

Abstract
The genetic basis of neurodevelopmental and neuropsychiatric diseases has been advanced by the discovery of large and recurrent copy number variants significantly enriched in cases when compared to controls. The pattern of this variation strongly implies that rare variants contribute significantly to neurological disease; that different genes will be responsible for similar diseases in different families; and that the same “primary” genetic lesions can result in a different disease outcome depending potentially on the genetic background. Next-generation sequencing technologies are beginning to broaden the spectrum of disease-causing variation and provide specificity by pinpointing both genes and pathways for future diagnostics and therapeutics.
Introduction
High-throughput genetic analyses, including current advances in detecting copy number variants (CNVs) and single nucleotide variants (SNVs), are leading to an explosion in the number of candidate genes and genomic regions contributing to neurodevelopmental disease [1-3]. This has been accompanied by a change in focus from a genetic model involving common genetic variants (>1 % frequency) to rare variants of high impact that are collectively common. CNV analyses, in particular, have led to the identification of numerous genomic regions, which, when deleted or duplicated, increase risk for autism, schizophrenia, epilepsy, and numerous intellectual disability phenotypes. Several themes have emerged from these and recent genome sequencing studies. 1) Every human carries a surprisingly large number of essentially disruptive mutations that are extremely rare (estimated at 250-300 genes per individual) [4]. 2) For certain neurological diseases there is an emerging view that there is an overall increase in the burden of the most disruptive mutations (i.e., larger CNVs). 3) Dozens of mutations in different regions and different genes have been identified for seemingly identical neurodevelopmental disorders. This is in stark contrast to earlier Mendelian disease models where one gene was primarily responsible for diseases such as Huntington disease, Duchenne muscular dystrophy, and familial Parkinsonism. 4) Finally, mutations in the same gene or seemingly identical large CNVs may result in very different disease outcomes. Interpretations of these findings are compounded by the lack of a consensus phenotyping criteria and the notion of various subtypes for a given “umbrella disorder.” These observations have suggested a more complicated genetic model underlying the etiology of neurodevelopmental disorders. Given their population prevalence and the cost to the healthcare system, genetics provides the best prospect to furthering our understanding of their cause, genetic counseling options, and eventually improved treatments. In this review we will highlight the current status of whole-genome efforts to identify genes and discuss their implications in the context of a common neurodevelopmental model for disease.
Copy number variation in neurodevelopmental disorders
Genetic linkage analysis and chromosome karyotype analyses initially played a key role in the discovery of genes important in neurological disease [5-9]. Many of these disorders were clinically well-defined and relatively quite rare facilitating their rapid genetic delineation. Other more common and complex phenotypes, including developmental delay, epilepsy, schizophrenia, and autism, in the general sense, have been genetically more elusive although successful linkage studies suggested a model where different genetic loci (each in a different family) contributed to disease. Genome-wide association studies (GWAS) have identified relatively few common variants of large effect, leading to increased interest in a rare-variant common-disease model to help explain the missing heritability [10].
Strong support for this model arose from the initial studies of large copy number variation of idiopathic cases of disease [11-15]. CNV studies using array comparative genomic hybridization (arrayCGH) or single nucleotide polymorphism (SNP) microarrays identified numerous large deletions and duplications among patients with neurodevelopmental disease. While very few specific loci initially reached statistical significance, a consistent pattern of increased CNV burden, particularly for large CNVs affecting multiple genes, was reported. This heterogeneity meant that sample sizes of a several hundred patients were insufficient to claim significance at the individual CNV level. Two studies recently examined CNVs in some of the largest cohorts (~15,000) of children with developmental delay and intellectual disability [1,2]. This has led to the discovery of new sites of potentially pathogenic variation and refinement of the smallest region of overlap of previously identified loci [1]. There are now 59 CNVs corresponding to 39 distinct genomic regions that show enrichment in CNVs among cases when compared to controls. The majority are large (>300 kbp), comprising on average 10-15 genes. Each CNV locus is extremely rare, with the most recurrent flanked by segmental duplications, which elevate local CNV mutation rates as a result of unequal crossover [14,16,17]. For Intellectual Disability/Developmental Delay (ID/DD), 25.7% of ascertained cases carry a CNV greater than 400 kbp in size, compared to only 11.5% of the control population (Figure 1A) [1]. It is, thus, predicted that 14.2% of disease may be associated with large CNVs. This matches well with the diagnostic yield of 14.7% described by Kaminsky et al., which also focused on large CNVs (primarily >500 kbp) [2], and previous studies reviewed in Hochstenbach et al. 2011 [18].
