

Abstract
Background. Metabolic syndrome has been linked to an increased cancer risk, but the role of dyslipidaemia in gastrointestinal malignancies is unclear. We aimed to assess the risk of oesophageal, stomach, colon, and rectal cancers using serum levels of lipid components. Methods. From the Swedish Apolipoprotein Mortality Risk (AMORIS) study, we selected 540,309 participants (> 20 years old) with baseline measurements of total cholesterol (TC), triglycerides (TG), and glucose of whom 84,774 had baseline LDL cholesterol (LDL), HDL cholesterol (HDL), apolipoprotein B (apoB), and apolipoprotein A-I (apoA-I). Multivariate Cox proportional hazards regression was used to assess glucose and lipid components in relation to oesophageal, stomach, colon, and rectal cancer risk. Results. An increased risk of oesophageal cancer was observed in persons with high TG (e.g. HR: 2.29 (95% CI: 1.42–3.68) for the 4th quartile compared to the 1st) and low LDL, LDL/HDL ratio, TC/HDL ratio, log (TG/HDL), and apoB/apoA-I ratio. High glucose and TG were linked with an increased colon cancer risk, while high TC levels were associated with an increased rectal cancer risk. Conclusion. The persistent link between TC and rectal cancer risk as well as between TG and oesophageal and colon cancer risk in normoglycaemic individuals may imply their substantiality in gastrointestinal carcinogenesis.
1. Introduction
The prevalence of dyslipidaemia is increasing worldwide [1, 2], either alone or as part of the metabolic syndrome [3]. Many epidemiological studies have consistently linked the metabolic syndrome as well as its separated components, most importantly impaired glucose metabolism and obesity, with increased risk of cancer [4–6]. However, the association between abnormal levels of serum lipid components as the main features of dyslipidaemia and the risk of individual cancers is unclear. For gastrointestinal malignancies, a positive linear association has been reported between triglycerides (TG) and risk of colon and rectal cancer [7–9]. In contrast, the role of total cholesterol (TC) in oesophageal, stomach, colon, and rectal cancer development remains inconsistent in observational studies [9–11]. Little is known about how other specific lipid markers such as high-density lipoprotein cholesterol (HDL) and low-density lipoprotein cholesterol (LDL) and their components, apolipoprotein A-I (apoA-I) and apolipoprotein B (apoB), affect cancer risk [12, 13].
To date, the precise mechanism by which serum lipids contribute to the development of cancer is unknown. Dyslipidaemia enhances oxidative stress as well as chronic inflammation [14–16] and is highly related to insulin resistance [17], all of which have been linked to carcinogenesis. Besides circulating lipids, an experimental study has shown that dietary lipids may induce local inflammation in alimentary tract which is thought to enhance development of gastrointestinal malignancies [18]. Nevertheless, evidence for a link between dietary fat and cholesterol in the development of oesophageal, stomach, colon, and rectal cancer is still inconclusive [19–21]. In this study we explored the link between serum lipid components and risk of oesophageal, stomach, colon, and rectal cancers in a large Swedish population-based cohort.
2. Methods
2.1. Study Population and Data Collection
The Swedish AMORIS database has been described in detail elsewhere [22–28]. Briefly, this database is based on the linkage of the Central Automation Laboratory (CALAB) database (1985–1996) to several Swedish national registries such as the National Cancer Register, the National Patient Register, the Cause of Death Register, the consecutive Swedish Censusesduring 1970–1990, and the National Register of Emigration by using the Swedish 10-digit personal identity number to provide information on socio-economic status (SES), vital status, cancer diagnosis, and emigration. The CALAB database includes data from 351,487 male and 338,101 female healthy individuals having clinical laboratory testing as part of a general health checkup or outpatients referred for laboratory testing. No individuals were inpatients at the time their blood samples were taken and none were excluded for disease symptoms or because of treatment. This study complied with the Declaration of Helsinki, and the ethics review board of the Karolinska Institute approved the study.
From the AMORIS cohort, we selected all participants aged > 20 years old whose serum levels of TC (mmol/L), TG (mmol/L) and glucose (mmol/L) were measured at baseline, that is, at time of study entry (n = 540, 309). Of those, a total of 89,153 participants had baseline measurements of body mass index (BMI, kg/m2) (Subgroup A) and a total of 84,774 had baseline measurement of more detailed lipid markers including LDL (mmol/L), HDL (mmol/L), apoB (g/L), and apoA-I (g/L) (Subgroup B) (Figure 1). All participants were free from cancer at time of study entry and none were diagnosed with cancer within the first three months of follow-up. Followup time was defined as time from baseline examination until date of diagnosis of oesophageal, stomach, colon or rectal cancer, date of death, emigration out of Sweden, or study closing date (December 31, 2002), whichever occurred first.
Figure 1.
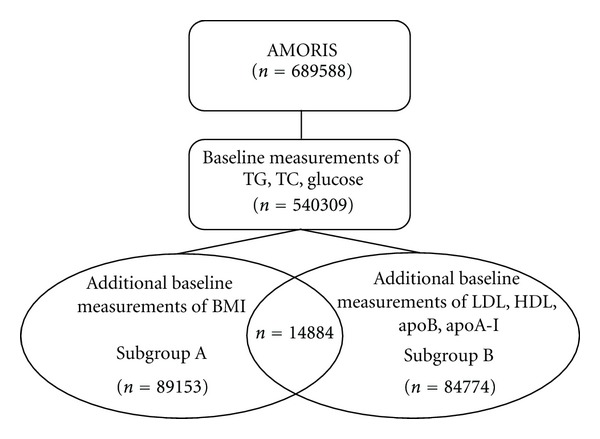
Overview of the study population.
Information on age and fasting status was also obtained from the CALAB database. Diagnosis of oesophageal (ICD-7: 150), stomach (ICD-7: 151), colon (ICD-7: 153), and rectal cancer (ICD-7: 154) was taken from the National Cancer Registry. Data for death and migration were obtained from the Swedish Cause of Death Register and the National Register of Emigration. From the Population and Housing Census, information about socioeconomic status (SES) was also collected. SES is based on occupational groups and classifies gainfully employed subjects into manual workers and nonmanual employees, below designated blue-collar and white-collar workers [29].
TC and TG were measured enzymatically, whereas apoB and apoA-I were measured by immunoturbidimetric methods [22, 30]. The concentrations of LDL and HDL were calculated, and the validation procedures have been reported previously [23]. The balance of cholesterol can be reflected by the ratio of TC to HDL (TC/HDL) or LDL to HDL cholesterol (LDL/HDL). This balance can also be assessed by using a ratio of apoB/apoA-I, as these particles are components of LDL and HDL to bind and transport cholesterol [31]. Glucose was measured enzymatically with a glucose oxidase/peroxidase method. All methods were fully automated with automatic calibration and performed at one accredited laboratory [22].
2.2. Data Analysis
Pearson correlation coefficients were calculated to assess the relation between glucose and different lipid components (TC, TG) and BMI. Multivariate Cox proportional hazards regression was used to assess the association between quartiles of lipid components as well as their ratios in relation to the risk of oesophageal, stomach, colon, and rectal cancer. To gain insight into the possible role of insulin resistance, we also assessed glucose in relation to gastrointestinal cancer risk. A test for trend was conducted by using assignment to quartiles as an ordinal scale. All models were adjusted for glucose, TC and TG levels (if not strongly correlated with the covariate of interest), as well as age, gender, SES, and fasting status.
We repeated the above analyses using log transformation of the continuous lipid components as well as dichotomized values based upon the cutoffs used in cardiovascular prevention and the National Cholesterol Education Programme (NCEP) (cutoffs: 6.11 mmol/L, 6.50 mmol/L, 1.71 mmol/L, 1.03 mmol/L, 4.10 mmol/L, 5.00, 3.50, 1.50, 1.05, 1.00, and 0.50 for glucose, TC, TG, HDL, LDL, TC/HDL, LDL/HDL, apoB, apoA-I, apoB/apoA-I, and log (TG/HDL), resp.) [32–34].
