

Abstract
Calcium binding to the regulatory domain of cardiac troponin C (cNTnC) causes a conformational change that exposes a hydrophobic surface to which troponin I (cTnI) binds, prompting a series of protein-protein interactions that culminate in muscle contraction. A number of cTnC variants that alter the Ca2+-sensitivity of the thin filament have been linked to disease. Tikunova and Davis have engineered a series of cNTnC mutations that altered Ca2+ binding properties and studied the effects on the Ca2+ sensitivity of the thin filament and contraction [Tikunova and Davis (2004) J Biol Chem279, 35341–35352]. One of the mutations they engineered, the L48Q variant, resulted in a pronounced increase in cNTnC Ca2+ binding affinity and Ca2+ sensitivity of cardiac muscle force development. In this work, we sought structural and mechanistic explanations for the increased Ca2+ sensitivity of contraction for the L48Q cNTnC variant, using an array of biophysical techniques. We found that the L48Q mutation enhanced binding of both Ca2+ and cTnI to cTnC. NMR chemical shift and relaxation data provided evidence that the cNTnC hydrophobic core is more exposed with the L48Q variant. Molecular dynamics simulations suggest that the mutation disrupts a network of crucial hydrophobic interactions so that the closed form of cNTnC is destabilized. The findings emphasize the importance of cNTnC's conformation in the regulation of contraction and suggest that mutations in cNTnC that alter myofilament Ca2+ sensitivity can do so by modulating Ca2+ and cTnI binding.
Keywords: Troponin C, Troponin I, Calcium binding, Fluorescence spectroscopy, Molecular dynamic simulation, NMR spectroscopy, Isothermal Titration Calorimetry
Striated muscle contraction is triggered by a transient increase in intracellular Ca2+, which binds to troponin C (TnC), the Ca2+-binding subunit of the cardiac troponin (cTn) complex on thin filaments. TnC is a dumbbell shaped protein that consists of N-terminal and C-terminal EF-hand motifs connected by a long flexible linker (1). There are two isoforms of TnC in striated muscle: skeletal (sTnC) and cardiac (cTnC). The regulatory lobe of sTnC (sNTnC) undergoes a large structural “opening” when it binds two Ca2+ ions(2). The structural change is much smaller in cNTnC upon binding Ca2+ and it remains essentially “closed” (3). This difference may be the result of cNTnC having only one functional Ca2+ binding site (site II) (4). The C-terminus of cTnC (cCTnC) contains high affinity Ca2+ binding sites, III and IV. Although these sites are thought to play primarily a structural role by anchoring the Tn complex to the thin filament, they may also be involved in the Ca2+ signaling pathway, since disease-related mutations in this region of cTnC affect cardiac muscle function (5–7). cTnC interacts with the other two components of cTn: cardiac troponin I (cTnI) and cardiac troponin T (cTnT). Following Ca2+ binding to site II of cNTnC, the “switch” region of cTnI (residues 147-163, cTnI147-163) binds to cNTnC and consequently the “inhibitory” region of cTnI (residues 112–146) dissociates from actin. The detachment of cTnI112–146 from actin permits increased mobility of tropomyosin over the surface of the thin filament, providing exposure of the myosin binding sites on actin and subsequently actomyosin cross-bridge formation that results in contractile force generation and cell shortening (8, 9).
A growing number of genetically identified variants (mutations) in cTn subunits associated with cardiomyopathies have been shown to alter protein-protein interactions involved in thin filament activation (10). Thus far, at least 84 mutations in cTn proteins have been identified in patients with hypertrophic, restrictive, and dilated cardiomyopathy (HCM, RCM, and DCM, respectively) (11, 12). Functional studies of HCM-associated mutations, in most cases, result in increased Ca2+ sensitivity of contraction of skinned myocardium, and at least three variants are located in cTnC (A8V, C84Y and D145E). While the increase in Ca2+-sensitivity may not cause HCM, it is possible the augmented contractility is associated with the progression and severity of HCM over time (13). Thus, understanding how altered Ca2+ binding influences cTn subunit interactions and signaling of thin filament activation could have considerable clinical relevance.
In this study we have focused on a cTnC variant, cTnC(L48Q), which was engineered by site-directed mutagenesis to enhance the Ca2+-sensitivity of cTnC. This cTnC variant has not been identified in HCM patients to date, thus it may have value in determining whether all mutations that increase the Ca2+ sensitivity of contraction contribute to the development of HCM. Additionally, the influence of the cTnC(L48Q) variant on contraction and relaxation of cardiac muscle have been studied in detail. Davis and Tikunova showed that human cTnC(L48Q) increased the Ca2+ affinity of the Tn complex and thin filaments (14, 15). Parvatiyar et al. (16) showed it increased both Ca2+ sensitivity of skinned porcine papillary contraction and ATPase sensitivity. Recently, Kreutziger et al. (17) reported that the rat L48Q variant of cTnC had similar effects in solution and increased Ca2+ sensitivity of contraction in rat trabeculae and myofibrils. It also prolonged the initial, slow phase of relaxation. Despite this wealth of functional data, the molecular mechanism for the effect of this mutation on activation and relaxation of cardiac muscle are not known. Additionally, little is known regarding how muations of cTnC that affect Ca2+ binding alter interactions with cTnI and influence thin filament activation signaling.
Leu48 makes a number of crucial hydrophobic contacts that contribute to stabilizing a closed form of cNTnC (1) – in both the apo and Ca2+-saturated states (3). We have employed an integrative approach to understand how cTnC(L48Q) results in increased myofilament Ca2+ sensitivity. We used fluorescence spectroscopy, isothermal titration calorimetry (ITC), and nuclear magnetic resonance (NMR) spectroscopy to confirm that an increase in both Ca2+ and cTnI affinity occurs by mutating L48 to glutamine. The isolated cNTnC L48Q variant, cNTnC(L48Q), dimerized in a concentration-dependent manner consistent with a more open conformation of the hydrophobic domain. The majority of cNTnC(L48Q) amide chemical shifts were located between cNTnC (predominantly closed conformation) and the cNTnC-cTnI147-163 complex (open), suggesting that cNTnC(L48Q) is more open than cNTnC. Molecular dynamics (MD) simulations of the same partial protein constructs were employed to probe the detailed relationship between structure, dynamics and function. In particular multiple 70ns simulations of cNTnC in the apo, Ca2+ saturated and cTnI147-163 bound states were performed. The MD results suggest that L48Q increases the binding affinity of cTnI147-163 for cNTnC by stabilizing its open conformation. Overall, the various results described here are consistent with L48Q stabilizing a more open conformation of cNTnC, which in turn enhances the Ca2+ and cTnI binding to cNTnC.