Figure 1. CNV burden and de novo rates.
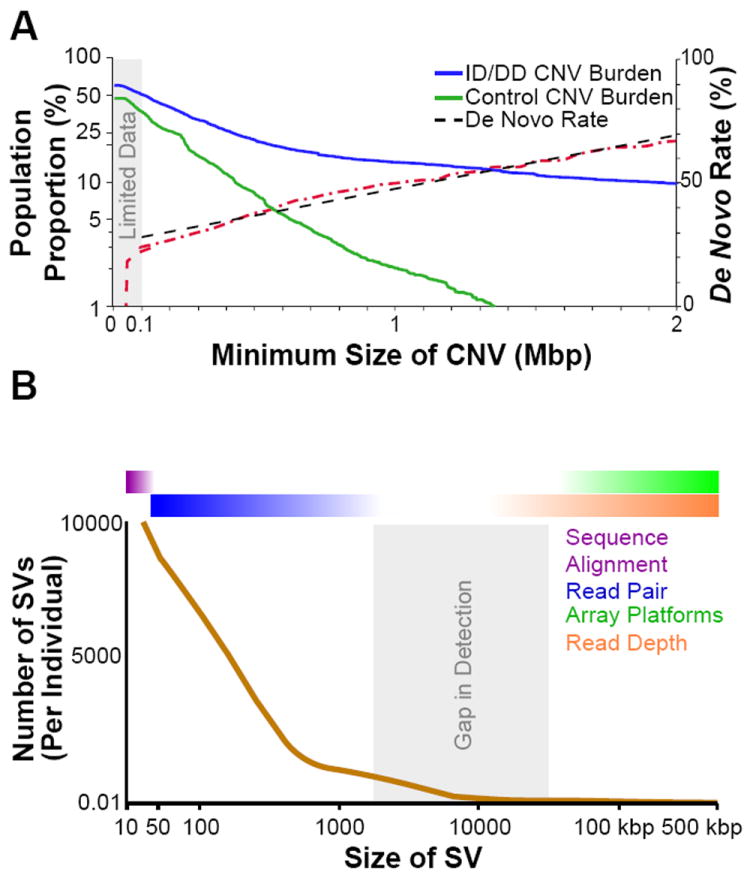
(A) CNV burden becomes significantly greater at >100 kbp in cases (blue line) of developmental delay when compared to controls (green). The observed size of a large CNV (>100 kbp) is linearly proportional (black line) to the odds that it originates sporadically. We anticipate that this trend (red line) will hold below 100 kbp (dropping exponentially as it approaches the size of an indel). In conjunction with an increase in CNV rates for smaller events, this implies that a significant amount of de novo CNV load remains to be discovered in the under-ascertained 10–50 kbp range. (B) The relative number of structural variants (SVs) detected in a normal individual is displayed as a function of SV size. The number of SVs begins to climb exponentially below ~500 bp reaching on the order of 10,000 events per person at 50 bp. Conversely, large events are significantly rarer with 500 kbp events detected in only a subset (9%) of individuals. Current paired-end sequence methodologies lose detection sensitivity above ~10 kbp, while array-based platforms rapidly lose sensitivity below 50–100 kbp. As a result, CNVs in this moderate size range are under-ascertained by the majority of methodologies.
Comparisons of CNV burden for different diseases suggest increasing CNV burden in disorders with greater phenotypic severity (defined here in terms of increasing comorbid cognitive impairment and congenital abnormalities) [1,19,20]. Cases with congenital malformations demonstrate the greatest increase in CNVs followed by ID/DD. Intermediate burdens were observed for idiopathic generealized epilepsy (IGE), autism spectrum disorder (ASD) and schizophrenia. While CNV studies for children with ID/DD show an increase of 14.2% for large CNVs, the burden is lower for ASD [1,21,22] with a diagnostic yield around 6-8% predominantly from de novo CNVs [18,21,22]. Studies of adults with schizophrenia and epilepsy demonstrate lower diagnostic yields of ~5% [11,12,18,23,24]. For other neurodevelopmental conditions there is either conflicting evidence for increased CNV burden, such as for bipolar disorder [25,26], or no evidence, as for Tourette’s syndrome and dyslexia [19,20,27]. In all cases burden statistics are more significant when considering de novo events exclusively [15,18,21,22,26].