Considering the putative link between obesity, dyslipidaemia, and cancer, in Subgroup A the associations between glucose, TC, TG, and risk of gastrointestinal cancers were assessed while adjusting for BMI. Furthermore, to assess the role of glucose, we performed a stratified analysis using dichotomized levels of glucose (cut-off: 6.11 mmol/L) for both the log transformation and dichotomized values of TC and TG. Adjustment for BMI and stratification by glucose levels were not done for Subgroup B due to the small number of gastrointestinal cancers in this subgroup.
We evaluated reverse causation between dyslipidaemia and cancer through a sensitivity analysis in which those with follow-up time < 3 years were excluded (n = 12,750). All analyses were conducted with Statistical Analysis Systems (SAS) release 9.1.3 (SAS Institute, Cary, NC, USA).
3. Results
During a mean followup of 12 years, a total of 318 oesophageal cancers, 776 stomach cancers, 2472 colon cancers, and 1510 rectal cancers were diagnosed (Table 1). The majority of the population was gainfully employed because most measurements were taken as part of health examinations done at company health checkups. Pearson Correlation Coefficients showed that glucose, TC, and TG were associated with BMI, with TG being most strongly linked to BMI (r = 0.32, P value < 0.0001) and glucose the weakest (r = 0.19, P value < 0.0001),
Table 1.
Baseline characteristics of study population.
| Oesophageal cancer (n = 318) |
Stomach cancer (n = 776) |
Colon cancer (n = 2472) |
Rectal cancer (n = 1510) |
All (n = 540309) |
|
|---|---|---|---|---|---|
| Age | |||||
| Mean (SD) | 55.75 (10.46) | 57.83 (11.80) | 57.93 (11.60) | 56.75 (10.82) | 43.84 (13.83) |
| Gender | |||||
| Male | 234 (73.58) | 497 (64.05) | 1375 (55.62) | 938 (62.12) | 290294 (53.73) |
| Female | 84 (26.42) | 279 (35.95) | 1097 (44.38) | 572 (37.88) | 250015 (46.27) |
| Socioeconomic status | |||||
| White collar | 110 (34.59) | 248 (31.96) | 915 (37.01) | 577 (38.21) | 200231 (37.06) |
| Blue collar | 154 (48.43) | 372 (47.94) | 1059 (42.84) | 700 (46.36) | 256413 (47.46) |
| Not gainfully employed or missing | 54 (16.98) | 156 (20.10) | 498 (20.15 | 233 (15.43) | 83665 (15.48) |
| Mean follow-up time (year) | 8.09 (4.73) | 7.62 (4.68) | 8.50 (4.67) | 8.53 (4.54) | 12.31 (4.13) |
| Fasting status | |||||
| Fasting | 183 (57.55) | 473 (60.95) | 1534 (62.06) | 953 (63.11) | 298337 (55.22) |
| Not fasting | 15 (4.72) | 48 (6.19) | 147 (5.95) | 96 (6.36) | 78318 (14.50) |
| Missing | 120 (37.74) | 255 (32.86) | 791 (32.00) | 461 (30.53) | 163654 (30.29) |
| Glucose (mmol/L) | |||||
| Mean (SD) | 5.49 (2.07) | 5.36 (1.81) | 5.27 (1.50) | 5.26 (1.56) | 4.97 (1.25) |
| Total cholesterol (mmol/L) | |||||
| Mean (SD) | 5.90 (1.17) | 6.00 (1.26) | 6.05 (1.16) | 6.11 (1.17) | 5.55 (1.16) |
| Triglycerides (mmol/L) | |||||
| Mean (SD) | 1.72 (1.37) | 1.48 (0.94) | 1.49 (0.95) | 1.47 (1.03) | 1.31 (0.99) |
| LDL cholesterol (mmol/L)a | |||||
| Mean (SD) | 3.40 (1.15) | 3.90 (1.11) | 3.88 (1.03) | 3.93 (1.05) | 3.59 (1.08) |
| HDL cholesterol (mmol/L)a | |||||
| Mean (SD) | 1.49 (0.42) | 1.52 (0.45) | 1.55 (0.48) | 1.57 (0.49) | 1.53 (0.44) |
| Apolipoprotein B (mmol/L)a | |||||
| Mean (SD) | 1.23 (0.35) | 1.33 (0.30) | 1.35 (0.34) | 1.33 (0.33) | 1.22 (0.34) |
| Apolipoprotein A-I (mmol/L)a | |||||
| Mean (SD) | 1.46 (0.22) | 1.43 (0.25) | 1.46 (0.25) | 1.46 (0.26) | 1.43 (0.23) |
| LDL/HDLa | |||||
| Mean (SD) | 2.76 (2.17) | 2.86 (1.27) | 2.84 (1.42) | 2.84 (1.48) | 2.68 (1.59) |
| Total cholesterol/HDLa | |||||
| Mean (SD) | 4.04 (1.69) | 4.37 (1.68) | 4.39 (1.87) | 4.38 (1.95) | 4.15 (2.13) |
| Triglycerides/HDLa | |||||
| Mean (SD) | 1.81 (3.93) | 1.18 (1.12) | 1.25 (1.32) | 1.21 (1.28) | 1.16 (1.74) |
| ApoB/ApoA-Ia | |||||
| Mean (SD) | 0.86 (0.28) | 0.96 (0.26) | 0.96 (0.31) | 0.94 (0.30) | 0.88 (0.30) |
| BMI (kg/m2)b | |||||
| <18.5 | 3 (6.82) | 2 (1.77) | 6 (1.58) | 1 (0.43) | 1608 (1.80) |
| 18.5–25 | 17 (38.64) | 54 (47.79) | 196 (51.58) | 123 (52.34) | 52509 (58.90) |
| 25–30 | 20 (45.45) | 40 (35.40) | 139 (36.58) | 85 (36.17) | 28339 (31.79) |
| ≥30 | 4 (9.09) | 17 (15.04) | 39 (10.26) | 26 (11.06) | 6697 (7.51) |
aMeasured in Subcohort B.
bMeasured in Subcohort A.
3.1. Oesophageal Cancer
A positive association was found between quartiles of TG, HDL, and risk of oesophageal cancer (e.g., HR for TG: 1.47 (95% CI: 0.91–2.36), 1.64 (95% CI: 1.02–2.65), 2.29 (95% CI: 1.42–3.68) for the 2nd, 3rd, and 4th quartiles, compared to the 1st, with P-value for trend: < 0.001). In addition, there was a negative trend between quartiles of LDL, LDL/HDL ratio, TC/HDL ratio, and apoB/apoA ratio and oesophageal cancer risk (Table 2). Similar associations were found when we used log transformations and dichotomized values of the lipid components (Table 3). When we stratified by glucose levels, high TG levels (≥1.71 mmol/L) and glucose <6.11 mmol/L increased the risk of oesophageal cancer (HR: 1.38 (95% CI: 1.05–1.83) compared to low TG) (Table 4).
Table 2.