METHODS
Protein Mutagenesis and Purification
Construction and expression of wild-type rat cTnC, cTnI and cTnT in pET24a vector has been described in a previous publication(18). cTnCC35S and cTnC(L48Q)C35S were constructed from the rat wild-type cTnC plasmid by a primer based site-directed mutagenesis kit and confirmed by DNA sequence analysis. The plasmids for cTnC variants were then transformed into Escherichia Coli BL21 cells and expressed and purified. The DNA encoding cNTnC (residues 1–89) was inserted into pET3a expression vector as previously described (19). The L48Q mutation was engineered using a quikchange site-directed mutagenesis kit from stratagene (using paired 30-mer oligonucleotides, 5'-AAG GTG ATG AGA ATG CAA GGC CAG AAC CCC-3' and 5'-GGG GTT CTG GCC TTG CAT TCT CAT CAC CTT-3'). The construct of cNTnC(L48Q) was transformed into Escherichia Coli BL21 cells, expressed, and purified. 15N-labeled cNTnC(L48Q) protein was expressed in minimal media enriched with (15NH4)2SO4 (20).
Fluorescent Labeling of Protein
The labeling procedure used here was as previous described(21, 22). The C35S mutation was introduced to allow site-specific attachment of a fluorescent probe at C84. Briefly, cTnCC35S and cTnC(L48Q)C35S, respectively, were first dialyzed against 1mM DTT in a buffer containing 6M urea, 25mM TRIS, 1mM ethylenediamine-N,N,N9,N9-tetraacetic acid (EDTA) at pH8.0. 5mM DTT was added and the proteins were then dialyzed against the same buffer but without DTT for at least 12h with 3 buffer changes. 100mM IANBD (N-(2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diozole Mw=406.14) (in dimethylformamide) was added in 3-fold molar excess over TnCC35S or cTnC(L48Q)C35S and the protein solutions were gently shaken in the dark for >4hr at 4°C. The labeling reaction was terminated by addition of 10mM DTT and the labeled protein solution was dialyzed against buffer containing 20mM MOPS,150mM KCl,3mM MgCl2,2mM EGTA,1mM DTT, pH7.0 to remove unreacted IANBD (3 times for at least 12h). Finally, cTnCC35S and cTnC(L48Q)C35S were labeled at C84 of cTnC with IANBD. We have demonstrated that the fluorescence probe at this position monitors cTnC N-terminal Ca2+ binding (21). Labeling efficiency was calculated by determination of the IANBD fluorophore to protein molar concentration ratio. The IANBD concentration in the labeled protein was determined by dividing the absorbance of the labeled protein at the maximum absorbance for the fuorophore by the extinction coefficient of IANBD (21000M−1cm−1) at 481nm wavelength. All protein concentrations were determined using Bio-rad protein assay. The final labeling efficiency was then determined as 90%.
Reconstitution of Tn Complexes
The Tn subunits cTnI, and cTnT were first dialyzed separately against 6M urea, 25mM TRIS, 1mM EDTA at pH8. After dialysis, IANBD-cTnCC35S/cTnI/cTnT were mixed at the molar ratio of 1:1:1. After incubating at room temperature for 30 min, the protein solution was dialyzed through a series of steps against (1) 2M urea, 0.75M KCl, 20mM MOPS, 3mM MgCl2, 1mM CaCl2, pH 7.0 (2) 1M urea, 0.75M KCl, 20mM MOPS, 3mM MgCl2, 2mM EGTA, pH 7.0 (3) 0.75M KCl, 20mM MOPS, 3mM MgCl2, 2mM EGTA, pH 7.0 (4) 0.5M KCl, 20mM MOPS, 3mM MgCl2, 2mM EGTA, pH 7.0 (5) 0.25M KCl, 20mM MOPS, 3mM MgCl2, 2mM EGTA, pH 7.0 (6) Finally, 150mM KCl, 20mM MOPS, 3mM MgCl2, 2mM EGTA, ,1mM DTT, pH 7.0. All dialysis was done in the dark (without stirring) at 4°C. Proteins that precipitated during the dialysis with decreasing KCl concentration were removed by centrifugation (23).
Steady-state Fluorescence Measurements
All steady-state fluorescence measurements were performed using a Perkin Elmer Luminescence Spectrometer LS50B at 15°C. IANBD fluorescence was excited at 490nm and monitored at ~530 nm (Both bandwidths set at ~8nm). Protein buffer solutions contained 20mM MOPS, 150mM KCl, 3mM MgCl2, 2mM EGTA, 1mM DTT (pH 7.0). The fluorescence signal of 2 ml IANBD-cTnCC35S or IANBD-cTnC35S (0.6 μM) was monitored with titration of microliter amounts of Ca2+ or cTnI in the presence (100 μM) or absence of Ca2+. The free Ca2+ concentration was calculated using the Maxchelator program (http://maxchelator.stanford.edu) (24). Ca2+ dependence of conformational changes (pCa value at half maximal fluorescence signal change) were obtained by fitting the binding curve with the sigmoid Hill equation as previously described (25). The reported values are the mean of three to five successive titrations. Data were presented as mean ± S.E.M. Statistical significance was determined by Student's t-test using SigmaPlot Software (Systat SoftwareInc.). p < 0.05 was considered as statistical significance.
Isothermal Titration Microcalorimetry
All experiments were performed using a Microcal, Inc isothermal titration microcalorimeter (ITC-200) in the Analytical Biopharmacy Core at the University of Washington. Experimental conditions were 30 °C, 20 mM MOPS, pH 7.0, 150mM KCl, 3 mM MgCl2, 2 mM EGTA, 1mM CaCl2. The sample cell was filled with 200 μl 3μM cTnI (with 1mM Ca2+) and titrated with 2μl per injection of 50–70μM cTnC (WT or L48Q, Ca2+ saturated). Control titration of cTnC (WT or L48Q) to buffer was performed for each independent experiment. Binding parameters were calculated by the Origin-ITC data analysis software package using single set of sites mode. All data is shown as a mean value ± S.E.M.
