beadstudio v2.2.22 (Illumina Inc.). The two metrics visualized were B allele frequency and log R ratio. The B allele frequency is the number of times an allele at a particular position is called A or B. Therefore, an individual homozygous for the B allele would have a score of ∼1, an individual homozygous for the A allele would have a score of ∼0 and an individual heterozygous for the A and B allele would have a score of ∼0.5. Log R ratio is the log2 ratio of the observed normalized R value the SNPs theta value, where theta is the ratio of signal at each polymorphism for beads recognizing an A allele to beads recognizing a B allele. An R > 1 indicates an increase in copy number and a value <1 indicates a deletion (Simon-Sanchez et al., 2008).
Autozygosity mapping
Autozygosity mapping was performed by visual inspection of Genome Viewer genotypes for extended regions of homozygosity (>200 kb) shared between affected individuals and not shared with unaffected individuals. Once all regions of overlapping homozygosity across cases were determined, genotypes at those loci were exported to determine whether individuals were identical by descent at these loci.
Exome sequencing
Exome sequencing was performed on Case VI-11. DNA (3 µg) was fragmented using a Covaris E210 (Covaris Inc.). Following fragmentation, DNA was end-repaired by way of 5′ phosphorylation using the enzyme Klenow polymerase. Adenine bases were added to the 3′ end of the phosphorylated fragment and ligated to Illumina adapters. After purification using an AMPure DNA Purification kit, adapter-ligated products were amplified. The DNA library was then hybridized to a biotinylated RNA exome capture library (SureSelect Human Exome Kit, Agilent Technologies) and the exome selected using streptavidin-coated magnetic beads (Dynal Magnetic Beads, Invitrogen). RNA was digested and the exome-enriched library was amplified. Clusters were generated using 8 pM of library on a single-read v.4 flow cell. The sample was sequenced for 61 cycles on the Illumina Genome Analyzer.
Exome data analysis
Sequence alignment and variant calling was performed against the reference human genome (UCSC hg18). The apparent PCR/optical duplicate rate was 8.7%. Removal of duplicate reads was performed using Picard (picard.sourceforge.net/index.shtml). Alignment was performed using BWA (version 0.5.7); 93.6% of reads, or 2.816 billion base pairs of sequence, aligned to the genome. The Genome Analysis Toolkit was then used to recalibrate base quality scores, call and filter the variants. In total, 20 787 single-nucleotide variants and 615 indels were called. Of the 20 787 single-nucleotide variants, 6461 were non-synonymous. Variants previously identified in the 1000 Genomes project or within dbSNP (www.ncbi.nlm.nih.gov/projects/SNP/build130) were removed; this resulted in a list of 692 single-nucleotide variants and 20 indels. The protein coding effects of these remaining potentially novel variants was predicted using SIFT and SeattleSeq Annotation (gvs.gs.washington.edu/SeattleSeqAnnotation). This resulted in 264 potentially damaging variants that were not in 1000 genomes or dbSNP. We then restricted our list to include variants within the segregating region defined by homozygosity mapping and genes that contained homozygous variants.
Figure 2.

Cross-species comparison of SLC52A2 protein sequence. Blue letters indicate the amino acid residue is not conserved. Red letters indicate position 306. Residue 306 is highly conserved.
Sanger sequencing
SLC52A2 PCR primers were designed using ExonPrimer so that PCR products would span whole exons and ∼35 bp of flanking introns (http://ihg.gsf.de/ihg/ExonPrimer.html). PCR primers were also designed for SLC52A1 and SLC52A3 as above and previously described (Green et al., 2010; Johnson et al., 2010) Primer sequences are shown in Supplementary Table 1. PCR amplification was performed using 25 ng of genomic DNA, 12 µl 2× PCR Master Mix (Roche), 1 µl each of 10 µM forward and reverse primer and 2 µl H2O. Touchdown PCR was performed as follows: 95°C for 5 min, followed by eight cycles of denaturing at 95°C for 20 s, annealing at 60°C for 20 s, and extending at 72°C for 30 s. Following these eight cycles, 20 cycles of the same conditions were run, except the annealing temperature was decreased by 0.5°C per cycle until the final annealing temperature reached 50°C. Finally, 12 cycles were run with denaturing at 95°C for 20 s, annealing at 50°C for 20 s and extending at 72°C for 30 s. The reaction underwent a final extension at 72°C for 5 min. PCR amplification products were cleaned with AMPure® SPRI beads as per the manufacturer’s protocol (Agencourt).