Hazard ratios and 95% confidence intervals for GI cancer for quartiles of lipid components, adjusted for age, gender, SES, fasting status, glucose (continuous), total cholesterol (continuous), and triglycerides (continuous).
| Oesophageal cancer | Stomach cancer | Colon cancer | Rectal cancer | |||||
|---|---|---|---|---|---|---|---|---|
| n | HR (95% CI) | n | HR (95% CI) | n | HR (95% CI) | n | HR (95% CI) | |
| Glucose (mmol/L)a | ||||||||
| <4.40 | 40 | 1.00 (Ref) | 99 | 1.00 (Ref) | 310 | 1.00 (Ref) | 242 | 1.00 (Ref) |
| 4.40–4.80 | 65 | 1.01 (0.68–1.50) | 185 | 1.12 (0.88–1.44) | 572 | 1.14 (1.00–1.32) | 338 | 0.85 (0.72–1.00) |
| 4.80–5.20 | 65 | 0.88 (0.59–1.32) | 201 | 1.06 (0.83–1.35) | 622 | 1.12 (0.98–1.29) | 346 | 0.77 (0.65–0.91) |
| ≥5.20 | 148 | 1.27 (0.89–1.87) | 291 | 0.96 (0.74–1.23) | 968 | 1.21 (1.05–1.40) | 584 | 0.88 (0.74–1.05) |
| P value for trend | 0.16 | 0.41 | 0.03 | 0.23 | ||||
| Total cholesterol (mmol/L)a | ||||||||
| <4.70 | 41 | 1.00 (Ref) | 89 | 1.00 (Ref) | 266 | 1.00 (Ref) | 150 | 1.00 (Ref) |
| 4.70–5.50 | 72 | 0.93 (0.63–1.37) | 172 | 1.01 (0.78–1.31) | 499 | 0.96 (0.83–1.11) | 316 | 1.11 (0.91–1.35) |
| 5.50–6.30 | 99 | 0.99 (0.69–1.44) | 212 | 0.96 (0.74–1.23) | 718 | 1.04 (0.90–1.21) | 405 | 1.10 (0.91–1.33) |
| ≥6.30 | 106 | 0.79 (0.54–1.15) | 303 | 1.03 (0.80–1.32) | 989 | 1.03 (0.90–1.19) | 639 | 1.32 (1.09–1.59) |
| P value for trend | 0.28 | 0.54 | 0.19 | 0.0003 | ||||
| Triglycerides (mmol/L)b | ||||||||
| <0.70 | 22 | 1.00 (Ref) | 74 | 1.00 (Ref) | 248 | 1.00 (Ref) | 168 | 1.00 (Ref) |
| 0.70–1.03 | 79 | 1.47 (0.91–2.36) | 225 | 1.17 (0.90–1.52) | 682 | 1.09 (0.94–1.26) | 424 | 0.97 (0.81–1.16) |
| 1.03–1.60 | 83 | 1.64 (1.02–2.65) | 223 | 1.19 (0.91–1.56) | 710 | 1.21 (1.05–1.41) | 410 | 0.97 (0.80–1.16) |
| ≥1.60 | 134 | 2.29 (1.42–3.68) | 254 | 1.17 (0.90–1.55) | 832 | 1.30 (1.12–1.52) | 508 | 1.03 (0.86–1.25) |
| P value for trend | 0.0001 | 0.44 | <0.0001 | 0.49 | ||||
| LDL cholesterol (mmol/L)c | ||||||||
| <2.82 | 18 | 1.00 (Ref) | 19 | 1.00 (Ref) | 59 | 1.00 (Ref) | 28 | 1.00 (Ref) |
| 2.82–3.51 | 18 | 0.61 (0.29–1.26) | 25 | 0.84 (0.44–1.60) | 101 | 1.05 (0.75–1.48) | 57 | 1.21 (0.75–1.96) |
| 3.51–4.25 | 9 | 0.22 (0.08–0.62) | 33 | 0.87 (0.42–1.77) | 119 | 0.92 (0.62–1.34) | 65 | 1.01 (0.60–1.72) |
| ≥4.25 | 12 | 0.22 (0.06–0.86) | 38 | 0.80 (0.30–2.08) | 136 | 0.78 (0.47–1.30) | 78 | 0.88 (0.45–1.75) |
| P value for trend | 0.007 | 0.73 | 0.26 | 0.36 | ||||
| HDL cholesterol (mmol/L)c | ||||||||
| <1.25 | 15 | 1.00 (Ref) | 33 | 1.00 (Ref) | 103 | 1.00 (Ref) | 55 | 1.00 (Ref) |
| 1.25–1.52 | 16 | 2.14 (0.97–4.71) | 29 | 0.88 (0.51–1.50) | 107 | 1.24 (0.93–1.67) | 50 | 1.04 (0.69–1.58) |
| 1.52–1.80 | 16 | 2.64 (1.16–6.03) | 27 | 0.76 (0.43–1.37) | 89 | 1.04 (0.75–1.43) | 50 | 1.04 (0.67–1.61) |
| ≥1.80 | 10 | 2.04 (0.79–5.27) | 26 | 0.62 (0.33–1.17) | 116 | 1.25 (0.89–1.74) | 73 | 1.40 (0.89–2.17) |
| P value for trend | 0.09 | 0.12 | 0.41 | 0.13 | ||||
| Apolipoprotein B (mmol/L)c | ||||||||
| <0.97 | 11 | 1.00 (Ref) | 13 | 1.00 (Ref) | 37 | 1.00 (Ref) | 28 | 1.00 (Ref) |
| 0.97–1.18 | 20 | 0.97 (0.45–2.10) | 20 | 0.88 (0.43–1.82) | 102 | 1.39 (0.94–2.05) | 48 | 0.76 (0.47–1.23) |
| 1.18–1.42 | 11 | 0.39 (0.15–1.03) | 42 | 1.51 (0.73–3.12) | 131 | 1.28 (0.85–1.93) | 83 | 0.84 (0.52–1.37) |
| ≥1.42 | 15 | 0.45 (0.14–1.44) | 40 | 1.45 (0.59–3.59) | 145 | 1.18 (0.72–1.92) | 69 | 0.48 (0.26–0.89) |
| P value for trend | 0.08 | 0.16 | 0.77 | 0.05 | ||||
| Apolipoprotein A-I (mmol/L)c | ||||||||
| <1.27 | 14 | 1.00 (Ref) | 31 | 1.00 (Ref) | 91 | 1.00 (Ref) | 57 | 1.00 (Ref) |
| 1.27–1.40 | 11 | 1.10 (0.49–2.43) | 32 | 1.07 (0.65–1.77) | 88 | 1.05 (0.78–1.41) | 40 | 0.74 (0.49–1.12) |
| 1.40–1.56 | 18 | 1.88 (0.91–3.86) | 22 | 0.63 (0.36–1.10) | 114 | 1.19 (0.89–1.58) | 54 | 0.87 (0.60–1.28) |
| ≥1.56 | 14 | 1.70 (0.78–3.70) | 30 | 0.75 (0.43–1.31) | 122 | 1.20 (0.89–1.61) | 77 | 1.15 (0.79–1.67) |
| P value for trend | 0.08 | 0.13 | 0.17 | 0.31 | ||||
| LDL/HDLc | ||||||||
| <1.74 | 18 | 1.00 (Ref) | 18 | 1.00 (Ref) | 80 | 1.00 (Ref) | 39 | 1.00 (Ref) |
| 1.74–2.32 | 13 | 0.58 (0.28–1.21) | 23 | 1.12 (0.60–2.10) | 98 | 0.99 (0.73–1.34) | 53 | 1.11 (0.72–1.68) |
| 2.32–3.16 | 13 | 0.43 (0.20–0.93) | 44 | 1.79 (0.99–3.23) | 117 | 0.92 (0.68–1.25) | 68 | 1.12 (0.73–1.71) |
| ≥3.16 | 13 | 0.28 (0.12–0.66) | 30 | 1.16 (0.57–2.35) | 120 | 0.79 (0.56–1.13) | 68 | 1.00 (0.62–1.63) |
| P value for trend | 0.003 | 0.37 | 0.17 | 0.99 | ||||
| Total cholesterol/HDLc | ||||||||
| <2.95 | 12 | 1.00 (Ref) | 18 | 1.00 (Ref) | 74 | 1.00 (Ref) | 35 | 1.00 (Ref) |
| 2.95–3.66 | 16 | 1.02 (0.47–2.17) | 26 | 1.14 (0.62–2.11) | 104 | 1.04 (0.77–1.40) | 61 | 1.28 (0.84–1.95) |
| 3.66–4.68 | 14 | 0.71 (0.31–1.59) | 36 | 1.43 (0.78–2.64) | 112 | 0.93 (0.68–1.27) | 64 | 1.13 (0.72–1.75) |
| ≥4.68 | 15 | 0.42 (0.17–1.05) | 35 | 1.33 (0.65–2.73) | 125 | 0.84 (0.58–1.20) | 68 | 1.03 (0.62–1.72) |
| P value for trend | 0.04 | 0.33 | 0.25 | 0.90 | ||||
| Triglycerides/HDLc | ||||||||
| <0.44 | 5 | 1.00 (Ref) | 21 | 1.00 (Ref) | 83 | 1.00 (Ref) | 48 | 1.00 (Ref) |