One possible interpretation of these results is that large CNVs, by virtue of affecting more genes by way of dosage imbalance, demonstrate a larger effect size than small CNVs and SNP variants. Consistent with this observation, the odds of a CNV being de novo is linearly proportional to its size with CNVs larger than 1 Mbp being more likely to arise sporadically than inherited from a parent (Figure 1A). There is compelling evidence, then, that CNVs are under strong purifying selection in the general population [24,26,28]. Remarkably, as much as 8-10% of the general population carries such extremely rare or private CNVs suggesting that they must play an important role in human health.
Size spectrum of copy number variation
The majority of CNV loci convincingly classified as pathogenic to date are large (>500 kbp). We posit that this reflects both a technological limitation and an ascertainment bias as a result of the mutation severity. Affordable whole-genome sequencing [29] has revealed a plethora of uncharacterized genetic variation below the lower limits of arrayCGH and SNP array platforms, which rapidly lose genome-wide sensitivity below 50 kbp for most commercial arrays [30]. The number of CNVs per individual increases linearly as sizes approach 100 kbp (closely related to the de novo rate and matching observations of selection against large CNVs), but then begins to increase exponentially for CNVs less than 10 kbp in size (Figure 1B) [31]. This poses a significant challenge in the study of smaller variants that are more likely to be inherited (Figure 1A) but present at a far greater count in any given individual (Figure 1B). Larger, ethnically matched control populations in conjunction with information regarding the inheritance will be required to assess significance as “the haystacks get larger and the needles become smaller.”
Whole-genome sequencing experiments focus primarily on detecting SNPs and indels (typically below ~20 bp in length). The analysis of copy number and structural variation is frequently an afterthought requiring specialized and computationally intensive methods [4,29,30]. No method is comprehensive and each differs in its sensitivity as a function of size and class of CNVs. Read-pair methodologies, for example, are most sensitive to events between 40 bp and 1 kbp (depending on library insert sizes and consistency) [30-33]. Read-depth methodologies are powerful for detecting copy number changes greater than 10 kbp and are dependent on sequence coverage, which limits the number of genomes that can be analyzed [29,30,34-37]. This leaves a gap in our detection abilities between about 1 kbp and 50 kbp where performance is suboptimal (Figure 1B).
Despite these challenges, studies are beginning to approach the gap in genetic variation from both ends. In addition to point mutations and indels [38,39], exome sequencing data can be used to discover larger events of potential relevance to disease [40,41]. Similarly, higher resolution and targeted array studies have identified numerous candidate CNVs well below 500 kbp [42-46]. Some recent prominent examples include a small duplication of VIPR2 in schizophrenia, and focal partial deletions of TMHLE in ASD [44,47,48]. Smaller CNVs also confer the advantage of reducing the smallest region of overlap for large CNV regions, such as the potential refinement of the 15q13.3 microdeletion to CHRNA7, the 17q21.31 microdeletion to MAPT/KIAA1267 [1], and the 16p11.2 deletion to SEZL6 [49-51]. Importantly, small CNVs are converging at sites where other events such as rare point mutations and translocation breakpoints have highlighted candidate genes. One such example is the 2q23.1 deletion syndrome, which has been associated with intellectual disability, seizures, microcephaly, and speech delay. Recently, the minimal region has been fine mapped via deletions as small as 37 kbp to a single gene—MBD5 [1,52-55]. This gene has also been discovered to contain both a rare mutation and common variants as well as translocation breakpoints by sequencing studies of ID, epilepsy, and ASD (Figure 2) [52,53].
Figure 2. 2q23.1 microdeletion syndrome refinement.