Sample Preparation and data analysis for NMR Spectroscopy
All NMR samples had starting volumes of 500 μL. The protein samples were dissolved in 100 mM KCl, 10 mM Imidazole, and 0.2–0.25 mM 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS) (Chenomx) with 0.01 % NaN3 in 95% H2O/5% D2O with 2–8 mM CaCl2 (Fluka). Concentrations of cNTnC(L48Q) varied from 0.5 mM for assignment experiments to 0.1 mM for cTnI147-163 titrations. The pH was kept between 6.7–7.0.All experiments were run on either a Varian Inova 500 MHz spectrometer or a Unity 600 MHz spectrometer. All data were collected at 30°C. All NMR data were processed with NMRPipe (26) and visualized with NMRViewJ (27). 15N-T1, 15N-T2, and NOE experiments were analyzed with the Rate Analysis and HetNOE modules in NMRViewJ. The analysis of titration data and dimerization data was done with xcrvfit (www.bionmr.ualberta.ca/bds/software/xcrvfit). Model-free analysis of the data was done using Mathematica notebooks (28). Models were chosen using Akaike's Information Criteria (AIC) (29), and Monte Carlo analysis was done on the chosen model to assess errors (30).
NMR Titrations
The titration of Ca2+ and cTnI147-163 into 15N-cNTnC(L48Q) was monitored by the 1H,15N-HSQC NMR experiment. At each titration point spectra were recorded, and the concentration dependent chemical shift perturbations were used to determine dissociation constants. For the Ca2+ titration, initially Ca2+-free buffer was prepared with Chelex 100 (Bio-Rad), which was used to chelate any free metal ions. The protein was run down a desalting column in the presence of EDTA. A 50 mM CaCl2 stock solution was prepared and titrated into the apo-cNTnC(L48Q) NMR sample to final concentrations of: 0.10, 0.15, 0.20, 0.25, 0.30, 0.35, 0.40, 0.45, 0.50, 0.60, 0.80, 1.41, 2.63 mM. A 3.8 mM stock of cTnI147-163 was prepared in DMSO-d6 (Cambridge Isotopes Inc.) and the concentration of cNTnC(L48Q) was determined to be 84 μM. cTnI147-163 was titrated into cNTnC(L48Q) to final concentrations of: 7, 14, 22, 29, 36, 43, 57, 71, 85, 99, 120, 140, 173, 205, and 269 μM. The pH was kept between 6.9 and 7.0 throughout the titration by adding 1M HCl or 1M NaOH.
NMR experiments for assignment
The 1H,15N-HSQC NMR spectra of 15N-labeled cNTnC(L48Q) were assigned for all three states of L48Q: Apo, Ca2+-bound, and cTnI147-163-bound. The 1H,15N-HSQC spectra for the different states of L48Q were assigned with the aid of the three-dimensional 15N-NOESYHSQC and 15N-TOCSYHSQC NMR experiments. The TOCSYHSQC experiment correlates intraresidue backbone amides with side-chain protons; and the NOESYHSQC experiment correlates backbone amide nuclei with nearby nuclei – either intraresidue or interresidue. Through the combination of these two NMR experiments, and previously published assignments for cNTnC (3, 31), we were able to completely assign the backbone amides of cNTnC(L48Q) (Figure 4).
FIGURE 4.
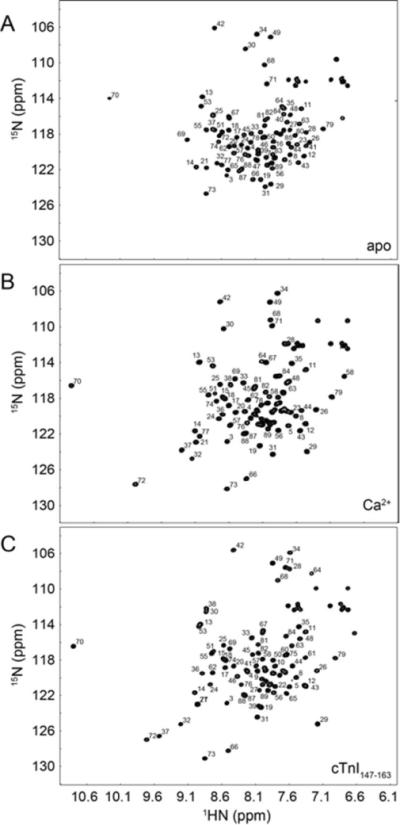
Assigned 1H,15N-HSQC spectra of A. Apo cNTnC(L48Q), B. cNTnC(L48Q)•Ca2+, and cNTnC (L48Q)•Ca2+•cTnI147-163. All well resolved backbone amide peaks are labeled (side chain NH2 were not assigned).
15N Backbone Relaxation experiments
To assess the concentration-dependent aggregation of cNTnC(L48Q)•Ca2+, T2 experiments were acquired at four protein concentrations: 0.15, 0.33, 0.7, and 1.2 mM. The T1 and NOE values were recorded with a cNTnC(L48Q)•Ca2+ concentration of 0.33 mM on the 500 and 600 MHz spectrometers; and with a concentration of 0.15 mM on the 500 MHz spectrometer. All experiments were recorded with the same experimental parameters: T1 values were determined using relaxation delays of 10, 50, 100, 200, 300, and 400 ms; T2 values were acquired using relaxation delays of 10, 30, 50, 70, 90, and 110 ms. Delays between transients for T1 and T2 experiments was set to 3s. The 1H-15N NOE experiments had a delay of 3s without the proton saturation and when proton saturation was on, it was set to 3s.