Approximately 25 ng (∼1 µl) of each PCR product was used as template per sequencing reaction. Sequencing reactions contained 5× Reaction Buffer (2 µl), BigDye® (0.5 µl) (Applied Biosystems), H2O (5.5 µl), and the forward or reverse primer that was used for amplification of the original product (1 µl). Conditions were as follows: 25 cycles of denaturation at 95°C, annealing at 50°C and extension at 60°C. Sequencing reactions were cleaned using CleanSEQ® SPRI beads according to the manufacturer’s protocol (Agencourt). Sequencing was performed using a 3730 DNA Analyzer (Applied Biosystems).
Mutation positions are based on NCBI reference sequences (www.ncbi.nlm.nih.gov), for complementary DNA, nucleotide 1 is the A in the translation initiation codon ATG. SLC52A2 mutation positions are based on sequences NM_024531.4 for the nucleotide sequence and NP_078807.1 for the protein sequence. SLC52A3 mutation positions are based on sequences NM_033409.3 for the nucleotide sequence, and NP_212134.3 for the protein sequence. MAPK15 variant positions are based on sequences NM_139021.2 for the nucleotide sequence, and NP_620590.2 for the protein sequence.
Results
Autozygosity mapping was performed using data from affected individuals VI-1, VI-8 and VI-11 and unaffected family members VI-7 and VI-12. Affected individuals share a region of homozygosity identical by descent from approximately rs2123758 to the telomere (last SNP = rs11992614) on chromosome 8q24.3 that was not shared by unaffected individuals (Supplementary Fig. 1). This 1.7 Mb region contains an estimated 83 genes (Supplementary Table 2). There were no other regions of homozygosity >200 kb that segregated with disease and were identical by descent. Disease segregating genomic duplications or deletions was not apparent at this or any other locus.
Exome sequencing was performed on Case VI-11. A total of 90.5% of baited regions had ≥10× coverage with a mean and median depth of coverage of 47× and 39×, respectively. Four hundred and fifty-eight potentially novel single-nucleotide variants were identified on chromosome 8. Of these variants, 13 are within the chromosome 8q24.3 locus and two of these are non-synonymous variants.
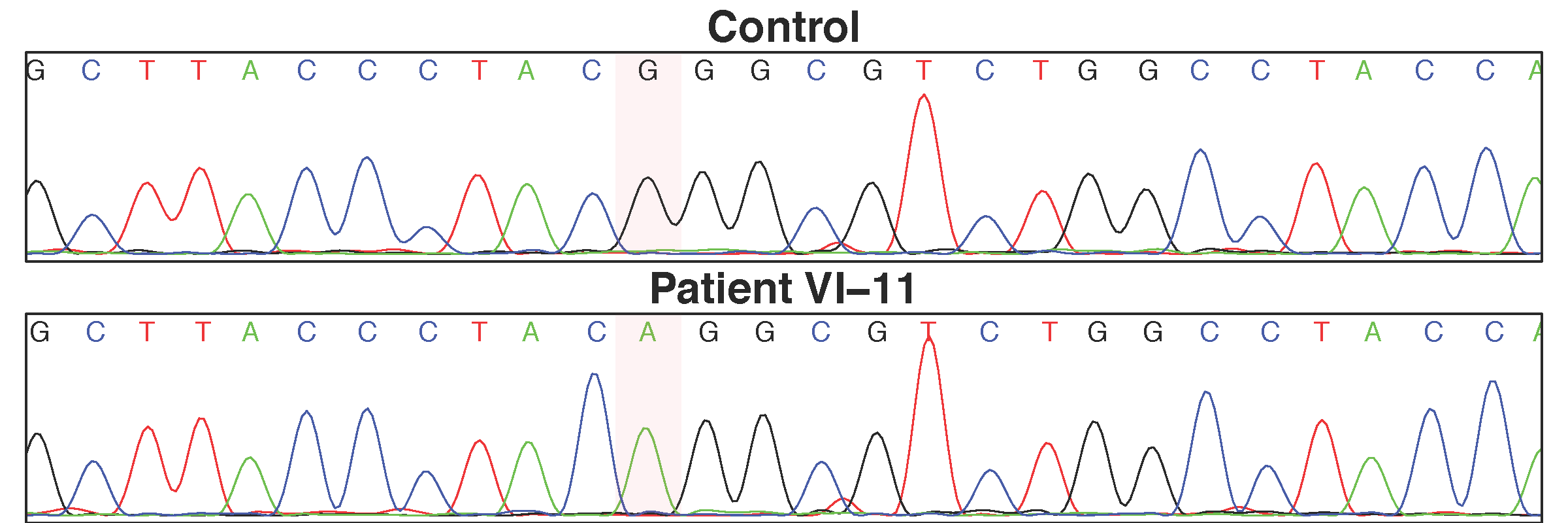
For the two novel non-synonymous variants found, individual VI-11 was homozygous for the non-reference allele in MAPK15 and SLC52A2. Sanger sequencing for p.P140L (c.419C>T) in MAPK15 and p.G306R (c.916G>A) in SLC52A2 was performed on all DNA samples collected from the family (Supplementary Fig. 2). Both p.P140L and p.G306R segregate with disease within this family. We screened both of these variants in control samples and found both were absent in 1400 chromosomes from neurologically normal subjects.