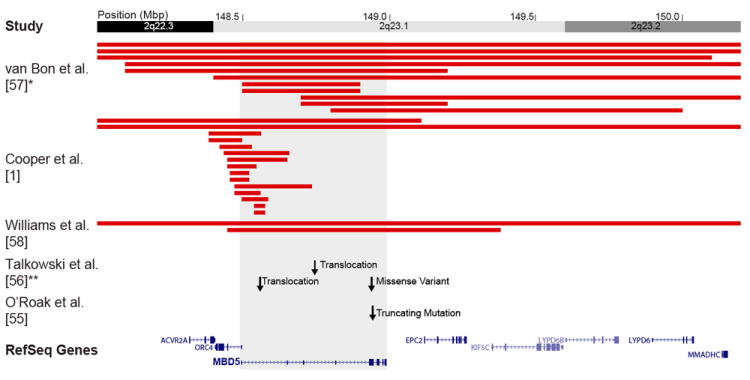
Highlighted are deletion cases from three recent studies that detected focal deletions (37.7 kbp to >1 Mbp) and point mutations converging on MBD5 as the critical gene for the 2q23.1 microdeletion syndrome. In a panel of 8,329 controls only two deletions were detected [1]. *Breakpoints for van Bon et al. [54] are rounded to the nearest 100 kbp. **CNVs from Talkowski et al. [53] overlap with cases from Cooper et al. [1] and are not shown.
These results hint that smaller CNVs are a promising direction for future studies complementing screens for disruptive point mutations by providing stronger priors for rare singleton mutations in the limited populations studied. This is especially important given our current abilities to apply arrayCGH in a clinical and basic research setting to tens of thousands of individuals, while sequencing is typically restricted to smaller cohorts (hundreds) for any given disease. Eventually, as we progress to more affordable sequencing of larger populations, a more comprehensive view of all forms of genetic variation will emerge. Integrating both CNV and sequence data will be mutually beneficial in pinpointing the most likely candidate genes.
A genetic model of neurodevelopmental disease
An interesting observation to emerge from CNV research has been the distinction between syndromic CNVs (e.g., Williams Syndrome deletion at 7q11.23) and CNVs that are much more variable in their outcome. The 15q13.3 microdeletion, for example, is significantly enriched in cases of autism, schizophrenia, and epilepsy being found in as many as 1% of IGE cases but absent in ethnically matched controls [1,2,22-24,56-59]. These and other observations imply that seemingly diverse disease states may share some common genetic, and perhaps neurodevelopmental, etiology. Moreover, the syndromic CNVs are most frequently de novo indicating that they are necessary and sufficient while the variable expressive CNVs are much more likely to be inherited from less severely affected parents. This indicates that they are, by themselves, insufficient to determine disease outcome [1,60]. The mechanisms for this variable expression are not clear but there is compelling evidence of an oligogenic model where multiple rare private variants of moderate to large effect compound to determine final phenotypic outcome [52,60,61]. This model is perhaps strongest for the 16p12.1 microdeletion. In this case, the deletions are almost always inherited; developmental delay patients carrying this microdeletion are significantly enriched for additional CNVs—so called second-site hits; and carrier parents of the 16p12.1 deletion are more likely to suffer from neuropsychiatric disease than noncarrier parents for the deletion [60]. These results suggest that the 16p12.1 microdeletion creates a sensitized state and that additional modifier loci (i.e., CNVs) are required to result in a child that is more severely developmentally disabled.
The implication of this model is both sobering and exciting. CNV and early genome sequencing experiments both implicate hundreds to thousands of different genes underlying various neurological phenotypes [22,39]. Analyses of candidate genes, however, are beginning to converge on a subset of protein-protein interaction networks or biological processes, many of which are strongly tied to neurodevelopment [18,52,62-64]. Genes involved in synaptic function have been repeatedly shown to play a role in neurological disorders [62,63,65,66]. Recent studies of ASD have identified overrepresentation of CNS development genes in addition to several other pathways [21,65,67]. In our own recent analysis of 209 exomes from patients with sporadic autism, we found that 39% of the de novo disruptive mutations formed a highly interconnected protein-protein interaction network involving beta-catenin upstream and downstream regulation (Figure 3) [52]. Notably, this pathway was not observed to be enriched in unaffected siblings and ranked significantly with respect to other autism candidate genes. Interestingly, several cases demonstrated multiple disruptive de novo events within this same network.
Figure 3. Protein-protein interaction network for ASD gene mutations.