Molecular Dynamics Simulations
The starting structure of the N-terminus of cTnC (cNTnC, from residue 1 to 89) and cTnI147-163 complex was taken from model 18 of the NMR structure (PDB entry 1mxl) (31). The starting structure of cNTnC in the Ca2+ saturated form was from model 14 of the NMR structure (PDB entry 1ap4) (3). Model 13 of the NMR structure (PDB entry 1spy) (3) was used for the apo state cNTnC starting structure. The L48Q mutation was created in silico using UCSF Chimera (32) in all three structures. All-atom, explicit solvent MD simulations were performed at 15°C (to match solution measurements in this study and previous mechanical measurements in cardiac muscle and myofibrils (17)) in the microcanonical (NVE, constant number of particles, volume, and total energy) ensemble using the in lucem molecular mechanics (ilmm) program (33) and the Levitt et al. force field (34). Starting structures, minimized for 1000 steps of steepest descent minimization, were solvated in a rectangular box of flexible three-center (F3C) water (35) with walls located at least 10 Å from any protein atom. The solvent density of the box was adjusted to 0.999129 g/mL, the experimental density for the simulation temperature of 15°C (36). A 2 fs time step was used, and structures were saved every 1 ps for analysis. Multiple (n ≥ 3) simulations for the structures of cNTnC•Ca2+•cTnI147-163, cNTnC•Ca2+and cNTnC (both WT and L48Q), respectively, were performed of up to 70ns each. Analysis of MD trajectories was performed with ilmm (33). Contacts between residues were defined as having a distance between two carbon atoms of ≤ 5.4 Å or any other non-carbon atoms of ≤ 4.6 Å. Distances were measured between specific atom pairs or between the centers of mass of groups of atoms (e.g. two helices). Protein images were generated using UCSF Chimera (32). Interhelical angles were calculated using the program interhlx (K. Yap, University of Toronto). The solvent accessible surface area (SASA) was calculated using Lee and Richards (37) algorithm. For all WT and L48Q measurements the cNTnĊCa2+˙cTnI147-163 simulations, cTnI147-163 was removed from the MD trajectories so that the SASA of the hydrophobic patch residues in cNTnC could be then calculated. All error bars for results from the WT and L48Q MD simulation are based on the standard deviation in the average values of the multiple runs of simulations.
RESULTS
Ca2+ and cTnI titrations into cTnC(L48Q) and cTnC monitored by fluorescence spectroscopy
To assess the Ca2+ binding affinity of the L48Q variant, we attached the fluoroprobe IANBD at C84 of cTnC. IANBD is an environment-sensitive and sulfhydryl-reactive extrinsic fluorophore that has been widely used to label biological molecules for studies of intra molecule interactions (22, 38, 39). Fluorescence labeling at C84 reports on conformational changes in cNTnC due to both Ca2+ and cross-bridge binding (40). We found that Ca2+ caused a dose-dependent increase in fluorescence from the IANBD-labeled cTnC, suggesting Ca2+ promotes a conformational change in the cTnC that leads to a decrease in the polarity of the environment around IANBD-labeled cysteine.
We compared the Ca2+-dependent conformational changes of cTnC(L48Q) and cTnC . As shown in the inset graph of Figure 1, cTnC(L48Q) underwent ~1.33 fold maximal increase in IANBD fluorescence when saturated with Ca2+ versus a ~1.25 fold increase for cTnC. The enhanced magnitude of total fluorescence change for cTnC(L48Q) implies a larger structural change of the regulatory domain of cTnC(L48Q) upon Ca2+ binding. We next added cTnC(L48Q) or cTnC to wild type cTnI and cTnT to form whole cTn complexes. Consistent with a previous report using recombinant human cTnC variants, the L48Q variant had an enhanced Ca2+ binding affinity (15). Ca2+ sensitivity of the fluorescence signal (reported as pCa at half-fluorescence increase) was shifted +0.32 pCa units, from 6.99±0.03 (cTn) to 7.31±0.03 (cTn(L48Q)) (curves in Figure 1). This matches well the 0.38 pCa unit increase in Ca2+ sensitivity of contraction we recently reported upon exchanging cTn(L48Q)C35S into skinned rat trabeculae (17). It is also consistent with the work of Tikunova and Liu (14), who found a 0.39 pCa units leftward shift of pCa at half-maximal decrease in IAANS fluorescence for L48Q cTn (from ~pCa 6.20 for control protein to ~pCa 6.59 for L48Q). The decreased pCa value in their results compared with ours likely results from differences in the fluoroprobe (IAANS vs IANBD), labeling strategy (at T53C of TnC C35S, T53C, C84S vs. at C84 of TnCC35S) and species (human cTnC vs. rat cTnC) exchanged into rat cardiac muscle.
FIGURE 1.

Effects of L48Q on the Ca2+ dependent changes in the fluorescence of complexes. (◯) Ca2+binding to L48Q ; (•) Ca2+ binding to . Inset graph, effects of L48Q on the total magnitude IANBD fluorescence increase. Excitation was at 490 nm and the emission was monitored at 530 nm. The error bars represent the standard error of 3–5 experiments.p<0.05
Interactions between cTnC and cTnI play a critical role in transferring the Ca2+-signal to other myofilament proteins to initiate cardiac muscle contraction. Thus, in addition to examining Ca2+ affinity, we tested whether the L48Q variant also altered cTnI binding affinity. Binding of cTnI to cTnC was measured by titrating labeled cTnC with cTnI in the presence or absence of Ca2+. Figure 2 shows the magnitude of IANBD fluorescence change as the cTnI concentration is increased up to 0.8 μM in solutions containing 0.6 μM cTnC(L48Q) or cTnC (control). The data demonstrate no further change in fluorescence signal beyond 0.6 μM cTnI for either cTnC, suggesting 1:1 binding of cTnC:cTnI was achieved. For both cTnC(L48Q) and control the magnitude of maximal IANBD fluorescence change was greater for the Ca2+ saturated states than the apo state, indicating a larger conformational change. The amplitude of the fluorescence signal change is an indicator of the magnitude of conformational change. This magnitude increase was compared for cTnC(L48Q) versus control in the apo and Ca2+ saturated states. Maximal fluorescence increase did not differ for the apo state, but was significantly increased for cTnC(L48Q) (3.73 ± 0.18 fold) compared with control (3.29 ± 0.11 fold), suggesting the regulatory domain of cTnC(L48Q) is more open when bound to cTnI. cTnI appeared to bind to cTnC(L48Q) •3Ca2+ more tightly than to cTnC •3Ca2+, with dissociation constants of 174 ± 8 nM and 198 ± 5 nM (derived from fitting the curves of normalized IANBD fluorescence change vs cTnI concentration with sigmoid Hill equation), respectively, but these values did not differ statistically.
FIGURE 2.