| 0.44–0.70 | 11 | 1.71 (0.59–4.94) | 26 | 1.14 (0.63–2.06) | 89 | 0.91 (0.68–1.24) | 57 | 0.99 (0.67–1.47) |
| 0.70–1.23 | 21 | 2.48 (0.91–6.75) | 30 | 1.25 (0.67–2.32) | 113 | 0.98 (0.72–1.33) | 58 | 0.84 (0.55–1.27) |
| ≥1.23 | 20 | 1.37 (0.43–4.42) | 38 | 1.88 (0.83–4.27) | 130 | 1.00 (0.67–1.49) | 65 | 0.81 (0.46–1.42) |
| P value for trend | 0.52 | 0.18 | 0.97 | 0.36 | ||||
| ApoB/ApoA-Ic | ||||||||
| <0.67 | 15 | 1.00 (Ref) | 14 | 1.00 (Ref) | 70 | 1.00 (Ref) | 37 | 1.00 (Ref) |
| 0.67–0.84 | 16 | 0.68 (0.33–1.39) | 22 | 1.16 (0.59–2.29) | 88 | 0.85 (0.62–1.17) | 52 | 0.93 (0.60–1.42) |
| 0.84–1.05 | 13 | 0.39 (0.18–0.86) | 46 | 2.00 (1.06–3.78) | 120 | 0.89 (0.65–1.21) | 76 | 1.00 (0.66–1.52) |
| ≥0.85 | 13 | 0.31 (0.13–0.74) | 33 | 1.37 (0.66–2.84) | 137 | 0.86 (0.61–1.21) | 63 | 0.67 (0.41–1.10) |
| P value for trend | 0.005 | 0.19 | 0.61 | 0.16 | ||||
aNot adjusted for TC.
bNot adjusted for TG.
cMeasured in Subcohort B.
Table 3.
Hazard ratios and 95% confidence intervals for GI cancer for lipid components as continuous variables and based on cut-off points used in cardiovascular disease prevention, adjusted for age, gender, SES, fasting status, glucose (continuous), total cholesterol (continuous), and triglycerides (continuous).
| Oesophageal cancer | Stomach cancer | Colon cancer | Rectal cancer | |||||
|---|---|---|---|---|---|---|---|---|
| n | HR (95% CI) | n | HR (95% CI) | n | HR (95% CI) | n | HR (95% CI) | |
| Glucose (mmol/L)a | ||||||||
| Unit increase in log | 2.03 (0.45–9.12) | 1.09 (0.41–2.88) | 2.30 (1.18–4.48) | 1.14 (0.54–2.44) | ||||
| <6.11 | 271 | 1.00 (Ref) | 686 | 1.00 (Ref) | 2218 | 1.00 (Ref) | 1353 | 1.00 (Ref) |
| ≥6.11 | 47 | 1.33 (0.87–2.01) | 90 | 1.02 (0.76–1.38) | 254 | 1.00 (0.84–1.19) | 157 | 1.10 (0.88–1.37) |
| Total cholesterol (mmol/L)a | ||||||||
| Unit increase in log | 0.63 (0.35–1.13) | 0.99 (0.67–1.47) | 1.10 (0.88–1.37) | 1.83 (1.38–2.44) | ||||
| <6.50 | 224 | 1.00 (Ref) | 523 | 1.00 (Ref) | 1647 | 1.00 (Ref) | 972 | 1.00 (Ref) |
| ≥6.50 | 94 | 0.90 (0.70–1.15) | 253 | 1.04 (0.89–1.22) | 825 | 1.01 (0.92–1.10) | 538 | 1.20 (1.07–1.34) |
| Triglycerides (mmol/L)b | ||||||||
| Unit increase in log | 1.62 (1.31–2.01) | 1.04 (0.90–1.21) | 1.17 (1.08–1.27) | 0.99 (0.89–1.09) | ||||
| <1.71 | 214 | 1.00 (Ref) | 569 | 1.00 (Ref) | 2337 | 1.00 (Ref) | 1117 | 1.00 (Ref) |
| ≥1.71 | 104 | 1.37 (1.06–1.76) | 207 | 1.04 (0.87–1.23) | 135 | 1.15 (1.05–1.27) | 393 | 1.00 (0.88–1.13) |
| LDL cholesterol (mmol/L)c | ||||||||
| Unit increase in log | 0.13 (0.03–0.49) | 1.40 (0.28–6.91) | 0.82 (0.37–1.80) | 0.80 (0.27–2.37) | ||||
| <4.10 | 44 | 1.00 (Ref) | 69 | 1.00 (Ref) | 248 | 1.00 (Ref) | 137 | 1.00 (Ref) |
| ≥4.10 | 13 | 0.70 (0.29–1.65) | 46 | 1.08 (0.62–1.88) | 167 | 1.00 (0.75–1.34) | 91 | 0.84 (0.57–1.24) |
| HDL cholesterol (mmol/L)c | ||||||||
| Unit increase in log | 3.66 (1.41–9.49) | 0.82 (0.39–1.71) | 1.44 (0.98–2.12) | 1.24 (0.73–2.12) | ||||
| <1.03 | 6 | 1.00 (Ref) | 10 | 1.00 (Ref) | 54 | 1.00 (Ref) | 32 | 1.00 (Ref) |
| ≥1.03 | 51 | 4.02 (1.43–11.32) | 105 | 1.81 (0.85–3.86) | 361 | 1.12 (0.79–1.59) | 196 | 0.84 (0.53–1.31) |
| Apolipoprotein B (mmol/L)c | ||||||||
| Unit increase in log | 0.25 (0.06–1.01) | 1.72 (0.42–7.03) | 1.30 (0.63–2.68) | 0.44 (0.18–1.09) | ||||
| <1.50 | 43 | 1.00 (Ref) | 84 | 1.00 (Ref) | 299 | 1.00 (Ref) | 176 | 1.00 (Ref) |
| ≥1.50 | 14 | 1.38 (0.62–3.06) | 31 | 1.01 (0.58–1.75) | 116 | 0.92 (0.69–1.22) | 52 | 0.56 (0.38–0.84) |
| Apolipoprotein A-I (mmol/L)c | ||||||||
| Unit increase in log | 7.56 (1.58–36.10) | 0.58 (0.18–1.91) | 1.54 (0.83–2.85) | 1.41 (0.61–3.27) | ||||
| <1.05 | 0 | N/A | 3 | 1.00 (Ref) | 14 | 1.00 (Ref) | 10 | 1.00 (Ref) |
| ≥1.05 | 57 | N/A | 112 | 1.28 (0.40–4.08) | 401 | 0.98 (0.57–1.68) | 218 | 0.68 (0.36–1.30) |
| LDL/HDLc | ||||||||
| Unit increase in log | 0.41 (0.20–0.81) | 1.16 (0.66–2.01) | 0.78 (0.59–1.04) | 0.82 (0.55–1.21) | ||||
| <3.50 | 48 | 1.00 (Ref) | 87 | 1.00 (Ref) | 322 | 1.00 (Ref) | 180 | 1.00 (Ref) |
| ≥3.50 | 9 | 0.44 (0.20–0.98) | 28 | 1.17 (0.70–1.95) | 93 | 0.88 (0.67–1.16) | 48 | 0.82 (0.56–1.19) |
| Total cholesterol/HDLc | ||||||||
| Unit increase in log | 0.18 (0.07–0.49) | 1.18 (0.56–2.48) | 0.73 (0.50–1.08) | 0.80 (0.47–1.38) | ||||
| <5.00 | 47 | 1.00 (Ref) | 87 | 1.00 (Ref) | 309 | 1.00 (Ref) | 169 | 1.00 (Ref) |
| ≥5.00 | 10 | 0.32 (0.14–0.74) | 28 | 0.97 (0.57–1.66) | 106 | 0.92 (0.70–1.21) | 59 | 1.02 (0.70–1.48) |
| Log (triglycerides/HDL)c | ||||||||
| Unit increase | 0.84 (0.47–1.50) | 1.32 (0.75–2.35) | 0.94 (0.72–1.23) | 0.95 (0.65–1.40) | ||||
| <0.5 | 45 | 1.00 (Ref) | 92 | 1.00 (Ref) | 329 | 1.00 (Ref) | 183 | 1.00 (Ref) |
| ≥0.5 | 12 | 0.39 (0.16–0.97) | 23 | 1.22 (0.61–2.44) | 86 | 1.02 (0.72–1.45) | 45 | 1.13 (0.70–1.85) |
| ApoB/ApoA-Ic | ||||||||
| Unit increase in log | 0.29 (0.12–0.70) | 1.49 (0.68–3.24) | 0.91 (0.61–1.35) | 0.66 (0.39–1.13) | ||||
| <1.00 | 42 | 1.00 (Ref) | 73 | 1.00 (Ref) | 258 | 1.00 (Ref) | 151 | 1.00 (Ref) |
| ≥1.00 | 15 | 0.51 (0.27–1.00) | 42 | 0.95 (0.61–1.48) | 157 | 0.90 (0.71–1.13) | 77 | 0.71 (0.52–0.98) |
aNot adjusted for total cholesterol.
