

Abstract
X-linked Infantile Spasms Syndrome (ISSX) is a catastrophic epilepsy of early childhood with intractable seizures, intellectual disability, and poor prognosis. A spectrum of mutations in the Aristaless-Related Homeobox gene (ARX) has been linked to ISSX, and downstream targets of this interneuron-expressed transcription factor are being defined. Recent advances combining in vitro and in vivo methods have unveiled complex interactions between Arx and its binding partners and their effects on cell migration and maturation that can help explain the diversity of ARX phenotypes. New mutant mouse models of Arx-induced pathology, including a recent human triplet-repeat expansion mutation with a phenotype of infantile spasms and electrographic seizures, provide valuable tools for exploring the pathophysiology of Arx and substrates for testing novel therapies.
Introduction
X-linked Infantile Spasms Syndrome (ISSX; OMIM ID: 308350) is a rare and catastrophic epilepsy syndrome of childhood that consists of early-onset myoclonic spasms, irregular high voltage EEG waves termed hypsarrhythmia, followed by intractable seizures and intellectual disability (ID) associated with autistic features. ISSX is one of over a dozen infantile spasms syndromes for which single genes have been discovered in the last decade, triggering a molecular reclassification of this major category of pediatric epilepsy [1*]. While the clinical spasms usually subside by age 5, epilepsy and ID persist through adulthood. Although a brief course of adrenocorticotropic hormone (ACTH) therapy can control spasms in the short term, there are concerns about long-lasting side effects and its overall efficacy [2, 3, 4]. In addition, no current therapies have been shown to prevent epilepsy or improve the intellectual outcome of patients with ISSX [5, 6, 7, 8, 9]. Therefore, understanding the mechanisms underlying ISSX and developing novel therapies remain critical scientific and clinical priorities.
The last decade has witnessed critical advancements in our understanding of the pathophysiology of the syndrome, beginning with the finding that expansions in the first and second poly-alanine tracts (pA1 and pA2) in the transcription factor gene Aristaless-Related Homeobox (ARX) (GenBank ID: NG_008281.1) cause ISSX [10, 11, 12, 13]. ARX mutations cause a spectrum of clinical disorders, from X-linked Lissencephaly and Ambiguous Genitalia (XLAG; OMIM ID: 300004) to ISSX and non-syndromic mental retardation (OMIM ID: 300419) [14]. In the CNS, ARX is expressed in subpallial proliferative zones and in developing and adult interneurons, where, under the influence of DLX1/2 and a downstream network of transcription factors, it plays a pivotal role in migration and differentiation [15, 16, 17, 18, 19, 20, 21].
The first mouse model to be generated, the constitutive Arx knockout mouse (ArxKO), displays XLAG-like characteristics with a small brain and severe interneuron migration deficit. ArxKO mice die soon after birth, preventing the study of the role of Arx in postnatal neurodevelopment [22] (Table 1). To overcome this limitation, several groups have developed postnatally viable rodent models of Arx mutations featuring full longevity. Also, the past two years have seen significant progress in exploring the molecular pathology of Arx mutations and their impact on gene expression during development.
Table 1.
Current Arx mouse models
| Model | Mutation | Viability | Spasms | Epilepsy | Brain | Comments | Ref. |
|---|---|---|---|---|---|---|---|
| ArxKO | Stop codon inserted in exon 2 | Perinatal lethal | No data | No data | Gross malformation, DG dysgenesis, CC agenesis, cortical Cb+ interneurons | Phenotype resembles human XLAG | 22 |
| Arx-/Y | cKO of exon 2 in interneurons expressing Dlx5/6 | ≥ 120 days | Only in adult mice; no spasms in infant mice | Myoclonic seizures in males and females at P14-17 and P90-120 | No gross malformations; ↓ Cb+ interneurons | Seizures with features resembling human infantile spasms in adult mice only | 43 |
| Arx(GCG)7 | 7 GCG triplets inserted in pA1 | ≥ 5 mos. | No spasms in infancy noted | Tonic clonic seizures in 1-month-old males, no interictal spikes detected | No gross malformation; ↓ Arx+ and striatal ChAT+ interneurons | Phenotype resembles human ISSX | 44 |