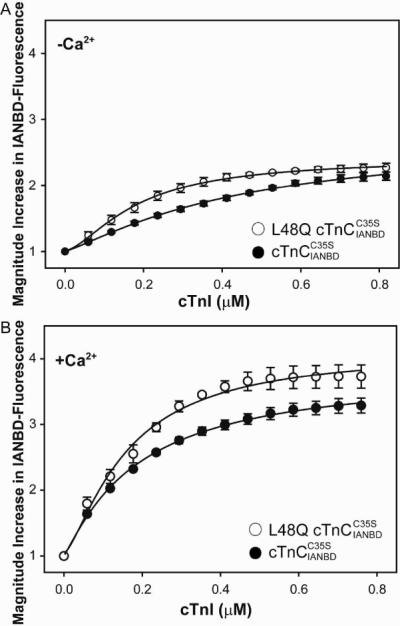
Effect of L48Q on the binding of cTnI to . The binding was determined by measuring the changes in IANBD fluorescence emission intensity of titrating with cTnI in A. the absence of Ca2+ (p<0.05) and B. the presence of Ca2+: (◯) L48Q ; (•) . Excitation was at 490nm and the emission was monitored at 530nm. The error bars represent the standard error of 3–5 experiments.
Binding of cTnI to cTnC bound to calcium by ITC
Isothermal titration calorimetry (ITC) was used to obtain a more comprehensive picture of cTnI binding to cTnC. ITC permits monitoring of protein-protein interactions without the need for chemical modifications that may modify the interaction surface. Representative ITC data from the titration of cTnI with Ca2+ saturated cTnC(L48Q) are shown in Figure 3. For each titration point, the quantity of heat released (as indicated by the negative deflection) is directly proportional to the amount of binding between the two proteins. The complete binding isotherm was obtained by plotting the integrated heat against the molar ratio of cTnC added to cTnI in the reaction cell. The stoichiometry (n), dissociation constant (KD) and enthalpy (ΔH) of binding were obtained by fitting these data using the Origin-ITC package. The results from a minimum of three independent ITC binding experiments for cTnC or cTnC(L48Q) binding to cTnI, in the presence of Ca2+, suggested the binding stoichiometry was approximately 1:1 for both WT and cTnC(L48Q). Consistent with the fluorescence data, the affinity of cTnC (L48Q) for cTnI was higher than cTnC, KD 132 ± 59 nM for cTnC(L48Q) and KD 159 ± 91 nM for cTnC. Furthermore, the total heat released upon binding to cTnI (ΔH) for cTnC(L48Q) was −22.1 ± 1.5 kJ̇mol−1, and −16.1 ± 3.9 kJ̇mol−1 for cTnC.
FIGURE 3.
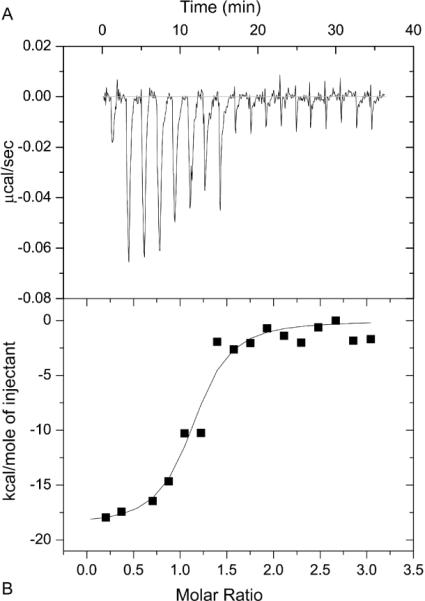
Microcalorimetric titration of cTnI with cTnC(L48Q) in the presence of Ca2+. A, an example trace of the titration of 3 μM cTnI with 50 μM – 70 μM cTnC(L48Q) at 30 °C. B, Integrated heats for each injection obtained from the raw data in panel A versus the molar ratio of cTnC(L48Q) to cTnI. The data were fit to the data using a 1:1 binding model, the fit is shown by the solid line.
Ca2+ and cTnI147-163 titrations into cNTnC(L48Q) by NMR spectroscopy
To investigate the structural significance of L48Q on the N-lobe regulatory domain of cTnC, we used 15N-labeled cNTnC(L48Q) for NMR experiments. The 1H,15N-HSQC NMR spectrum of 15N-labeled cNTnC(L48Q) was assigned for apo, Ca2+ saturated and Ca2+ + cTnI147-163 bound states using the three-dimensional 15N-NOESYHSQC and 15N-TOCSYHSQC NMR experiments (Figure 4). NMR was used to measure the affinity of cNTnC(L48Q) for Ca2+ and the affinity of Ca2+ saturated cNTnC(L48Q) for cTnI147-163 (Figures 5A,B). Ca2+ was titrated into a sample containing apo cNTnC(L48Q) and 1H,15N-HSQC spectra were acquired at each point. The concentration-dependent chemical shift perturbations of five well-resolved resonances were used to determine the dissociation constant of Ca2+, based on the global fitting method developed by Hoffman and Sykes (41). The protocol determined a global dissociation constant that fit all the data with a minimum sum of squared error (SSE). Ca2+ bound to cNTnC(L48Q) with a dissociation constant of 0.6 μM (SSE=0.055) (Figures 5C,D), ~2-fold lower than the dissociation constant of 2.6 ± 1 μM for cNTnC(wt) (42). This result is consistent with our findings in intact troponin and with those of Tikunova and Davis (2004) for cTnC(L48Q) , and it supports the use of the isolated domain to ascertain the effects of the L48Q mutation.
FIGURE 5.

1H,15N-HSQC spectra of A. the Ca2+ titration into Apo-cNTnC(L48Q) and B. the cTnI147-163 titration into cNTnC(L48Q)•Ca2+. C. Binding curves of the Ca2+ titration into cNTnC(L48Q) that were used in the global fit to determine the dissociation constant and D. the corresponding sum of squared error (SSE) for the global fit. E. Representative binding curves of the cTnI147-163 titration into cNTnC(L48Q)•Ca2+ that were used in the global fit to determine the dissociation constant and F. the corresponding SSE for the global fit.
We next measured the affinity of the cTnI147-163 switch peptide for cNTnC(L48Q)•Ca2+, which is the sequence of cTnI responsible for triggering contraction through specific binding to cNTnC•Ca2+ (43). The global fit yielded a cTnI147-163 dissociation constant of 61 μM (SSE=0.2) (Figures 5E,F), which is approximately 2× tighter than the dissociation constant of 150 ± 10 μM measured for cNTnC by NMR(31). The significantly weaker interaction between the fragment of cTnI (cTnI147-163) and the isolated N-terminal domain of cTnC than what was measured by ITC and fluorescence for the full-length constructs is expected since full-length cTnI binds to both domains of cTnC with the interaction between cTnI34-71 and cCTnC being in the nanomolar range (44).