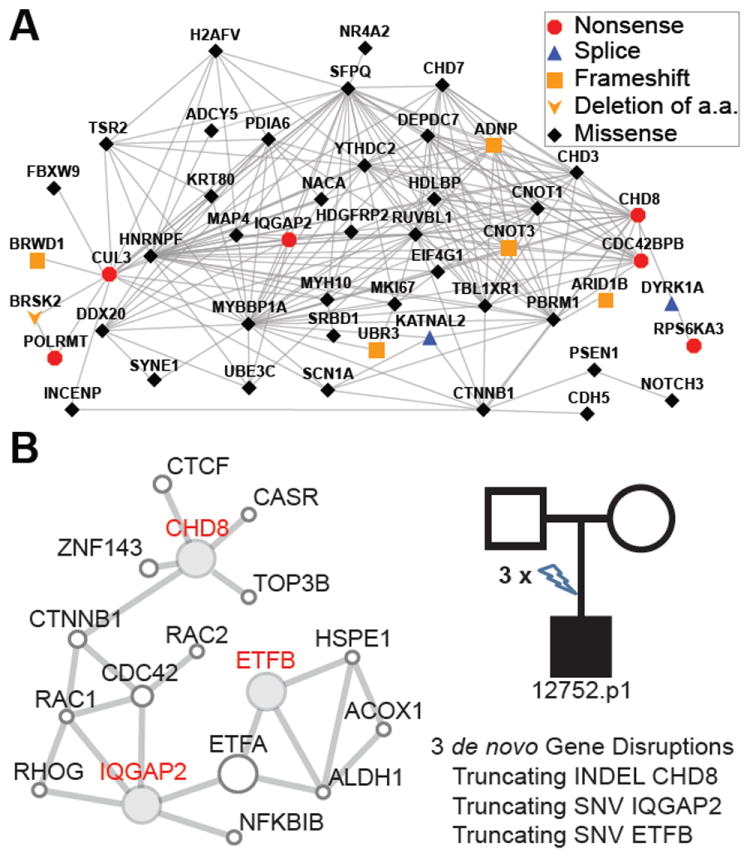
(A) Shown is a highly interconnected protein-protein interaction network consisting of 49/125 severe de novo gene mutations identified from 209 children with simplex autism described in O’Roak et al. [52]. The pathway is enriched in upstream and downstream regulators of beta-catenin. It ranks significantly with respect to autism genes and is not identified in similarly characterized unaffected siblings. (B) An example of a patient with autism who carries three de novo truncating mutations in genes that encode proteins that are part of a beta-catenin linked network.
Future Directions
The development of the human brain is complex involving thousands of different genes. With respect to disease, we envision that this process can be perturbed either by individual mutations of singularly large effect or by a few disruptive mutations in different genes (oligogenic) that compound at the molecular level to lead to variable outcomes with respect to neurodevelopmental disease. The former mutations are largely sporadic in origin while the latter are more likely to be inherited. In both cases, we propose that individual pathogenic or modifier loci are extremely rare but collectively common in the population. Under this model, the focus should be on the discovery of disruptive mutations in cases that are largely absent from the general population. The extreme locus heterogeneity implies that most initial sequencing studies will be woefully underpowered to prove pathogenicity of any given gene [52]. Power will arise from 1) the integration of both CNV and SNV data to triage specific regions or candidate genes; 2) consideration of disruptive mutations across different diseases (e.g., meta-analyses of epilepsy, schizophrenia, autism cohorts, etc.); and 3) pathway and functional analyses that converge on specific pathways (Figure 4). Once identified, pathways of clinical significance will become the target of both better diagnostics and, ultimately, therapeutics.
Figure 4. A model for neurodevelopmental disease.