Sanger sequencing of SLC52A3 and SLC52A1 was performed in the Lebanese family and the 44 European and Asian patients with Brown–Vialetto–Van Laere syndrome features and childhood motor neuron disease. This revealed no mutations in the Lebanese family, but two cases that carried mutations in the previously linked gene SLC52A3. These were a novel compound heterozygous change p.G54R/p.I75T (c.160G>A and c.224T>C), and a previously described mutation, p.F457L (c.1371C>G) that was present as a homozygous change (Green et al., 2010). No mutations in SLC52A1 were identified. Sequencing of SLC52A2 in these 44 subjects revealed a mutation in a patient (affected member of Family D) from the United Kingdom with a similar phenotype to that seen in the Lebanese Brown–Vialetto–Van Laere syndrome family. This mutation was the same homozygous SLC52A2 p.G306R (c.916G>A) as that seen in the Lebanese family. Notably, this case did not have the p.P140L (c.419C>T) mutation in MAPK15 that was in cis with the SLC52A2 mutation in the Lebanese family. This adds significant weight to the notion that p.G306R, rather than p.P140L, is the pathogenic change. It would also suggest that either p.G306R has occurred independently on two different haplotypes or, perhaps less likely, that the families share a common founder. This latter case would require that a recent recombination event has occurred between the two variants or the MAPK15 mutation has arisen subsequent to the p.G306R mutation. We screened the p.G306R mutation in an additional series of UK controls, 516 samples from the Wellcome trust 1958 cohort. This mutation was absent from this series.
Family D consists of a single affected female of Scottish heritage. Her parents are unaffected and upon questioning were reported to be unrelated. There is no family history of any neurological problems. The proband is presently 11 years of age. She presented at 18 months of age with an ataxic gait. At 6 years of age, she had evidence of weakness of pincer grasp, which rapidly progressed to severe upper limb weakness (subgravity) over the course of a few weeks. She was also diagnosed with bilateral sensorineural hearing loss at age 6 years. By 7 years of age, she was wheelchair dependent due to axial hypotonia and weakness but maintained the ability to walk with full truncal and head support, as she retained full strength in her lower limbs. She was also diagnosed with optic atrophy and sleep hypoventilation, requiring the initiation of bilevel positive airway pressure at age 7 years. At 8 years of age, she was formally diagnosed with severe axonal sensorimotor neuropathy. She has developed an apparent hypermetabolic syndrome, which has developed since 6 years of age. Her present caloric intake exceeds 1000 calories over her calculated metabolic demand and a weight equivalent to <0.4% for age. She underwent extensive metabolic testing at the age of 10 years, which revealed evidence of carnitine deficiency and a urinary organic acid profile mimicking that of MADD. Following the identification of the SLC52A2 mutation and baseline metabolic testing including plasma riboflavin, flavin mononucleotide and flavin adenine dinucleotide, she was initiated on high-dose oral riboflavin therapy. After 5 months of riboflavin therapy (at an initial dose of 150 mg/day for duration of 3 months and a further increased dose of 450 mg/day for the past 2 months), she has demonstrated a normalization of her acylcarnitine profile, urinary organic acid profile and riboflavin/flavin mononucleotide/flavin adenine dinucleotide levels. Concomitantly, she has demonstrated quantitative improvements in her pulmonary function, brainstem auditory-evoked potential and visual-evoked potential testing as well as clinical evidence of improved upper limb strength. In particular, she now has antigravity thumb abduction and extension (previously subgravity).