Amide chemical shift comparison of cNTnC(L48Q)•Ca2+ with other troponin states
The apo state of cNTnC is in a closed conformation, with its hydrophobic residues buried in the core of the protein. The NMR structure of cNTnC•Ca2+ indicates that a minor opening occurs when Ca2+-binds to site II (3). However, cTnI147-163 binding is required to achieve the open state of cNTnC (Figure 6). The conformation of cNTnC is described by the A/B and C/D interhelical angles (90° means the helices are orthogonal and thus the protein is open; and an angle near 180° indicates a more closed conformation – Figure 6). Although it is convenient to discuss the conformation of cNTnC in terms of angles, it must be stressed that an NMR structure is a static representation of cNTnC. cNTnC•Ca2+ appears to be in a dynamic equilibrium, fluxing between closed and open states (45–48).
FIGURE 6.
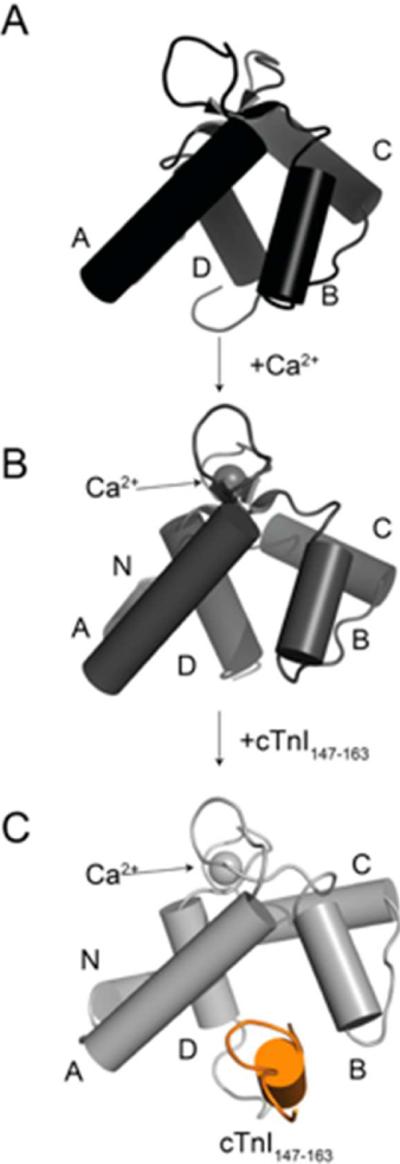
Conformational change of cNTnC from A. closed in the apo state with an interhelical angle of helices A and B of ~140°. (pdb: 1spy) through B. slightly open in the Ca2+-bound state with an interhelical angle of helices A and B of ~130° (pdb: 1ap4) to C. fully open when cTnI147-163 is bound (pdb: 1mxl) with an interhelical angle of helices A and B of ~102°. cNTnC is depicted in cartoon mode with the helices shown in cylinder representation and are labeled (the N helix is pointing directly into the page in panel A. and is therefore not visible).
Chemical shift differences between nuclei are caused by a change in the local magnetic environment, such as by local structure or solvent exposure. So, in addition to their applicability in discerning the affinity and stoichiometry of Ca2+ or cTnI binding, chemical shifts may be able to provide insight into the conformation of cNTnC. The Ca2+-sensitizing agent, bepridil, stabilizes the open form of cNTnC in a similar manner as cTnI147-163 (PDB:1DTL), with an A/B interhelical angle of ~92° (49). Although they are structurally distinct molecules (bepridil is a small hydrophobic molecule and cTnI147-163 is an amphipathic peptide), they induce similar amide chemical shift perturbations in the 1H,15N-HSQC spectrum of cNTnC, most likely because they both stabilize the open state of cNTnC•Ca2+. We overlaid the 1H,15N-HSQC spectra of cNTnC•Ca2+, cNTnC•Ca2+•cTnI147-163, cNTnC•Ca2+•bepridil, and cNTnC(L48Q)•Ca2+ to estimate the conformation of the L48Q variant (Figure 7A). The chemical shifts of cNTnC(L48Q)•Ca2+ were intermediate between those for cNTnCs•Ca2+ and the bepridil or cTnI147-163 cNTnC•Ca2+ complexes. This result suggests the L48Q mutation moves the conformational equilibrium of cNTnC•Ca2+ towards the open state; however not as much as bepridil or cTnI147-163. We recently developed a program, ORBplus, to analyze the relationship between the interhelical angles of cNTnC and amide chemical shifts (50). ORBplus predicted that the A/B interhelical angle of cNTnC(L48Q)•Ca2+ is approximately 10° more open than cNTnC•Ca2+ (~120° versus ~130°) (50).
FIGURE 7.
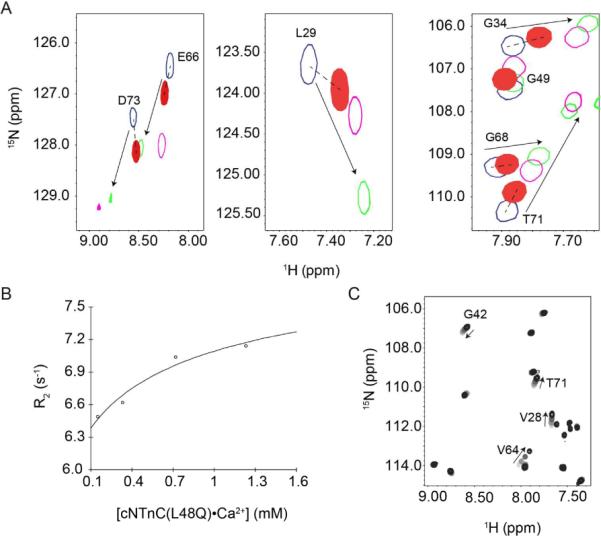
Characterization of the conformation of cNTnC(L48Q)•Ca2+ by NMR. (A) Comparison of 1H,15N-HSQC spectra from cNTnC(L48Q)•Ca2+ (filled red) with cNTnC•Ca2+ (blue), cNTnC•Ca2+•cTnI147-163 (green) and cNTnC•Ca2+•bepridil (magenta). The resonances of cNTnC(L48Q)•Ca2+ are shifted intermediately between cNTnC(wt)•Ca2+ and the cTnI147-163 or bepridil bound forms of cNTnC. Dependence of the average 15N-T2 (B) and 1H,15N-HSQC (C) as a function of cNTnC(L48Q)•Ca2+ concentration. In the 1H,15N-HSQC spectrum, contours are colored from light grey to black as cNTnC(L48Q)•Ca2+ concentration increases. Several representative residues are labeled and arrows indicate the direction of chemical shift as concentration was increased.