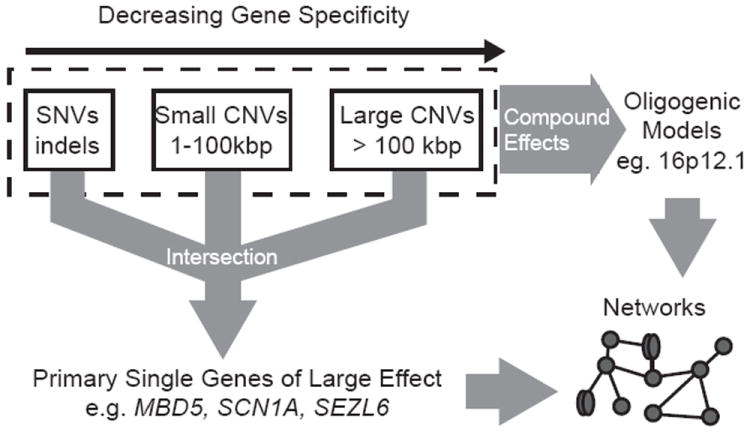
Two paths are shown involving either 1) mutation events of singularly large effect (e.g., a de novo CNV) resulting in disease or 2) rare mutations that compound to lead to disease of varying severity and expressivity. The intersection of CNV, indel, and point mutation can be used to readily identify primary genes where imbalance is sufficient to cause disease. In the second pathway, multiple events will need to co-exist in an individual to elicit a disease phenotype. Mutations in both converge on common protein interaction networks and genetic pathways important in neurodevelopment.
Highlights.
Current mutation discovery efforts for neurodevelopmental disorders are focused on large CNVs and small protein-coding SNVs and indels. Between these extremes lies an under-ascertained range of genetic variants between 1 and 50 kbp, representing a promising range for future expansion of gene discovery.
Extensive phenotypic variability has been documented for large CNVs with seemingly similar breakpoints. There is evidence in support of an oligogenic model, where multiple rare variants of large effect may aggregate within individuals and underlie this variability.
Despite extreme locus heterogeneity and seemingly disparate phenotypes, genetic studies are beginning to converge on a core set of biological pathways across a diverse array of neurodevelopmental conditions. This suggests that a large component of the genetic etiology may be shared. These pathways are important targets for diagnosis and therapeutic development.
Acknowledgments
We also thank Tonia Brown and Brian O’Roak for useful discussions and editing the manuscript. B.P.C. is supported by a fellowship from the Canadian Institutes of Health Research. E.E.E. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1**.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–846. doi: 10.1038/ng.909. The authors analyze 15,767 individuals with developmental delay and 8,329 control individuals. This study provided evidence for pathogenicity for 59 genomic regions, refined critical interval for a known genomic disorder region, and shows concordance of phenotypic severity with large CNV burden. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2**.Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13:777–784. doi: 10.1097/GIM.0b013e31822c79f9. The authors analyzed 15,749 individuals with developmental delay and 10,118 controls. This study showed significant enrichment for 14 deletions and 7 duplications among cases compared to controls. Overall, this study provided with evidence for pathogenicity of CNVs diagnosed routinely in laboratories. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet. 2010;11:161–187. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 4.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camerino G, Mattei MG, Mattei JF, Jaye M, Mandel JL. Close linkage of fragile X-mental retardation syndrome to haemophilia B and transmission through a normal male. Nature. 1983;306:701–704. doi: 10.1038/306701a0. [DOI] [PubMed] [Google Scholar]
- 6.Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5:11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- 7**.Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304:325–329. doi: 10.1056/NEJM198102053040604. This is the one of the first manuscripts to report on a Prader-Willi syndrome, a genomic disorder, resulting from a deletion of chromosome 15q11.2q13.1. [DOI] [PubMed] [Google Scholar]
- 8.Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, St Clair DM, Muir WJ, Blackwood DH, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- 9.Ottman R, Risch N, Hauser WA, Pedley TA, Lee JH, Barker-Cummings C, Lustenberger A, Nagle KJ, Lee KS, Scheuer ML, et al. Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet. 1995;10:56–60. doi: 10.1038/ng0595-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, Franke A, Malafosse A, Genton P, Thomas P, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. This is the first study to report the role of rare copy number variants in schizophrenia. The authors analyzed 150 cases and 268 ancestry-matched controls. They find about that 15% of cases carry a novel deletion or duplication and genes within these CNVs are enriched for signaling networks involved in neurodevelopment. [DOI] [PubMed] [Google Scholar]
- 13.de Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, Janssen IM, Reijmersdal S, Nillesen WM, Huys EH, Leeuw N, et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 15.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 17.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 18.Hochstenbach R, Buizer-Voskamp JE, Vorstman JA, Ophoff RA. Genome arrays for the detection of copy number variations in idiopathic mental retardation, idiopathic generalized epilepsy and neuropsychiatric disorders: lessons for diagnostic workflow and research. Cytogenet Genome Res. 2011;135:174–202. doi: 10.1159/000332928. [DOI] [PubMed] [Google Scholar]