bNot adjusted for triglycerides.
cMeasured in Subcohort B.
Table 4.
Hazard ratios and 95% confidence intervals for GI cancer for lipid components as continuous variables and based on cut-off points used in cardiovascular disease prevention, stratified by glucose levels, adjusted for age, gender, SES, fasting status, glucose (continuous), total cholesterol (continuous), and triglycerides (continuous).
| Oesophageal cancer | Stomach cancer | Colon cancer | Rectal cancer | |||||
|---|---|---|---|---|---|---|---|---|
| n | HR (95% CI) | n | HR (95% CI) | n | HR (95% CI) | n | HR (95% CI) | |
| Glucose < 6.11 mmol/L | ||||||||
|
| ||||||||
| Total cholesterol (mmol/L)a | ||||||||
| Unit increase in log | 0.53 (0.28–1.02) | 1.00 (0.65–1.53) | 1.05 (0.83–1.34) | 1.75 (1.29–2.38) | ||||
| <6.50 | 192 | 1.00 (Ref) | 463 | 1.00 (Ref) | 1483 | 1.00 (Ref) | 889 | 1.00 (Ref) |
| ≥6.50 | 79 | 0.89 (0.68–1.18) | 223 | 1.03 (0.87–1.22) | 735 | 1.00 (0.91–1.10) | 464 | 1.15 (1.02–1.29) |
| Triglycerides (mmol/L)b | ||||||||
| Unit increase in log | 1.62 (1.28–2.05) | 1.08 (0.92–1.26) | 1.19 (1.09–1.30) | 0.97 (0.87–1.09) | ||||
| <1.71 | 193 | 1.00 (Ref) | 521 | 1.00 (Ref) | 1678 | 1.00 (Ref) | 1044 | 1.00 (Ref) |
| ≥1.71 | 78 | 1.38 (1.05–1.83) | 165 | 1.07 (0.89–1.28) | 540 | 1.16 (1.05–1.29) | 309 | 0.98 (0.86–1.12) |
|
| ||||||||
| Glucose ≥ 6.11 mmol/L | ||||||||
|
| ||||||||
| Total cholesterol (mmol/L)a | ||||||||
| Unit increase in log | 1.02 (0.23–4.47) | 0.68 (0.23–2.05) | 1.26 (0.65–2.45) | 2.79 (1.19–6.56) | ||||
| <6.50 | 32 | 1.00 (Ref) | 60 | 1.00 (Ref) | 164 | 1.00 (Ref) | 83 | 1.00 (Ref) |
| ≥6.50 | 15 | 0.90 (0.70–1.15) | 30 | 1.04 (0.66–1.64) | 90 | 1.01 (0.77–1.32) | 74 | 1.73 (1.25–2.40) |
| Triglycerides (mmol/L)b | ||||||||
| Unit increase in log | 1.59 (0.98–2.60) | 0.95 (0.65–1.38) | 1.17 (0.93–1.46) | 1.11 (0.84–1.47) | ||||
| <1.71 | 21 | 1.00 (Ref) | 48 | 1.00 (Ref) | 124 | 1.00 (Ref) | 73 | 1.00 (Ref) |
| ≥1.71 | 26 | 1.27 (0.69–2.32) | 42 | 0.96 (0.62–1.48) | 130 | 1.20 (0.93–1.55) | 84 | 1.15 (0.83–1.60) |
aNot adjusted for TC.
bNot adjusted for TG.
3.2. Stomach Cancer
Quartiles of HDL were negatively associated with risk of stomach cancer, albeit not statistically significant (Table 2). In contrast, a positive association was found between the 2nd quartile of the apoB/apoA-I ratio and risk of stomach cancer, although no clear trend was apparent (HR: 2.00 (95% CI: 1.06–3.78) compared to the 1st quartile, P value for trend 0.19). No association was observed when using log transformations and dichotomized values of lipid components (Table 3). Similarly, stratification by glucose levels (Table 4) did not show any relation between glucose, lipid components and stomach cancer risk.
3.3. Colon Cancer
We found a positive trend between quartiles of glucose, TG and colon cancer risk (e.g., HR for TG: 1.09 (95% CI: 0.94–1.26), 1.21 (95% CI: 1.05–1.41), and 1.30 (95% CI: 1.12–1.52) for 2nd, 3rd, and 4th quartile, compared to the 1st with P value for trend < 0.001) (Table 2). This linear association was confirmed when we assessed log transformations and dichotomized values of TG (Table 3). Stratification by glucose levels resulted in a positive association between high TG levels and colon cancer risk in persons with glucose <6.11 mmol/L (Table 4).
3.4. Rectal Cancer
We observed a positive association between quartiles of TC and the risk of rectal cancer (HR: 1.11 (95% CI: 0.91–1.35), 1.10 (95% CI: 0.91–1.33), 1.32 (95% CI: 1.09–1.59) for 2nd, 3rd, and 4th quartile, compared to the 1st with P value for trend <0.001) (Table 2). Similar results were found when we used log transformations and dichotomized values of lipid components (Table 3). Glucose-stratified analysis showed that TC was consistently positively associated with rectal cancer risk in both those with high and normal glucose levels (Table 4).
We repeated all the above analyses in Subgroup A and found no statistically significant results, with or without additional adjustment for BMI (results not shown). Finally, we performed a sensitivity analysis by excluding those with follow-up time < 3 years, but this did not substantially alter our findings (results not shown).