The metabolic profiles of the three riboflavin genes are also different and can form another useful way to identify each patient group (Table 1). Case 4 from the Lebanese family has not as yet been started on riboflavin, but given the response in patients with SLC52A3 mutations and the early response in Family D with the same SLC52A3 mutation, we plan to start high-dose oral riboflavin therapy after baseline metabolic measurements are obtained.
Table 1.
Plasma carnitine and acylcarnitine profiles in patients with riboflavin transporter deficiencies and multiple acyl-CoA dehydrogenase deficiency
| Plasma acylcarnitine species | Riboflavin transporter deficiency |
MADD deficiencya | ||
|---|---|---|---|---|
| SLC52A3 | SLC52A1 deficiency in mother | SLC52A2 | ||
| C0 | 18.3, 35.1, 37.6 (22–55) | 11 (4–33) | 18.3 (18–52) | 4.2 (22–55) |
| C2 | 3.2, 4.0, 0.74 (3.4–13) | 19.3 (<11.5) | 5.1 (3.4–13) | |
| C4 | 0.65, 2.97, 0.43 (0.07–0.58) | 1.72 (<0.6) | 8.55 (0.07–0.58) | |
| C5b | 0.29, 1.99, 0.91 (0.04–0.22) | 0.34 (<0.24) | 0.31 (0.04–0.22) | |
| C6 | 0.27, 0.87, 0.02 (0.02–0.12) | 0.2 (0–0.1) | 0.79 (<0.12) | 0.14 (0.02–0.12) |
| C8 | 0.18, 0.70, 0.53 (0.04–0.22) | 0.4 (0–0.4) | 1.64 (<0.24) | 0.25 (0.04–0.22) |
| C10 | 0.25, 0.86, 0.77 (0.04–0.30) | 0.9 (0–0.4) | 2.15 (<0.40) | 0.49 (0.04–0.30) |
| C12 | 0.15, 0.44, 0.64 (0.04–0.12) | 1.15 (0–0.34) | 1.64 (<0.22) | 0.49 (0.04–0.12) |
| C14 | 0.14, 0.23, 0.46 (<0.08) | 0.77 (0–0.19) | 0.94 (<0.12) | 0.39 (<0.08) |
| C14:1 | 1.08 (0–0.27) | 2.19 (<0.25) | ||
| C16 | 0.19, 0.19, 0.32 (0.06–0.24) | 0.91 (0–0.74) | 0.61 (<0.28) | 0.37 (0.06–0.24) |
| C18:1 | 0.16, 0.13, 0.42 (0.06–0.28) | 1.45 (<0.40) | 0.53 (0.06–0.28) | |
| C18:2 | 0.44 (<0.23) | |||
| Reference | (Bosch et al., 2011) | (Ho et al., 2011) | This report | |
a Results for a single patient; profiles vary considerably.
b Derived from impaired dehydrogenation of isovaleryl-CoA (intermediate in leucine metabolism). All other acylcarnitine species can be explained by impaired fatty acyl-CoA dehydrogenation steps.
Concentrations are in micromolarity per litre. Abnormal results in bold, reference range for the laboratory within parentheses.
Discussion
SLC52A2 contains five exons and spans ∼3.4 kb across chromosome 8. It produces a 1952-nucleotide transcript in which the longest open reading frame is 1338 nucleotides, encoding solute carrier family 52, riboflavin transporter member 2, a protein of 445 amino acids. Both SLC52A3 and SLC52A1 are highly expressed in the small intestine (and testis and placenta, respectively), suggesting that these are riboflavin transporters in these tissues. SLC52A2 is also expressed in the small intestine, although at a much lower level compared with brain tissue, where its expression is particularly pronounced (Yao et al., 2010). SLC52A2 shares 86.7 and 44.1% amino acid identity with SLC52A1 and SLC52A3, respectively (Yao et al., 2010). The variant we identified, p.G306R, lies in a highly conserved region of exon 3 in SLC52A2 (Figs 3–5).
Figure 3.

Comparison of SLC52A2 and SLC52A1 protein sequence. Red letters indicate amino acid residue 306.
Figure 4.