- 19*.Girirajan S, Brkanac Z, Coe BP, Baker C, Vives L, Vu TH, Shafer N, Bernier R, Ferrero GB, Silengo M, et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 2011;7:e1002334. doi: 10.1371/journal.pgen.1002334. The authors in this study analyzed three neurological disorders including autism, developmental delay, and dyslexia and show an increase in large CNV burden correlating with the severity of the phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Girirajan S, Eichler EE. De Novo CNVs in Bipolar Disorder: Recurrent Themes or New Directions? Neuron. 2011;72:885–887. doi: 10.1016/j.neuron.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 21.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grozeva D, Kirov G, Ivanov D, Jones IR, Jones L, Green EK, St Clair DM, Young AH, Ferrier N, Farmer AE, et al. Rare copy number variants: a point of rarity in genetic risk for bipolar disorder and schizophrenia. Arch Gen Psychiatry. 2010;67:318–327. doi: 10.1001/archgenpsychiatry.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26**.Malhotra D, McCarthy S, Michaelson JJ, Vacic V, Burdick KE, Yoon S, Cichon S, Corvin A, Gary S, Gershon ES, et al. High Frequencies of De Novo CNVs in Bipolar Disorder and Schizophrenia. Neuron. 2011;72:951–963. doi: 10.1016/j.neuron.2011.11.007. Previous studies on bipolar disorder did not show a strong effect of common variants on bipolar disorder. This manuscript is one of the first studies to report enrichment of rare de novo CNVs in bipolar disorder and overlap with genes associated with schizophrenia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernandez TV, Sanders SJ, Yurkiewicz IR, Ercan-Sencicek AG, Kim YS, Fishman DO, Raubeson MJ, Song Y, Yasuno K, Ho WS, et al. Rare Copy Number Variants in Tourette Syndrome Disrupt Genes in Histaminergic Pathways and Overlap with Autism. Biol Psychiatry. 2011 doi: 10.1016/j.biopsych.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, Eichler EE. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE. Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hormozdiari F, Alkan C, Eichler EE, Sahinalp SC. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi J, Zhao F. inGAP-sv: a novel scheme to identify and visualize structural variation from paired end mapping data. Nucleic Acids Res. 2011;39:W567–575. doi: 10.1093/nar/gkr506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Wu Y. SVseq: an approach for detecting exact breakpoints of deletions with low-coverage sequence data. Bioinformatics. 2011;27:3228–3234. doi: 10.1093/bioinformatics/btr563. [DOI] [PubMed] [Google Scholar]
- 34.Campbell PJ, Stephens PJ, Pleasance ED, O’Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xi R, Hadjipanayis AG, Luquette LJ, Kim TM, Lee E, Zhang J, Johnson MD, Muzny DM, Wheeler DA, Gibbs RA, et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc Natl Acad Sci U S A. 2011;108:E1128–1136. doi: 10.1073/pnas.1110574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang ZD, Gerstein MB. Detection of copy number variation from array intensity and sequencing read depth using a stepwise Bayesian model. BMC Bioinformatics. 2010;11:539. doi: 10.1186/1471-2105-11-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38**.Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. This study systematically studied 136 consanguineous families to identify causal loci for autosomal-recessive intellectual disability. Strikingly, the authors identified 23 known genes and 50 novel genes across multiple cellular functions. These results support a model of oligogenic heterogeneity in intellectual disability and provide additional insights into disease processes. [DOI] [PubMed] [Google Scholar]
- 39*.O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. This study is the first report of mutations in sporadic autism using exome sequencing. The authors analyzed 20 trios and identified potentially causative mutations within FOXP1, GRIN2B, SCN1A, and LAMC3 in four probands. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karakoc E, Alkan C, O’Roak BJ, Dennis MY, Vives L, Mark K, Rieder MJ, Nickerson DA, Eichler EE. Detection of structural variants and indels within exome data. Nat Methods. 2011 doi: 10.1038/nmeth.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sathirapongsasuti JF, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, Quackenbush J, Nelson SF. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27:2648–2654. doi: 10.1093/bioinformatics/btr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Behnecke A, Hinderhofer K, Bartsch O, Numann A, Ipach ML, Damatova N, Haaf T, Dufke A, Riess O, Moog U. Intragenic deletions of IL1RAPL1: Report of two cases and review of the literature. Am J Med Genet A. 2011;155A:372–379. doi: 10.1002/ajmg.a.33656. [DOI] [PubMed] [Google Scholar]