4. Discussion
The present study showed that risk of oesophageal cancer was positively associated with serum levels of TG, and negatively with LDL, LDL/HDL ratio, TC/HDL ratio, log (TG/HDL), and apoB/apoA-I ratio. Meanwhile, we observed a higher risk of colon cancer with high levels of glucose and TG, and an increased risk of rectal cancer in those with high TC levels. Stratification by glucose levels showed that risk of colon cancer was positively associated with TG levels in primarily those with glucose < 6.11 mmol/L.
Several mechanisms have been suggested to link altered levels of serum lipids and development of cancer. Lipid peroxides in lipoproteins are known to induce cellular damage and inflammatory response [14, 35], which may promote malignant transformation of normal cells. Furthermore, elevated levels of LDL as well as reduced levels of HDL have been linked to an increased activity of proinflammatory markers such as TNF-α and IL-6 [36, 37]. On the other hand, TG and TC biosyntheses are essentially regulated by insulin [38], and hyperinsulinaemia occurring has been shown to promote carcinogenesis in animal studies [39]. Therefore, it is also possible that lipid components mediate the role of insulin in cancer development instead of acting as an independent risk factor.
Alternatively, lipid components may act as a marker of other exposure such as lifestyle that actually modifies the risk of gastrointestinal cancer, and thus it may not be fruitful for prevention to target lipids per se. Nevertheless, the use of hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors or statins, a widely used lipid-lowering agent, have been suggested to lower the risk of colorectal and gastric cancer in several Western and Asian studies [40–42]. This early evidence, albeit requiring further confirmation, indicates that lipids are directly involved in cancer causative pathways, either as an independent culprit or a mediator of other factors responsible for carcinogenesis.
4.1. Oesophageal Cancer
We found a positive linear association between TG levels and risk of oesophageal cancer. Prior studies have indicated the positive link between dietary fat as well as obesity and the risk of oesophageal adenocarcinoma [43–45], but little evidence has been reported regarding the role of serum lipids. However, high levels of TG and TC have been reported to be related to increased incidence of lymph node metastasis in patients with resectable oesophageal adenocarcinoma [46]. The present study also observed a negative association between oesophageal cancer risk and several other lipid components. LDL is easily oxidized and may serve as a marker for lipid peroxidation [47], which is thought to enhance carcinogenesis. Interestingly, in a population-based study in China, the levels of LDL as well as antibody targeting oxidized LDL (ox-LDL) were lower in the groups with more advanced stage of oesophageal squamous cell carcinoma compared to those with normal oesophageal squamous epithelium [48]. As oesophageal squamous cell carcinoma remains the majority of oesophageal cancer in Eastern countries [49], such finding may not necessarily reflect the association between lipid components and oesophageal cancer in general. However, our findings, which are based on a European population, also suggest a protective role of LDL, which suggests a more complex etiology for oesophageal cancer in the context of the lipid metabolism.
4.2. Stomach Cancer
We observed a weak negative trend between HDL levels and risk of stomach cancer. Low levels of HDL have been associated with increased inflammatory activity [50], which may contribute to cancer development. A higher incidence of stomach dysplasia has also been reported for lower HDL levels in a Korean case-control study [51]. There was no association between TG levels and stomach cancer risk in the large European cohort Me-Can [8, 52], which is consistent with what we found in AMORIS. There was also no association between TC and stomach cancer, which is in contrast with the inverse relation between TC levels and stomach cancer risk in Asian studies [10, 53]. Differences in diet and ethnicities related to stomach cancer risk may explain these opposite findings [54].
4.3. Colorectal Cancer
We observed an increased risk of colon cancer in those with high glucose and TG levels, and an increased risk of rectal cancer in those with high TC levels. Both in vitro and animal studies have shown that high dietary fat and cholesterol increase lipid peroxidation and alter immune responses in colon [15, 55, 56]. Furthermore, fat intake has been shown to modify Ras and ERK signaling activities, which play an important role in the development of colorectal cancer [57]. Additionally, insulin resistance which results in elevated glucose levels have also been suggested to greatly impact the relation between dyslipidemia and gastrointestinal cancer [17]. Higher levels of glucose and TG have been linked to increased colon and rectal cancer risks in several European cohorts [8, 52, 54]. Meanwhile, a Korean study showed elevated TC to be positively associated with a marginally increased risk of colon cancer in men [53], but a nested case-control study in the European Prospective Investigation into Cancer and Nutrition (EPIC) study showed an inverse association between TC and colon cancer risk [9]. As Western diet has been linked to increased colorectal cancer risk [58], cultural-related lifestyle may contribute to such discrepancy in the relation between lipid metabolism and risk of colorectal cancer in different ethnic groups.
4.4. Strengths and Limitations
The major strength of this study is the large number of subjects with baseline measurements of lipid biomarkers and glucose, all measured at the same clinical laboratory. The use of national registers provided detailed follow-up information on diagnosis of cancer, time of death, and emigration for all subjects. The AMORIS population was mainly selected by analyzing blood samples from healthy checkups in nonhospitalized individuals. However, this healthy cohort effect would not affect the internal validity of the current study and is likely to be minor since it has been shown that the AMORIS cohort is similar to the general working population of Stockholm County in terms of SES and ethnicity [59]. A limitation of this study is that there was no record of lipid-lowering medications, and only limited data on BMI was available which confined the statistical power when also studying BMI. However, lipid-lowering medication was not used widely in Sweden during the earlier part of the observation period for this study (1985–1996) [60]. Additionally, there was no available information on other possible confounders such as smoking status, alcohol consumption, nor was there on tumor grade, stage or histology. As lipid metabolism is altered as cancer develops [60, 61], reverse causation may be another reason for some of the inconsistencies with other studies. However, exclusion of the first three years of followup did not alter our findings.
5. Conclusion
The current study provides new evidence linking the lipid metabolism to oesophageal cancer etiology and showed a positive association between TC and rectal cancer risk as well as between TG and oesophageal and colon cancer risk in normoglycaemic individuals. To consider the role of dietary fat as well as serum lipids in carcinogenesis, more detailed studies are needed to clarify the effects of both systemic and local effect of lipid constituents on gastrointestinal cancer development. Further investigations in this subject should also take into account other metabolic determinants including insulin and obesity.