Structure of riboflavin and metabolites. RF and its cofactor forms containing additional ADP or phosphate groups at the 5′ hydroxyl side chain terminus of riboflavin to produce FMN or FAD. Riboflavin can freely diffuse in and out of the cell, FAD and FMN are metabolically trapped within the cell. FMN = flavin mononucleotide; FAD = flavin adenine dinucleotide; RF = riboflavin.
Figure 5.

Riboflavin transport and metabolic trapping of FMN and FAD. The RFK product, riboflavin kinase, catalyzes the phosphorylation of riboflavin (vitamin B2) to form FMN. The FLAD1 product is thought to catalyze the adenylation of FMN to form FAD. ETF = electron transport flavoprotein; CoQ = coenzyme Q (ubiquinone); ETFDH = electron transport flavoprotein dehydrogenase; FMN = flavin mononucleotide; FAD = flavin adenine dinucleotide; RF = riboflavin. Note the three riboflavin transporters are variably expressed in disparate tissues (Yao et al., 2010).
When mutations in SLC52A3 were found in patients with Brown–Vialetto–Van Laere syndrome (Green et al., 2010; Johnson et al., 2010), the significance of the fact that the gene product was a riboflavin transporter was unclear (Green et al., 2010). For children with this untreatable progressive condition, it was important to consider that correction of intracellular riboflavin deficiency might offer hope for stabilization of the disorder. Bosch et al. (2011) showed that children with Brown–Vialetto–Van Laere syndrome and mutations in SLC52A3 did indeed have biochemical evidence of riboflavin deficiency. They observed in these patients urine organic acid and plasma acylcarnitine profiles suggestive of a mild form of MADD (glutaric aciduria type II). MADD is an inherited condition that develops as a result of defective dehydrogenation reactions in mitochondrial fatty acid beta-oxidation and branched-chain amino acid catabolism. In classical MADD, electron donors to flavin adenine dinucleotide, electron transport flavoprotein or electron transport flavoprotein dehydrogenase are absent. Flavin adenine dinucleotide is a metabolite of riboflavin; therefore, riboflavin deficiency impairs the same dehydrogenation reactions as those in MADD and leads to the characteristic MADD biochemical profile. Bosch et al. (2011) treated patients with Brown–Vialetto–Van Laere syndrome with high dose riboflavin and observed a rapid improvement in muscle weakness and correction of biochemical abnormalities and this has also been seen in a patient from the United Kingdom (Houlden, personal communication). There has recently been another report of a patient with a complex neonatal multisystem neurological disorder and a biochemical profile suggestive of MADD. Screening of SLC52A1 was undertaken because it had been identified as a riboflavin transporter. Analysis revealed that the affected infant’s mother had a heterozygous deletion of SLC52A1 spanning exons 2 and 3. The authors postulated that dietary riboflavin deficiency during pregnancy, coupled with defective transport of riboflavin in the mother led to riboflavin deficiency in the infant. The clinical symptoms and biochemical abnormalities observed in the infant resolved completely on treatment with riboflavin (Ho et al., 2011). Taken together, these reports and our current data on SLC52A2 support a role for riboflavin transport defect and the development of Brown–Vialetto–Van Laere syndrome and provide support for riboflavin supplementation as a treatment (Figs 4 and 5); however, we would note that given the severity of this disorder we are still cautious on the overall response that can be achieved.
In this study, we identified a missense mutation p.G306R in SLC52A2 that segregated with disease in the Lebanese family. An additional variant P140L in MAPK15, was observed segregating in cis with this mutation in the Lebanese family. Although we were unable to test DNA from the patient’s father, the autozygosity data would suggest a low level of parental consanguinity. Screening of SLC52A2 in a cohort of patients with childhood onset motor neuron disease revealed one further case with the same p.G306R mutation but a different ethnic background and genetic haplotype that did not contain the MAPK15 variant. This confirms the pathogenic nature of the SLC52A2 gene mutation but does suggest that such defects are rare. We did not identify mutations in SLC52A1 in any of our cases with childhood motor neuron disease. The fact that two genes have been identified in riboflavin receptor genes, where defects are associated with childhood motor neuron disease, suggests that other genes in riboflavin transport or metabolism are likely to be causative in this patient group.