| ArxPL | Pro to Leu substitution at position 355 (353 in humans)* | ≥ 5 mos. | No spasms in infancy noted | Increased lethality after bicuculline-induced seizures | No gross malformation; ↓ tangential migration; ↓ striatal ChAT+ interneurons | *Missense mutation in the homeodomain | 44 |
| ArxPR | Pro to Arg substitution at position 355 (353 in humans)* | Perinatal lethal | No data | No data | Gross brain malformation; ↓ radial and tangential migration; ↓ striatal ChAT+ interneurons | *Missense mutation in the homeodomain. Phenotype resembles human XLAG | 44 |
| Arx(GCG)10+7 | 8 GCT triplets inserted in pA1* | Normal life span | Transient spasms detected between P7 and P11 | Frequent spontaneous interictal spikes and electrographic seizures with behavioral arrest | No gross malformation; ↓ cortical Cb+ interneurons; ↓ striatal ChAT+ and NPY+ and Cb+ interneurons | *The expanded mouse pA1 replicates the human-like mutation. Phenotype resembles human ISSX | 31 |
Abbreviations: DG (Dentate Gyrus), CC (Corpus Calosum), Cb (Calbindin), NPY (Neuropeptide Y), ChAT (Choline Acetyl Transferase), ↓ (decrease in expression)
Mutations in ARX domains lead to distinct phenotypes
ARX has several conserved domains: the octapeptide domain, 3 nuclear localization sequences (NLS1-3), 4 poly-alanine tracts (pA1-4), the DNA-binding homeodomain (HD) and the Aristaless domain [11,15] (Fig. 1). Truncations and HD mutations commonly cause severe phenotypes, whereas missense mutations and in-frame expansions of the first two pAs associate with the more neurologically restricted ARX phenotypes, such as ISSX [14]. Despite this apparently consistent genotype-phenotype relationship, precise details on the complex molecular pathology of different ARX mutations are poorly understood. In recent years, several attempts to reproduce the most common ARX mutations and investigate their downstream effects have appeared and are reviewed here.
Figure 1.
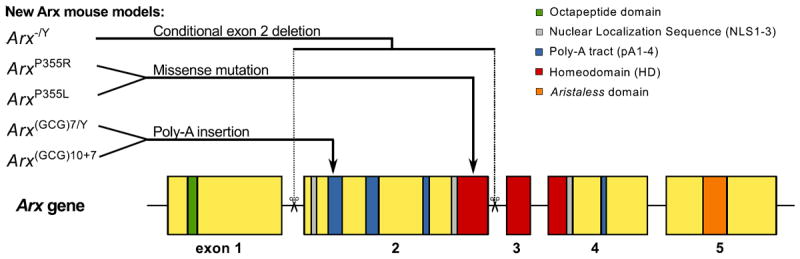
Diagram of Arx gene and its major conserved functional domains. These domains include the Octapeptide domain, 3 Nuclear Localization Sequences (NLS1-3), 4 poly-alanine tracts (pA1-4), the DNA binding Homeodomain (HD), and the Aristaless domain. The general location of the mutations of current experimental Arx mouse models is shown.
The transfer of Arx into the nucleus is mediated by importins, which transiently bind to the NLS and release Arx inside the nucleus by a RanGTP-dependent mechanism [23,24]. In an in vitro trafficking study, Arx proteins with naturally occurring NLS mutations and overexpressed in HEK293 cells accumulated next to the nuclear membrane and co-localized with the importin, IPO3. In contrast, WT Arx diffusely stained throughout the nucleus and did not co-localize with IPO3, suggesting that IPO3 normally rapidly dissociates from Arx once inside the nucleus. Interestingly, binding of NLS mutated Arx to IPO3 was indistinguishable from WT Arx. However, mutant Arx proteins did not dissociate from IPO3 once inside the nucleus, suggesting that the principal defect is the inability of mutant Arx to uncouple from IPO3 [25*].
Arx directly represses the transcription of Lmo1, Ebf3, and Shox2 [26,27**] (Fig. 2). To investigate how missense HD mutations may affect this repression, Shoudbridge et al. (2011) measured transcript levels of Lmo1 and Shox2 in HEK293T cells transiently overexpressing either WT Arx, NLS, or HD mutants. Electrophoretic mobility shift assays (EMSA) and ChIP-qPCR data suggest that HD mutations lead to de-repression of Arx targets by loss of DNA binding. NLS mutations also result in transcriptional de-repression, which could be explained by the formation of peri-nuclear aggregates. Conversely, HD mutants would not be expected to mislocalize or form aggregates, yet some HD mutations (i.e. L343Q, P535R, and R358S) display double the rate of protein mislocalization relative to WT-Arx. Although below the 5-7 fold rate of abnormal protein localization observed in NLS mutants, this result raises the possibility that some HD mutations partially disrupt nuclear translocation [28*].
Figure 2.