References
- 1.Goff DC, Bertoni AG, Kramer H, et al. Dyslipidemia prevalence, treatment, and control in the Multi-Ethnic Study of Atherosclerosis (MESA): gender, ethnicity, and coronary artery calcium. Circulation. 2006;113(5):647–656. doi: 10.1161/CIRCULATIONAHA.105.552737. [DOI] [PubMed] [Google Scholar]
- 2.Ghandehari H, Kamal-Bahl S, Wong ND. Prevalence and extent of dyslipidemia and recommended lipid levels in US adults with and without cardiovascular comorbidities: the National Health and Nutrition Examination Survey 2003-2004. American Heart Journal. 2008;156(1):112–119. doi: 10.1016/j.ahj.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Wyszynski DF, Waterworth DM, Barter PJ, et al. Relation between atherogenic dyslipidemia and the Adult Treatment Program-III definition of metabolic syndrome (Genetic Epidemiology of Metabolic Syndrome Project) American Journal of Cardiology. 2005;95(2):194–198. doi: 10.1016/j.amjcard.2004.08.091. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed RL, Schmitz KH, Anderson KE, Rosamond WD, Folsom AR. The metabolic syndrome and risk of incident colorectal cancer. Cancer. 2006;107(1):28–36. doi: 10.1002/cncr.21950. [DOI] [PubMed] [Google Scholar]
- 5.Bjørge T, Lukanova A, Jonsson H, et al. Metabolic syndrome and breast cancer in the Me-Can (metabolic syndrome and cancer) project. Cancer Epidemiology Biomarkers and Prevention. 2010;19(7):1737–1745. doi: 10.1158/1055-9965.EPI-10-0230. [DOI] [PubMed] [Google Scholar]
- 6.Russo A, Autelitano M, Bisanti L. Metabolic syndrome and cancer risk. European Journal of Cancer. 2008;44(2):293–297. doi: 10.1016/j.ejca.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Tabuchi M, Kitayama J, Nagawa H. Hypertriglyceridemia is positively correlated with the development of colorectal tubular adenoma in Japanese men. World Journal of Gastroenterology. 2006;12(8):1261–1264. doi: 10.3748/wjg.v12.i8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ulmer H, Borena W, Rapp K, et al. Serum triglyceride concentrations and cancer risk in a large cohort study in Austria. British Journal of Cancer. 2009;101(7):1202–1206. doi: 10.1038/sj.bjc.6605264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Duijnhoven FJB, Bueno-De-Mesquita HB, Calligaro M, et al. Blood lipid and lipoprotein concentrations and colorectal cancer risk in the European Prospective Investigation into Cancer and Nutrition. Gut. 2011;60(8):1094–1102. doi: 10.1136/gut.2010.225011. [DOI] [PubMed] [Google Scholar]
- 10.Iso H, Ikeda A, Inoue M, Sato S, Tsugane S. Serum cholesterol levels in relation to the incidence of cancer: the JPHC study cohorts. International Journal of Cancer. 2009;125(11):2679–2686. doi: 10.1002/ijc.24668. [DOI] [PubMed] [Google Scholar]
- 11.Chyou PH, Nomura AMY, Stemmermann GN, Kato I. Prospective study of serum cholesterol and site-specific cancers. Journal of Clinical Epidemiology. 1992;45(3):287–292. doi: 10.1016/0895-4356(92)90089-6. [DOI] [PubMed] [Google Scholar]
- 12.Konradsen S, Ag H, Lindberg F, Hexeberg S, Jorde R. Serum 1,25-dihydroxy vitamin D is inversely associated with body mass index. European Journal of Nutrition. 2008;47(2):87–91. doi: 10.1007/s00394-008-0700-4. [DOI] [PubMed] [Google Scholar]
- 13.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. The Lancet. 2008;371(9612):569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 14.Ahotupa M, Suomela JP, Vuorimaa T, Vasankari T. Lipoprotein-specific transport of circulating lipid peroxides. Annals of Medicine. 2010;42(7):521–529. doi: 10.3109/07853890.2010.510932. [DOI] [PubMed] [Google Scholar]
- 15.Erdelyi I, Levenkova N, Lin EY, et al. Western-style diets induce oxidative stress and dysregulate immune responses in the colon in a mouse model of sporadic colon cancer. Journal of Nutrition. 2009;139(11):2072–2078. doi: 10.3945/jn.108.104125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moustafa T, Fickert P, Magnes C, et al. Alterations in lipid metabolism mediate inflammation, fibrosis, and proliferation in a mouse model of chronic cholestatic liver injury. Gastroenterology. 2012;142(1):140–151.e12. doi: 10.1053/j.gastro.2011.09.051. [DOI] [PubMed] [Google Scholar]
- 17.Hsieh J, Hayashi AA, Webb J, Adeli K. Postprandial dyslipidemia in insulin resistance: mechanisms and role of intestinal insulin sensitivity. Atherosclerosis Supplements. 2008;9(2):7–13. doi: 10.1016/j.atherosclerosissup.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 18.Ji Y, Sakata Y, Tso P. Nutrient-induced inflammation in the intestine. Current Opinion in Clinical Nutrition and Metabolic Care. 2011;14(4):315–321. doi: 10.1097/MCO.0b013e3283476e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayne ST, Navarro SA. Diet, obesity and reflux in the etiology of adenocarcinomas of the esophagus and gastric cardia in humans. Journal of Nutrition. 2002;132(11, supplement):3467S–3470S. doi: 10.1093/jn/132.11.3467S. [DOI] [PubMed] [Google Scholar]
- 20.Järvinen R, Knekt P, Hakulinen T, Rissanen H, Heliövaara M. Dietary fat, cholesterol and colorectal cancer in a prospective study. British Journal of Cancer. 2001;85(3):357–361. doi: 10.1054/bjoc.2001.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lucenteforte E, Bosetti C, Gallus S, et al. Macronutrients, fatty acids and cholesterol intake and stomach cancer risk. Annals of Oncology. 2009;20(8):1434–1438. doi: 10.1093/annonc/mdp009. [DOI] [PubMed] [Google Scholar]
- 22.Jungner I, Marcovina SM, Walldius G, Holme I, Kolar W, Steiner E. Apolipoprotein B and A-I values in 147 576 Swedish males and females, standardized according to the World Health Organization-International Federation of Clinical Chemistry First International Reference Materials. Clinical Chemistry. 1998;44(8 I):1641–1649. [PubMed] [Google Scholar]
- 23.Walldius G, Jungner I, Holme I, Aastveit AH, Kolar W, Steiner E. High apolipoprotein B, low apolipoprotein A-I, and improvement in the prediction of fatal myocardial infarction (AMORIS study): a prospective study. The Lancet. 2001;358(9298):2026–2033. doi: 10.1016/S0140-6736(01)07098-2. [DOI] [PubMed] [Google Scholar]
- 24.Holme I, Aastveit AH, Jungner I, Walldius G. Relationships between lipoprotein components and risk of myocardial infarction: age, gender and short versus longer follow-up periods in the Apolipoprotein MOrtality RISk study (AMORIS) Journal of Internal Medicine. 2008;264(1):30–38. doi: 10.1111/j.1365-2796.2008.01925.x. [DOI] [PubMed] [Google Scholar]
- 25.Holme I, Aastveit AH, Hammar N, Jungner I, Walldius G. Relationships between lipoprotein components and risk of ischaemic and haemorrhagic stroke in the Apolipoprotein MOrtality RISk study (AMORIS) Journal of Internal Medicine. 2009;265(2):275–287. doi: 10.1111/j.1365-2796.2008.02016.x. [DOI] [PubMed] [Google Scholar]
- 26.Walldius G, Jungner I, Kolar W, Holme I, Steiner E. High cholesterol and triglyceride values in Swedish males and females: increased risk of fatal myocardial infarction. First report from the AMORIS (Apolipoprotein related MOrtality RISk) study. Blood Pressure. 1992;4(supplement):35–42. [PubMed] [Google Scholar]
- 27.Van Hemelrijck M, Garmo H, Binda E, et al. Immunoglobulin e and cancer: a meta-analysis and a large Swedish cohort study. Cancer Causes and Control. 2010;21(10):1657–1667. doi: 10.1007/s10552-010-9594-6. [DOI] [PubMed] [Google Scholar]
- 28.Van Hemelrijck M, Garmo H, Holmberg L, et al. Prostate cancer risk in the Swedish AMORIS study: the interplay between triglycerides, total cholesterol, and glucose. Cancer. 2011;117:2086–2095. doi: 10.1002/cncr.25758. [DOI] [PubMed] [Google Scholar]
- 29.Central Bureau for Statistics. Statistics Sweden. Stockholm, Sweden: Central Bureau for Statistics; 2008. [Google Scholar]
- 30.Swanson CA, Potischman N, Barrett RJ, et al. Endometrial cancer risk in relation to serum lipids and lipoprotein levels. Cancer Epidemiology Biomarkers and Prevention. 1994;3(7):575–581. [PubMed] [Google Scholar]