15N-T2 and amide chemical shift as a function of concentration
The Ca2+-triggered conformational change of cNTnC is likely less dramatic than sNTnC because only one Ca2+ ion binds to cNTnC, whereas two Ca2+ ions bind to the skeletal isoform. The biological role of the exposed hydrophobic surface, in either isoform, is to promote binding of the switch region of troponin I. The exposed hydrophobic surface also serves as an interface for in vitro dimerization (in the absence of troponin I). The NMR relaxation rate R2 (1/T2) is proportional to the rotational correlation time of a protein and, hence, to its size. It has been shown that the apparent sizes of cNTnC•Ca2+ or sNTnC•2Ca2+ increase as a function of concentration (51). The dissociation constant for dimer formation (Kdimer=[monomer]2/[dimer]) was previously determined to be ~7.3 mM for cNTnC(wt)•Ca2+ and ~1.3 mM for sNTnC•2Ca2+ (51). To determine the dimerization constant of cNTnC(L48Q)•Ca2+ we measured the T2 relaxation time of L48Q at four different concentrations (1.2, 0.72, 0.33, and 0.14 mM) and plotted R2 as a function of protein concentration (52) (Figure 7B). The dimerization constant of cNTnC(L48Q)•Ca2+ was ~2 mM with an R2 (monomer) of 6.2 s−1 and a R2 (dimer) of 8.4 s−1 (Figure 7B). The decreased Kdimer of cNTnC(L48Q)•Ca2+ when compared to cNTnC•Ca2+ suggests that cNTnC(L48Q)•Ca2+ is more open than cNTnC•Ca2+, but not as open as sNTnC•2Ca2+. The concentration dependence is also shown in the amide chemical shifts at the same four concentrations (Figure 7C).
Backbone 15N relaxation data
NMR relaxation data is a valuable source of experimental evidence for protein dynamics. The 15N backbone NMR relaxation parameters T1, T2, and nuclear Overhauser effect (NOE) depend on the tumbling of the protein as a whole, as well as on internal motions within the protein. The measured relaxation data for cNTnC(L48Q)•Ca2+ at 0.33 mM are shown on a per residue basis in Figure S1 (supporting information). The results indicate that cNTnC(L48Q)•Ca2+ is a highly structured and rigid protein. The dramatic increase in T1 and T2 as well as a drop in NOE at the C-terminus of cNTnC(L48Q)•Ca2+, is consistent with disorder. The average T1500 (superscript indicates the magnetic field the relaxation data were acquired at, expressed as the frequency of 1H in MHz) for all residues was 408 ± 87 ms with an average error of 15 ms, and the average T1600 was 466 ± 62 ms with an average error of 27 ms. The average T2500 for all residues was 151 ± 102 ms with an average error of 8 ms, and the average T2600 was 142 ± 69 ms with an average error of 7 ms. The average NOE500 was 0.60 ± 0.52 (average error of 0.023) and the average NOE600 was 0.70 ± 0.31 (average error of 0.011).
The relaxation parameters for 0.15 mM cNTnC•Ca2+ have been previously published (53). The average T1500 was 440 ± 106 ms, with an average error of 12 ms; the average T2500 was 166 ± 82 ms, with an average error of 5 ms. The average NOE500 was 0.57 ± 0.27 with an average error of 0.04. To directly compare cNTnC(L48Q)•Ca2+ with cNTnC•Ca2+ we prepared a 0.15 mM sample and recorded the relaxation data at 500 MHz. The average T1500 was 408 ± 97 ms with an average error of 20 ms, the average T2500 was 154 ± 80 ms with an average error of 9 ms, and the average NOE was 0.70 ± 0.44 ms with an average error of 0.11 ms. We have superimposed the data from cNTnC•Ca2+ with the L48Q variant in Figure S2 (supporting information).
While most of the relaxation data between cNTnC•Ca2+ and cNTnC(L48Q)•Ca2+ do not differ, inspection of residues in site I suggests that the L48Q mutation modulates the dynamics of this loop (residues 29–40). We used a suite of Mathematica notebooks to perform model-free analysis of the 15N relaxation data to determine the order parameters for residues in site I (28). Models for each residue were chosen according to Akaike's information criteria (AIC) (29). The average S2 obtained for loop 1 is 0.85 ± 0.11 for cNTnC(L48Q)•Ca2+, and 0.77 ± 0.08 for cNTnC•Ca2+ implying, if anything, that the NH bond vectors in the loop are more rigid on the ps-ns timescale in cNTnC(L48Q)•Ca2+.
Effect of L48Q substitution on cNTnC-cTnI147-163 interaction and position of the helix B of cNTnC
We used MD simulations to further investigate structural changes caused by the L48Q variant that may explain the experimentally observed increase in cTnI binding to cTnC. Simulations were performed for both cNTnC(WT)•Ca2+ and cNTnC(L48Q)•Ca2+ complexed with cTnI147-163 (1MXL) at neutral pH and 15°C and sampled over 70ns for multiple simulations (≥5). We first investigated the residue contacts between cNTnC and cTnI147-163. L48 is located at the end of helix B of cNTnC and makes multiple contacts with cTnI147-163 peptide (Figure 8A). . Residues from cNTnC and cTnI147-163 with more than 20% time in contact over entire simulations were chosen and counted as contact pairs between the two proteins. We plotted the number of contact pairs between cNTnC and cTnI147-163 for each residue position of cNTnC to determine the overall contact map for cNTnC interaction with cTnI147-163. Results are shown for WT in Figure 8B and L48Q in Figure 8C. The general pattern is similar for both cNTnC structures. The data show that residue 48 contacts the greatest number of residues in cTnI147-163 (Figure 8B) for both proteins, suggesting this position of helix B in cNTnC is important for cNTnC interaction with the switch region of cTnI. However, some cNTnC residues have increased or decreased number of contact pairs for L48Q compared to WT, which may significantly alter the interactions between cNTnC and cTnI switch peptide. While this merits further investigation, detailed analysis is beyond the scope of this study.