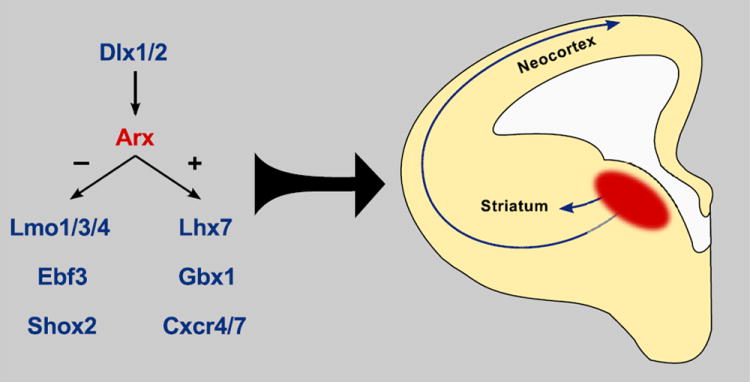
Arx regulates transcription of genes involved in interneuron development. The transcription factors Dlx1/2 upregulate Arx expression. Arx itself acts as both a transcriptional suppressor and an activator. Arx regulates transcription of several genes, including other transcription factors such as Lhx7, Gbx1, Ebf3, Lmo1/3/4, and Shox2, each of which play distinct roles in interneuron proliferation, migration, and differentiation. Other targets, such as Cxcr4, encode chemokine receptors directly involved in regulation of interneuron migration during embryonic and early postnatal development.
A 24-bp duplication in pA2 and a 7-alanine expansion in pA1 are both common causes of ISSX, although other mutations have been reported [10,11,13]. The connection between relatively small in-frame expansions in a poly-alanine tract and the cellular pathogenesis of infantile spasms, seizures, and intellectual disability is unclear. One hypothesis, that expansions of Arx lead to misfolding and aggregation, has gained traction after the finding that overexpression of Arx with expanded pA1 and pA2 in vitro caused Arx mislocalization and increased cell death [23,29]. Recently, heterologous overexpression of expanded Arx proteins in HEK293 cells produced similar results [30]. New mutations in pA1 and pA2 found in patients were introduced into HEK293T cells. The degree of protein mislocalization correlated with the length of the pA expansion and phenotypic severity. Protein mislocalization was also observed in Arx proteins containing point mutations in the HD [28*]. Although nuclear translocation defects and protein misfolding can result in mislocalization, it is possible that the degree of mislocalization and aggregation observed in heterologous overexpression systems may not be the same in developing neurons. In fact, the recently reported pA1 expansion mouse model does not show increased cell death, and protein mislocalization is nonuniform, appearing predominantly in a subset of interneurons in the somatosensory cortex [31**]. Therefore, protein mislocalization may only partially explain Arx pA1 expansion cellular pathology; and other defects in regulation of gene expression may be contributing factors. In vivo data from electroporation of ArxE (8-alanine expansion in pA1) in Arx-/Y embryos support this notion. While WT-Arx rescued radial and tangential migration, ArxE partially rescued radial, but not tangential, migration. This was accompanied by a selective transcriptional de-repression of Arx direct targets, raising the possibility that radial and tangential migration modes require expression of distinct gene subsets [32**]. These results disagree with in vitro data where increasing the length of pA1 and pA2 resulted in increased transcriptional repression as measured by a reporter vector [33].
Co-factors can refine the activity of transcription factors with cell-type specificity. In the case of Arx, the co-repressor Tle1 displayed diminished binding to ArxE in vitro. It is possible that differential expression of Tle1 and other unknown Arx co-factors across the forebrain may partially explain the different effects of ArxE on radial and tangential migration and the broad spectrum of ARX phenotypes [32**,33]. Overall, these studies paint a complex picture where protein mislocalization and selective gene deregulation both contribute to the molecular pathogenesis of Arx pA expansions.
Transcriptional control of Arx targets
Mutations that impair DNA binding, nuclear translocation, or interactions with co-repressors are likely to differentially impact the expression of Arx targets, which may partially explain the broad spectrum of ARX-related phenotypes. In fact, relatively subtle single nucleotide homeodomain mutations display significant differences in transcriptional repression [34]. In C. elegans, deletion of the Arx orthologue, alr-1, results in a highly variable touch-insensitive phenotype, suggesting a role for alr-1 in maintaining a high expression level of its targets by minimizing stochastic variation in their transcription [35*]. Although it is difficult to translate these results to mammalian brain, they may contribute to the clinical variability in humans with ARX mutation, notwithstanding environmental and genomic individual differences.