- 31.Walldius G, Jungner I. The apoB/apoA-I ratio: a strong, new risk factor for cardiovascular disease and a target for lipid-lowering therapy—a review of the evidence. Journal of Internal Medicine. 2006;259(5):493–519. doi: 10.1111/j.1365-2796.2006.01643.x. [DOI] [PubMed] [Google Scholar]
- 32.Jungner I, Walldius G, Holme I, Kolar W, Steiner E. Apolipoprotein B and A-I in relation to serum cholesterol and triglycerides in 43 000 Swedish males and females. International Journal of Clinical & Laboratory Research. 1992;21(2-4):247–255. doi: 10.1007/BF02591655. [DOI] [PubMed] [Google Scholar]
- 33.Millán J, Pintó X, Muñoz A, et al. Lipoprotein ratios: physiological significance and clinical usefulness in cardiovascular prevention. Vascular Health and Risk Management. 2009;5:757–765. [PMC free article] [PubMed] [Google Scholar]
- 34.Cleeman JI. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) Journal of the American Medical Association. 2001;285(19):2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 35.Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. British Journal of Pharmacology. 2008;153(1):6–20. doi: 10.1038/sj.bjp.0707395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mizia-Stec K, Zahorska-Markiewicz B, Mandecki T, et al. Hyperlipidaemias and serum cytokines in patients with coronary artery disease. Acta Cardiologica. 2003;58(1):9–15. doi: 10.2143/AC.58.1.2005253. [DOI] [PubMed] [Google Scholar]
- 37.Zuliani G, Volpato S, Blè A, et al. High interleukin-6 plasma levels are associated with low HDL-C levels in community-dwelling older adults: the InChianti study. Atherosclerosis. 2007;192(2):384–390. doi: 10.1016/j.atherosclerosis.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cianflone K, Paglialunga S, Roy C. Intestinally derived lipids: metabolic regulation and consequences-An overview. Atherosclerosis Supplements. 2008;9(2):63–68. doi: 10.1016/j.atherosclerosissup.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 39.Novosyadlyy R, Lann DE, Vijayakumar A, et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Research. 2010;70(2):741–751. doi: 10.1158/0008-5472.CAN-09-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng M-H, Chiu H-F, Ho S-C, Tsai S-S, Wu T-N, Yang C-Y. Statin use and the risk of colorectal cancer: a population-based case-control study. World Journal of Gastroenterology. 2011;17(47):5197–5202. doi: 10.3748/wjg.v17.i47.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiu H-F, Ho S-C, Chang C-C, Wu T-N, Yang C-Y. Statins are associated with a reduced risk of gastric cancer: a population-based case-control study. American Journal of Gastroenterology. 2011;106(12):2098–2103. doi: 10.1038/ajg.2011.277. [DOI] [PubMed] [Google Scholar]
- 42.Simon MS, Rosenberg CA, Rodabough RJ, et al. Prospective analysis of association between use of statins or other lipid-lowering agents and colorectal cancer risk. Annals of Epidemiology. 2012;22(1):17–27. doi: 10.1016/j.annepidem.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Doherty MG, Freedman ND, Hollenbeck AR, Schatzkin A, Abnet CC. A prospective cohort study of obesity and risk of oesophageal and gastric adenocarcinoma in the NIH-AARP Diet and Health Study. Gut. 2012;61(9):1261–1268. doi: 10.1136/gutjnl-2011-300551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Doherty MG, Cantwell MM, Murray LJ, Anderson LA, Abnet CC. Dietary fat and meat intakes and risk of reflux esophagitis, Barrett’s esophagus and esophageal adenocarcinoma. International Journal of Cancer. 2011;129(6):1493–1502. doi: 10.1002/ijc.26108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang ZF, Kurtz RC, Yu GP, et al. Adenocarcinomas of the esophagus and gastric cardia: the role of diet. Nutrition and Cancer. 1997;27(3):298–309. doi: 10.1080/01635589709514541. [DOI] [PubMed] [Google Scholar]
- 46.Sako A, Kitayama J, Kaisaki S, Nagawa H. Hyperlipidemia is a risk factor for lymphatic metastasis in superficial esophageal carcinoma. Cancer Letters. 2004;208(1):43–49. doi: 10.1016/j.canlet.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 47.Nishi K, Itabe H, Uno M, et al. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22(10):1649–1654. doi: 10.1161/01.atv.0000033829.14012.18. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Li H, Diao Y, et al. Relationship between oxidized LDL antibodies and different stages of esophageal carcinoma. Archives of Medical Research. 2008;39(8):760–767. doi: 10.1016/j.arcmed.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 49.Lagergren J, Lagergren P. Oesophageal cancer. British Medical Journal. 2010;341:p. c6280. doi: 10.1136/bmj.c6280. [DOI] [PubMed] [Google Scholar]
- 50.Rivera JJ, Nasir K, Campbell C, Carvalho JAM, Blumenthal RS, Santos RD. Relation of plasma lipoprotein levels with low-grade inflammation in white men without clinical evidence of myocardial ischemia. American Journal of Cardiology. 2007;100(3):450–454. doi: 10.1016/j.amjcard.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 51.Jung MK, Jeon SW, Cho CM, et al. Hyperglycaemia, hypercholesterolaemia and the risk for developing gastric dysplasia. Digestive and Liver Disease. 2008;40(5):361–365. doi: 10.1016/j.dld.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 52.Borena W, Stocks T, Jonsson H, et al. Serum triglycerides and cancer risk in the metabolic syndrome and cancer (Me-Can) collaborative study. Cancer Causes and Control. 2011;22(2):291–299. doi: 10.1007/s10552-010-9697-0. [DOI] [PubMed] [Google Scholar]
- 53.Kitahara CM, De González AB, Freedman ND, et al. Total cholesterol and cancer risk in a large prospective study in Korea. Journal of Clinical Oncology. 2011;29(12):1592–1598. doi: 10.1200/JCO.2010.31.5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bornschein J, Rokkas T, Selgrad M, Malfertheiner P. Helicobacter pylori and clinical aspects of gastric cancer. Helicobacter. 2009;14(1):41–45. doi: 10.1111/j.1523-5378.2009.00695.x. [DOI] [PubMed] [Google Scholar]
- 55.Angeli JPF, Garcia CCM, Sena F, et al. Lipid hydroperoxide-induced and hemoglobin-enhanced oxidative damage to colon cancer cells. Free Radical Biology and Medicine. 2011;51(2):503–515. doi: 10.1016/j.freeradbiomed.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 56.Tseng T-H, Hsu J-D, Chu C-Y, Wang C-J. Promotion of colon carcinogenesis through increasing lipid peroxidation induced in rats by a high cholesterol diet. Cancer Letters. 1996;100(1-2):81–87. doi: 10.1016/0304-3835(95)04073-0. [DOI] [PubMed] [Google Scholar]
- 57.Davidson LA, Lupton JL, Jiang YH, Chapkin RS. Carcinogen and dietary lipid regulate ras expression and localization in rat colon without affecting farnesylation kinetics. Carcinogenesis. 1999;20(5):785–791. doi: 10.1093/carcin/20.5.785. [DOI] [PubMed] [Google Scholar]
- 58.Kim MK, Sasaki S, Otani T, Tsugane S. Dietary patterns and subsequent colorectal cancer risk by subsite: a prospective cohort study. International Journal of Cancer. 2005;115(5):790–798. doi: 10.1002/ijc.20943. [DOI] [PubMed] [Google Scholar]
- 59.Van Hemelrijck M. Metabolic syndrome and prostate cancer: biomarkers and treatment side-effects [Ph.D. thesis] London, UK: King’s College London; 2010. [Google Scholar]
- 60.Magrini N, Einarson T, Vaccheri A, McManus P, Montanaro N, Bergman U. Use of lipid-lowering drugs from 1990 to 1994: an international comparison among Australia, Finland, Italy (Emilia Romagna Region), Norway and Sweden. European Journal of Clinical Pharmacology. 1997;53(3-4):185–189. doi: 10.1007/s002280050360. [DOI] [PubMed] [Google Scholar]
- 61.Patterson AD, Maurhofer O, Beyoğlu D, et al. Aberrant lipid metabolism in hepatocellular carcinoma revealed by plasma metabolomics and lipid profiling. Cancer Research. 2011;71(21):6590–6600. doi: 10.1158/0008-5472.CAN-11-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]