Although Brown–Vialetto–Van Laere syndrome is a rare disorder, it is extremely important to genetically characterize it to allow the accurate diagnosis of patients but more importantly to target those with riboflavin transport gene defects for treatment by supplementation. The identification of these genes will also improve our understanding of the related motor neuron diseases such as amyotrophic lateral sclerosis, where riboflavin transport gene defects may contribute to the common sporadic form of this disease and open up a potential therapeutic option. Given that several cases and families with Brown–Vialetto–Van Laere syndrome remain unexplained, the opportunity exists for further genetic studies in this area where a similar combined DNA array and exome sequencing approach offer promise of identifying these genes.
Funding
This work was supported, in part, by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke and the National Institute on Ageing, National Institutes of Health, Department of Health and Human Services; project number Z01 AG 000958-08. We thank the MRC (H.H. MRC fellowship G0802760 and MRC neuromuscular centre grant) and the Wellcome Trust for financial support. A.R.F. is a Muscular Dystrophy Campaign fellow. This study was supported by the NIHR UCLH/UCL Comprehensive Biomedical Research Centre. F.M. is supported by the Great Ormond Street Children’s Charity.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- MADD
multiple acyl-CoA dehydrogenase deficiency
- SNP
single-nucleotide polymorphism
References
- Bosch AM, Abeling NG, Ijlst L, Knoester H, van der Pol WL, Stroomer AE, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2011;34:159–64. doi: 10.1007/s10545-010-9242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CH. Infantile amyotrophic lateral sclerosis of the family type. J Nerv Ment Dis. 1894;21:707–16. [Google Scholar]
- Gallai V, Hockaday JM, Hughes JT, Lane DJ, Oppenheimer DR, Rushworth G, et al. Ponto-bulbar palsy with deafness (Brown-Vialetto-Van Laere Syndrome) J Neurol Sci. 1981;50:259–75. doi: 10.1016/0022-510x(81)90172-6. [DOI] [PubMed] [Google Scholar]
- Gibbs J, Singleton A. Application of genome-wide single nucleotide polymorphism typing: simple association and beyond. PLOS Genet. 2006;2:1511–7. doi: 10.1371/journal.pgen.0020150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green P, Wiseman M, Crow YJ, Houlden H, Riphagen S, Lin JP, et al. Brown-Vialetto-Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in C20orf54. Am J Hum Genet. 2010;86:485–9. doi: 10.1016/j.ajhg.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho G, Yonezawa A, Masuda S, Inui K, Sim KG, Carpenter K, et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum Mutat. 2011;32:E1976–84. doi: 10.1002/humu.21399. [DOI] [PubMed] [Google Scholar]
- Johnson JO, Gibbs JR, Van Maldergem L, Houlden H, Singleton AB. Exome sequencing in Brown-Vialetto-van Laere syndrome. Am J Hum Genet. 2010;87:567–9; author reply 569–70. doi: 10.1016/j.ajhg.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlenbaumer G, Hullmann J, Appenzeller S. Novel genomic techniques open new avenues in the analysis of monogenic disorders. Hum Mutat. 2011;32:144–51. doi: 10.1002/humu.21400. [DOI] [PubMed] [Google Scholar]
- Laere JV. Un nouveau cas de paralysie bulbo-pontine chronique progressive avec surdité. Rev Neurol. 1977;133:119–24. [PubMed] [Google Scholar]
- Mégarbané A, Desguerres I, Rizkallah E, Delague V, Nabbout R, Barois A, et al. Brown-Vialetto-Van-Laere syndrome in a large inbred Lebanese family: confirmation of autosomal recessive inheritance? Am J Med Genet. 2000;92:117–21. doi: 10.1002/(sici)1096-8628(20000515)92:2<117::aid-ajmg7>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Sathasivam S. Brown-Vialetto-Van Laere syndrome. Orphanet J Rare Dis. 2008;3:9. doi: 10.1186/1750-1172-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Scholz S, Matarin Mdel M, Fung HC, Hernandez D, Gibbs JR, et al. Genomwide SNP assay reveals mutations underlying Parkinson’s disease. Human Mut. 2008;29:315–22. doi: 10.1002/humu.20626. [DOI] [PubMed] [Google Scholar]
- Vialetto E. Contributo alla forma ereditaria della paralisi bulbare progresiva. Rive Sper Freniat. 1936;40:1–24. [Google Scholar]
- Yao Y, Yonezawa A, Yoshimatsu H, Masuda S, Katsura T, Inui K. Identification and comparative functional characterization of a new human riboflavin transporter hRFT3 expressed in the brain. J Nutr. 2010;140:1220–6. doi: 10.3945/jn.110.122911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}