Given the dimensions and severity of human ARX-related disease, the identification of novel ARX targets and elucidation of their individual roles in neurodevelopment are high priorities. Recent microarray and qPCR efforts have led to the discovery of 35-84 genes potentially regulated by Arx. Three of these genes have been confirmed as direct targets: Lmo1, Shox2 and Ebf3. In addition, Arx modifies the brain expression of Lmo3, Lmo4, Cxcr4, Cxcr7, and Lhx7, among others [26,27**]. Cxcr4 and Cxcr7 are receptors that bind stromal cell-derived factor α (SDF-α) and play distinct roles in interneuron migration [36, 37, 38, 39, 40, 41]. The transcription factor gene Ebf3 is strongly repressed by Arx and its ectopic expression in Arx null mutants halts the normal migration of interneurons, indicating that its gene targets are key regulators of cortical development. In contrast, ectopic expression of Lmo1, another transcription factor gene normally repressed by Arx, did not result in detectable migration deficits [27**]. Dozens of potential novel downstream targets have recently been identified using a high-throughput method to detect Arx binding to promoter regions, enriching the already complex pool of genes controlling interneuron development that may be regulated by Arx [42] (Fig. 2).
While in vitro methods have permitted a rapid growth in our understanding of Arx-driven molecular pathology and transcriptional defects, the creation of rodent models engineered with Arx mutations remains a necessary step for investigating how endogenously expressed mutant Arx proteins alter specific steps in interneuron development leading to the neural circuit deficits and clinical features of ISSX.
Engineered Arx poly-A expansions in mice reproduce human ARX/ISSX phenotypes
While the first constitutive Arx knockout mouse model highlighted the importance of Arx in development, the null mutant dies shortly after birth, limiting its usefulness for studying the role of Arx in adulthood [22]. To test the hypothesis that Arx deficiency in developing interneurons is sufficient to replicate features of ISSX, Marsh et al. (2009) created a viable conditional knockout model of Arx by deleting the gene only in cells within the medial ganglionic eminences. The conditional Arx knockout (Arx-/Y; Dlx5/6CIG) displays spontaneous seizures by day P14, abnormal EEG patterns, and loss of calbindin immunoreactive neocortical interneurons. The presence of infantile spasms was not noted in this model. Interestingly, heterozygous females also showed spontaneous seizures, but not as frequently as male hemizygotes [43*]. Although there are no known human null mutations reported, the Arx-/Y; Dlx5/6CIG mouse represents a unique opportunity to study the role of Arx in interneuron development in brain tissue with an otherwise wild-type background, which may provide additional clues regarding interneuron specific mechanisms underlying ISSX (Table 1).
Recently, two mouse models engineered with expansions in pA1 which reproduce the human 7-alanine expansion have been independently developed [31,44**]. The model published by Kitamura and colleagues, Arx(GCG)7/Y, survived for approximately 3 months, allowing the detection of spontaneous seizures, learning impairments, loss of cholinergic and GABAergic interneurons, and interneuron migration deficits in the developing brain. In their study, two other models with homeobox domain mutations, P355L and P355R, were also made for comparison. The ArxP355L (or ArxPL) line had a similar, but more subtle phenotype compared to Arx(GCG)7/Y. These features stood in striking contrast to the ArxP355R (or ArxPR) model whose mutation lies in the same position within the gene, yet causes perinatal death, gross brain malformations, and severe defects in interneuron migration similar to the Arx-/Y mouse and human XLAG [44**]. It has been shown that an expanded Arx protein at the pA1 results in misregulation of Arx transcriptional targets [23]. Therefore, it is possible that the P355L and the (GCG)7 mutations result in a similar defect in transcriptional regulation (Table 1, Fig.1).
The model developed in our laboratory has a similar 7-alanine (GCG)7 expansion mutation, but with a notable difference. Unlike the human ARX gene, where pA1 has 16 alanine codons (10 of which are coded by GCG), the pA1 in the mouse Arx contains 15 alanine codons (4 of which are coded by GCG). To achieve the same 23-alanine long pA1 tract mutation seen in humans, an expanded poly-alanine mutation was inserted in the pA1 of the mouse orthologue such that it contained a total of 23 alanine residues. This humanized mouse model of ARX pA1 expansion, denominated Arx(GCG)10+7, has a normal life span, and displays spontaneous seizures and frequent interictal spikes in the EEG. In addition, Arx(GCG)10+7 mutant pups display a transient pattern of dyskinetic motor spasms during the first two weeks of life resembling motor spasms observed in humans with ISSX [31**]. Notably, hypsarrhythmia, an EEG abnormality commonly seen in association with human infantile spasms, was not observed in Arx(GCG)10+7 mutants studied at 2 weeks postnatal, although mice were not examined for this EEG phenotype at earlier postnatal stages when spasms were present due to technical challenges. Hypsarrhythmia has not been observed in all ISSX cases with a known orthologous ARX triplet repeat expansion [45]. Arx(GCG)10+7 mutant mice do display impaired learning ability and social interactions, which parallel the human ID syndrome (Table 1).