- 43.Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, Kang SH, Yang Y, Wiszniewska J, Nowakowska BA, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Celestino-Soper PB, Shaw CA, Sanders SJ, Li J, Murtha MT, Ercan-Sencicek AG, Davis L, Thomson S, Gambin T, Chinault AC, et al. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet. 2011;20:4360–4370. doi: 10.1093/hmg/ddr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL, Korf BR, Carroll AJ. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet A. 2011;155A:2386–2396. doi: 10.1002/ajmg.a.34177. [DOI] [PubMed] [Google Scholar]
- 46.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, Zhang N, Mowry BJ, Olincy A, Amin F, et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vacic V, McCarthy S, Malhotra D, Murray F, Chou HH, Peoples A, Makarov V, Yoon S, Bhandari A, Corominas R, et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crepel A, Steyaert J, De la Marche W, De Wolf V, Fryns JP, Noens I, Devriendt K, Peeters H. Narrowing the critical deletion region for autism spectrum disorders on 16p11.2. Am J Med Genet B Neuropsychiatr Genet. 2011;156:243–245. doi: 10.1002/ajmg.b.31163. [DOI] [PubMed] [Google Scholar]
- 50.Konyukh M, Delorme R, Chaste P, Leblond C, Lemiere N, Nygren G, Anckarsater H, Rastam M, Stahlberg O, Amsellem F, et al. Variations of the candidate SEZ6L2 gene on Chromosome 16p11.2 in patients with autism spectrum disorders and in human populations. PLoS One. 2011;6:e17289. doi: 10.1371/journal.pone.0017289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51*.Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Jr, Dobyns WB, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. The first report of a single CNV locus associated with autism. Deletions of 16p11.2 account for about 1% of individual with autism. [DOI] [PubMed] [Google Scholar]
- 52.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith J, et al. Sporadic autism exomes reveal interconnected protein network and extreme locus heterogeneity. Nature. 2012 doi: 10.1038/nature10989. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, Repnikova EA, Gastier-Foster J, Thrush DL, Kathiresan S, Ruderfer DM, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011;89:551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Bon BW, Koolen DA, Brueton L, McMullan D, Lichtenbelt KD, Ades LC, Peters G, Gibson K, Moloney S, Novara F, et al. The 2q23.1 microdeletion syndrome: clinical and behavioural phenotype. Eur J Hum Genet. 2010;18:163–170. doi: 10.1038/ejhg.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williams SR, Mullegama SV, Rosenfeld JA, Dagli AI, Hatchwell E, Allen WP, Williams CA, Elsea SH. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet. 2010;18:436–441. doi: 10.1038/ejhg.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dibbens LM, Mullen S, Helbig I, Mefford HC, Bayly MA, Bellows S, Leu C, Trucks H, Obermeier T, Wittig M, et al. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet. 2009;18:3626–3631. doi: 10.1093/hmg/ddp311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57*.Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. This study identified that about 1% of individuals with idiopathic generalized epilepsy carry a deletion on 15q13.3. This locus has also been reported in individuals with intellectual disability, schizophrenia, and autism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. This study demonstrates an oligogenic model for neurodevelopmental disorders. The authors show that the 16p12.1 deletion is mostly inherited, associated with a milder phenotype, but in concert with another large CNV elsewhere in the genome, can result in severe developmental delay phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, Lewis L, Akbar H, Varghese R, Boerwinkle E, et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet. 2011;20:3366–3375. doi: 10.1093/hmg/ddr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64**.Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. This is one of the primary studies to suggest early neurodevelopment pathology interacting with normal brain maturation as a cause for adult onset schizophrenia. [DOI] [PubMed] [Google Scholar]
- 65.Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 67.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]