At the neuropathological level, the Arx(GCG)10+7 mutation causes a partial loss of neocortical calbindin- and NPY-positive GABAergic interneurons and loss of striatal NPY-, calbindin-, and ChAT-positive neurons. Parvalbumin- and calretinin-positive neurons are largely spared. This pattern of interneuronal loss suggests that specific regional patterns of synaptic dysinhibition may generate the complex phenotype of epilepsy, motor spasms, and cognitive impairment. Intracellularly, the molecular lesion also displays complexity, since cytoplasmic inclusions in the Arx mutant are seen in somatosensory cortex neurons but not observed elsewhere in the forebrain, suggesting that protein mislocalization may be cell-type specific rather than playing a uniform role in in the development of the Arx(GCG)10+7 phenotype [31**]. The viability of the Arx(GCG)10+7 mouse model containing the human-like 23-alanine expansion in pA1 with a phenotype closely resembling human ISSX makes it a valuable tool for studying the pathophysiology and potential future treatment strategies of ARX diseases in vivo.
Conclusion
The recent studies on how Arx mutations alter its function, the identification of novel transcriptional targets, and the creation of clinically relevant models of ARX/ISSX represent a significant advance toward understanding the pathogenesis of human ARX-mediated disease. Alterations in gene expression patterns driven by Arx mutations have highlighted genes whose precise roles in neurodevelopment and the rise of ISSX and other Arx-related disorders have yet to be described. While in vitro studies continue to provide a framework for exploring the molecular pathology and cellular consequences of various Arx mutations, the recent generation of several models of Arx deficiency based on naturally occurring human mutations has provided valuable tools to validate downstream gene changes in the context of the developing brain, and an opportunity to link these with human neurological phenotypes. These models are also likely to provide useful substrates for investigating novel therapies for Arx-related diseases, for which only limited treatment options exist.
Highlights.
ARX is a transcription factor deeply involved in interneuron development
ARX mutations can cause epilepsy, infantile spasms, and intellectual disability
Downstream consequences of Arx mutations reveal complex molecular pathogenesis
Mouse models of human ARX mutations are valuable tools to study role of Arx in vivo
New mouse models provide a substrate for the investigation of novel therapies
Acknowledgments
Supported by an American Epilepsy Society Predoctoral Fellowship (PRO), NINDS NS29709 (JLN), and The Blue Bird Circle Foundation for Pediatric Neurology Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1*.Paciorkowski AR, Thio LL, Dobyns WB. Genetic and biologic classification of infantile spasms. Pediatr Neurol. 2011;45:355–367. doi: 10.1016/j.pediatrneurol.2011.08.010. Based on the significant advances on the biological and genetic basis of infantile spasms in past decades and the latest recommendations from the International League Against Epilepsy, the authors propose a new and useful classification system of infantile spasms that reflects its many known genetic and biological causes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamano S-I, Yamashita S, Tanaka M, Yoshinari S, Minamitani M, Eto Y. Therapeutic efficacy and adverse effects of adrenocorticotropic hormone therapy in west syndrome: differences in dosage of adrenocorticotropic hormone, onset of age, and cause. J Pediatr. 2006;148:485–488. doi: 10.1016/j.jpeds.2005.11.041. [DOI] [PubMed] [Google Scholar]
- 3.Partikian A, Mitchell WG. Major adverse events associated with treatment of infantile spasms. Journal of Child Neurology. 2007;22:1360–1366. doi: 10.1177/0883073807310988. [DOI] [PubMed] [Google Scholar]
- 4.Ohya T, Nagai T, Araki Y, Yanagawa T, Tanabe T, Iyoda K, Kurihara M, Yamamoto K, Masunaga K, Iizuka C, et al. A pilot study on the changes in immunity after ACTH therapy in patients with West syndrome. Brain and Development. 2009;31:739–743. doi: 10.1016/j.braindev.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Frost J, Hrachovy R. Infantile spasms: diagnosis, management and prognosis. Kluwer; 2003. [Google Scholar]
- 6.Mackay MT, Weiss SK, Adams-Webber T, Ashwal S, Stephens D, Ballaban-Gill K, Baram TZ, Duchowny M, Hirtz D, Pellock JM, et al. Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology. 2004;62:1668–1681. doi: 10.1212/01.wnl.0000127773.72699.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gayatri NA, Ferrie CD, Cross H. Corticosteroids including ACTH for childhood epilepsy other than epileptic spasms. Cochrane Database Syst Rev. 2007 doi: 10.1002/14651858.CD005222.pub2. [DOI] [PubMed] [Google Scholar]
- 8.Chudomelova L, Scantlebury MH, Raffo E, Coppola A, Betancourth D, Galanopoulou AS. Modeling new therapies for infantile spasms. Epilepsia. 2010;51:27–33. doi: 10.1111/j.1528-1167.2010.02605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellock JM, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, Gaillard WD, Gibson PA, Holmes GL, Nordl DR, et al. Infantile spasms: a U.S. consensus report. Epilepsia. 2010;51:2175–2189. doi: 10.1111/j.1528-1167.2010.02657.x. [DOI] [PubMed] [Google Scholar]
- 10.Strømme P, Mangelsdorf ME, Scheffer IE, Gécz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain and Development. 2002;24:266–268. doi: 10.1016/s0387-7604(02)00079-7. [DOI] [PubMed] [Google Scholar]
- 11.Strømme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SME, Bruyere H, Lütcherath V, Gedeon ÁK, Wallace RH, Scheffer IE, et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet. 2002;30:441–445. doi: 10.1038/ng862. [DOI] [PubMed] [Google Scholar]
- 12.Turner G, Partington M, Kerr B, Mangelsdorf M, Gécz J. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identicalARX gene mutation. Am J Med Genet. 2002;112:405–411. doi: 10.1002/ajmg.10714. [DOI] [PubMed] [Google Scholar]
- 13.Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, Ben Jeema L, Zemni R, Vinet M-C, Francis F, et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Human Molecular Genetics. 2002;11:981–991. doi: 10.1093/hmg/11.8.981. [DOI] [PubMed] [Google Scholar]
- 14.Kato M, Das S, Petras K, Kitamura K, Morohashi K-I, Abuelo DN, Barr M, Bonneau D, Brady AF, Carpenter NJ, et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum Mutat. 2004;23:147–159. doi: 10.1002/humu.10310. [DOI] [PubMed] [Google Scholar]
- 15.Miura H, Yanazawa M, Kato K, Kitamura K. Expression of a novel aristaless related homeobox gene “Arx” in the vertebrate telencephalon, diencephalon and floor plate. Mech Dev. 1997;65:99–109. doi: 10.1016/s0925-4773(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 16.Ohira R, Zhang YH, Guo W, Dipple K, Shih SL, Doerr J, Huang BL, Fu LJ, Abu-Khalil A, Geschwind D, et al. Human ARX gene: genomic characterization and expression. Mol Genet Metab. 2002;77:179–188. doi: 10.1016/s1096-7192(02)00126-9. [DOI] [PubMed] [Google Scholar]
- 17.Colombo E, Galli R, Cossu G, Gécz J, Broccoli V. Mouse orthologue of ARX, a gene mutated in several X-linked forms of mental retardation and epilepsy, is a marker of adult neural stem cells and forebrain GABAergic neurons. Dev Dyn. 2004;231:631–639. doi: 10.1002/dvdy.20164. [DOI] [PubMed] [Google Scholar]
- 18.Poirier K, Van Esch H, Friocourt G, Saillour Y, Bahi N, Backer S, Souil E, Castelnau-Ptakhine L, Beldjord C, Francis F, et al. Neuroanatomical distribution of ARX in brain and its localisation in GABAergic neurons. Brain Res Mol Brain Res. 2004;122:35–46. doi: 10.1016/j.molbrainres.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 19.Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JLR. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci. 2005;8:1059–1068. doi: 10.1038/nn1499. [DOI] [PubMed] [Google Scholar]
- 20.Friocourt G, Poirier K, Rakić S, Parnavelas JG, Chelly J. The role of ARX in cortical development. Eur J Neurosci. 2006;23:869–876. doi: 10.1111/j.1460-9568.2006.04629.x. [DOI] [PubMed] [Google Scholar]
- 21.Colombo E, Collombat P, Colasante G, Bianchi M, Long J, Mansouri A, Rubenstein JLR, Broccoli V. Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. Journal of Neuroscience. 2007;27:4786–4798. doi: 10.1523/JNEUROSCI.0417-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- 23.Shoubridge C, Cloosterman D, Parkinson Lawerence E, Brooks D, Gécz J. Molecular pathology of expanded polyalanine tract mutations in the Aristaless-related homeobox gene. Genomics. 2007;90:59–71. doi: 10.1016/j.ygeno.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Lin W, Ye W, Cai L, Meng X, Ke G, Huang C, Peng Z, Yu Y, Golden JA, Tartakoff AM, et al. The roles of multiple importins for nuclear import of murine aristaless-related homeobox protein. J Biol Chem. 2009;284:20428–20439. doi: 10.1074/jbc.M109.004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25*.Shoubridge C, Tan M, Fullston T, Cloosterman D, Coman D, McGillivray G, Mancini GM, Kleefstra T, Gécz J. Mutations in the nuclear localization sequence of the Aristaless related homeobox; sequestration of mutant ARX with IPO13 disrupts normal subcellular distribution of the transcription factor and retards cell division. Pathogenetics. 2010;3:1. doi: 10.1186/1755-8417-3-1. In this in vitro study, the authors show that mutations in the nuclear localization sequence of Arx disrupt its ability to dissociate from an importin protein, IPO3, resulting in abnormal protein localization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fulp CT, Cho G, Marsh ED, Nasrallah IM, Labosky PA, Golden JA. Identification of Arx transcriptional targets in the developing basal forebrain. Human Molecular Genetics. 2008;17:3740–3760. doi: 10.1093/hmg/ddn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27**.Colasante G, Sessa A, Crispi S, Calogero R, Mansouri A, Collombat P, Broccoli V. Arx acts as a regional key selector gene in the ventral telencephalon mainly through its transcriptional repression activity. Developmental Biology. 2009;334:59–71. doi: 10.1016/j.ydbio.2009.07.014. The authors used microarray and qPCR experiments to identify Arx targets in Arx null mice. The functional role of some of the transcriptional targets, such as Lmo1 and Ebf3, was assessed, pointing the way to potential mechanisms underlying how each downstream Arx target may regulate interneuron development. [DOI] [PubMed] [Google Scholar]
- 28*.Shoubridge C, Tan MH, Seiboth G, Gécz J. ARX homeodomain mutations abolish DNA binding and lead to a loss of transcriptional repression. Human Molecular Genetics. 2011 doi: 10.1093/hmg/ddr601. The functional impact of ARX homeodomain mutations was measured with an in vitro luciferase assay and ChIP-qPCR. The authors showed that homeodomain mutations disrupt DNA binding and compromise Arx-mediated transcriptional repression. [DOI] [PubMed] [Google Scholar]
- 29.Nasrallah IM, Minarcik JC, Golden JA. A polyalanine tract expansion in Arx forms intranuclear inclusions and results in increased cell death. The Journal of Cell Biology. 2004;167:411–416. doi: 10.1083/jcb.200408091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fullston T, Finnis M, Hackett A, Hodgson B, Brueton L, Baynam G, Norman A, Reish O, Shoubridge C, Gécz J. Screening and cell-based assessment of mutations in the Aristaless-related homeobox (ARX) gene. Clinical Genetics. 2011;80:510–522. doi: 10.1111/j.1399-0004.2011.01685.x. [DOI] [PubMed] [Google Scholar]
- 31**.Price MG, Yoo JW, Burgess DL, Deng F, Hrachovy RA, Frost JD, Noebels JL. A Triplet Repeat Expansion Genetic Mouse Model of Infantile Spasms Syndrome, Arx(GCG)10+7, with Interneuronopathy, Spasms in Infancy, Persistent Seizures, and Adult Cognitive and Behavioral Impairment. Journal of Neuroscience. 2009;29:8752–8763. doi: 10.1523/JNEUROSCI.0915-09.2009. The first poly-alanine tract in a mouse Arx was expanded to 23 alanine residues, thus replicating an exact human mutation commonly linked with ISSX. The mouse reproduced key features of human ISSX, including spasms in infancy, seizures, behavioral and cognitive impairment, and selective loss of interneurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32**.Nasrallah MP, Cho G, Putt ME, Kitamura K, Golden JA. Differential effects of a polyalanine tract expansion in Arx on neural development and gene expression. Human Molecular Genetics. 2011 doi: 10.1093/hmg/ddr538. In this paper, the authors dissect the downstream transcriptional consequences of an expanded first poly-alanine tract of Arx, showing that the direct Arx targets are differentially affected by this mutation. This selective nature of how the mutation alters the normal transcriptional repression is attributed to impaired binding of Arx to Tle1, a co-repressor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKenzie O, Ponte I, Mangelsdorf M, Finnis M, Colasante G, Shoubridge C, Stifani S, Gécz J, Broccoli V. Aristaless-related homeobox gene, the gene responsible for West syndrome and related disorders, is a Groucho/transducin-like enhancer of split dependent transcriptional repressor. NSC. 2007;146:236–247. doi: 10.1016/j.neuroscience.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 34.Cho G, Nasrallah MP, Lim Y, Golden JA. Distinct DNA binding and transcriptional repression characteristics related to different ARX mutations. Neurogenetics. 2012 doi: 10.1007/s10048-011-0304-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35*.Topalidou I, van Oudenaarden A, Chalfie M. Caenorhabditis elegans aristaless/Arx gene alr-1 restricts variable gene expression. Proc Natl Acad Sci U S A. 2011;108:4063–4068. doi: 10.1073/pnas.1101329108. This an interesting study of the role of alr-1 orthologue of Arx in C. elegans, showing that, in this organism, alr-1 acts by restricting the natural stochastic variation of gene expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stumm RK, Zhou C, Ara T, Lazarini F, Dubois-Dalcq M, Nagasawa T, Höllt V, Schulz S. CXCR4 regulates interneuron migration in the developing neocortex. Journal of Neuroscience. 2003;23:5123–5130. doi: 10.1523/JNEUROSCI.23-12-05123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stumm R, Hollt V. CXC chemokine receptor 4 regulates neuronal migration and axonal pathfinding in the developing nervous system: implications for neuronal regeneration in the adult brain. Journal of Molecular Endocrinology. 2007;38:377–382. doi: 10.1677/JME-06-0032. [DOI] [PubMed] [Google Scholar]
- 38.Liapi A, Pritchett J, Jones O, Fujii N, Parnavelas JG, Nadarajah B. Stromal-derived factor 1 signalling regulates radial and tangential migration in the developing cerebral cortex. Dev Neurosci. 2008;30:117–131. doi: 10.1159/000109857. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka DH, Mikami S, Nagasawa T, Miyazaki JI, Nakajima K, Murakami F. CXCR4 Is Required for Proper Regional and Laminar Distribution of Cortical Somatostatin-, Calretinin-, and Neuropeptide Y-Expressing GABAergic Interneurons. Cerebral Cortex. 2010;20:2810–2817. doi: 10.1093/cercor/bhq027. [DOI] [PubMed] [Google Scholar]
- 40.Sánchez-Alcañiz JA, Haege S, Mueller W, Pla R, Mackay F, Schulz S, López-Bendito G, Stumm R, Marín O. Cxcr7 Controls Neuronal Migration by Regulating Chemokine Responsiveness. Neuron. 2011;69:77–90. doi: 10.1016/j.neuron.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Li G, Stanco A, Long JE, Crawford D, Potter GB, Pleasure SJ, Behrens T, Rubenstein JLR. CXCR4 and CXCR7 Have Distinct Functions in Regulating Interneuron Migration. Neuron. 2011;69:61–76. doi: 10.1016/j.neuron.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quillé M-L, Carat S, Quéméner-Redon S, Hirchaud E, Baron D, Benech C, Guihot J, Placet M, Mignen O, Férec C, et al. High-Throughput Analysis of Promoter Occupancy Reveals New Targets for Arx, a Gene Mutated in Mental Retardation and Interneuronopathies. PLoS ONE. 2011;6:e25181. doi: 10.1371/journal.pone.0025181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Marsh E, Fulp C, Gomez E, Nasrallah I, Minarcik J, Sudi J, Christian SL, Mancini G, Labosky P, Dobyns W, et al. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain. 2009;132:1563–1576. doi: 10.1093/brain/awp107. The conditional deletion of Arx in developing interneurons in a mouse resulted in spontaneous seizures and partial loss of cortical interneurons. This work provided further evidence of the cell-autonomous role of Arx in interneuron development and that deleting it only in interneurons is sufficient to cause developmental epilepsy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44**.Kitamura K, Itou Y, Yanazawa M, Ohsawa M, Suzuki-Migishima R, Umeki Y, Hohjoh H, Yanagawa Y, Shinba T, Itoh M, et al. Three human ARX mutations cause the lissencephaly-like and mental retardation with epilepsy-like pleiotropic phenotypes in mice. Human Molecular Genetics. 2009;18:3708–3724. doi: 10.1093/hmg/ddp318. The authors made several mice with point mutations in the homeodomain and a 7-alanine expansion in the first poly-alanine tract of Arx which displayed developmental epilepsy and loss of interneurons. These mice constitute valuable tools for studying the consequences of Arx mutations in vivo. [DOI] [PubMed] [Google Scholar]
- 45.Guerrini R, Moro F, Kato M, Barkovich AJ, Shiihara T, McShane MA, Hurst J, Loi M, Tohyama J, Norci V, et al. Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology. 2007;69:427–433. doi: 10.1212/01.wnl.0000266594.16202.c1. [DOI] [PubMed] [Google Scholar]