FIGURE 8.

Interactions between cNTnC and cTnI147-163. A. Structure of cNTnC(L48Q)•Ca2+•cTnI147-163 (modified from pdb: 1mxl) with residue Q48 shown in sticks, cTnI147-163 in white, helix A in cyan and helix B in green. B and C. Number of cTnI147-163 residues that contact cNTnC (number of contact pairs) for WT and L48Q. The error bars represent the standard error from 3–5 runs of simulations.
Li et al. determined that a structural deviation in WT cNTnĊCa2+ associated with binding of the cTnI147-163 peptide is the movement of the helix B away from the helix A(31). The simulations of the cNTnC(L48Q)•Ca2+•cTnI147-163 complex demonstrate a significant helix B movement for L48Q, as shown by the snapshots for simulations at 0 and 60ns in Figure 9A. (The MD movies are available in the supporting information.) This helix B movement occurred in all of the cNTnC(L48Q)•Ca2+•cTnI147-163 simulations but it was not observed for any of the 5 WT cNTnC•Ca2+•cTnI147-163 simulations. To investigate further, we analyzed the distances and interhelical angles between helices A and B. The distances between the centers of mass of helices A and B (COMdist AB), averaged over the last 25 ns of multiple simulations, were 17.7 ± 0.7 Å for L48Q and 16.5 ± 0.7 Å for WT. The differences in COMdist AB (ΔCOMdist AB) at the end (from 45–70ns) compared with the beginning of the simulations (10–25ns) show that helices A and B moved ~0.4 Å further apart from each other for L48Q compared to WT, but the change is subtle, particularly given the dynamic nature of these helices.
FIGURE 9.

Effects of L48Q on the mobility of helix B in cNTnC. A. Snapshots from cNTnC(L48Q)°Ca2+°cTnI147-163 and cNTnC°Ca2+°cTnI147-163 simulations at 0ns(white) and 60ns(rainbow). B. Comparisons of 0ns (white) vs. 60ns (rainbow) Ca2+ saturated structures for cNTnC(L48Q)°Ca2+ (upper) and cNTnC°Ca2+. C. Comparisons of 0ns (white) vs. 60ns (rainbow) apo state structures for cNTnC(L48Q) (upper)and cNTnC. Residues L48 and Q48 are shown as sticks. D. Helices A and B truncated from structures at 0 ns (white) and 60 ns (A helix: blue and B helix: green) for NTnC(L48Q)°Ca2+°cTnI147-163 (left),cNTnC(L48Q) °Ca2+ (middle), and cNTnC°Ca2+ (right). The helices are shown rotated 90° right from the structures in panels A–C to better view the A/B helix angle. E. Cα RMSD for helices A and B of L48Q (white) and WT (black) averaged over all simulations of cNTnC°Ca2+°cTnI147-163, cNTnC°Ca2+ and cNTnC apo. F. Surface rendering of cNTnC°Ca2+ for structures from the WT (0 ns, upper; and 60 ns,lower left) and L48Q (60 ns, lower right) simulations. The hydrophobic residues (F20, A23, F24, I26, F27, I36, L41, V44, L48, L57, M60, F77, M80, M81) are colored red with the rest of the protein in white.
To further validate the conformational change in cNTnC by the L48Q mutation, we performed MD simulations of apo and Ca2+-saturated cNTnC structures (PDB entries: 1SPY and 1AP4, respectively) for both WT and L48Q. In Figures 9B and C, the starting structures are compared with the structures at 60 ns for cNTnC(L48Q) and cNTnC. For both apo and Ca2+-saturated simulations, the helix B of cNTnC(L48Q) underwent a large movement away from the core of the domain. For WT cNTnC, no movement of the helix was observed. Figure 9D compares for helices A and B only, for all simulation structures at 60 ns versus the starting structures (90° counter clockwise rotation from y-axis of the structure shown in panels A, B and C). From this view, it is clear that the helices A and B are generally more open for cNTnC(L48Q). This movement of the helix B was not dependent on the binding of Ca2+ or cTnI147-163 in the Ca2+ saturated state. Generally, cNTnC(L48Q) had greater values of Cα-RMSD of helices A and B relative to the original NMR structure from all states populated in the simulations (Figure 9E), illustrating the dynamic character and increased mobility of these helices. The difference in the motion of the helix B between cNTnC(L48Q) and cNTnC is presumably due to Gln 48 at the end of the helix B in the variant, which disrupts key contacts L48 makes with residues on the helix A, such as F20, A23, and F27 (Figure S3, supporting information).
The interhelical angle between helices A and B (Figure 6 and Figure 9D) was used to quantify the degree of opening conformation in cNTnC. The A/B interhelical angles averaged from multiple simulations were ~87° for L48Q and ~107° for WT, suggesting a more open conformation for L48Q cNTnC in the Ca2+ and cTnI147-163 bound state. Similar results were found for both apo and Ca2+ saturated simulations, helices A/B were generally more open for L48Q than WT structures. In particular, our simulations suggest there is a decrease of about 10° in the AB interhelical angle of L48Q compared to WT in the Ca2+ saturated state. This magnitude of change is consistent with the prediction from ORBplus that the A/B interhelical angle of cNTnC(L48Q)•Ca2+ is ~10° more open than cNTnC•Ca2+ (50).
The distance between the backbone α-carbons of M81 (on helix D) and N50 (B-C loop) was used to quantify the opening of cNTnC (Figure S4, supporting information), as was previously done to monitor the opening of cNTnC when cTnI147-163 bound or when Ca2+ bound to sNTnC (31). M81-D50 distances were generally larger in the cNTnC(L48Q) compared to cNTnC, as calculated from all state simulations (apo, Ca2+saturated and cTnI147-163 bound) (Table S1,supporting information). The increase in distance between M81-D50 of L48Q is consistent with the results from the interhelical angle and the distances between helices A and B, which all indicate that the L48Q variant induced a more open state of cNTnC.
To quantify the exposure of the hydrophobic surface in cNTnC that associates with the binding with the switch region of cTnI, we selected hydrophobic residues F20, A23, F24, I26, F27, I36, L41, V44, L48, L57, M60, F77, M80, M81 as hydrophobic patch residues. In simulations these cNTnC residues made contacts for more than 40% of time with the switch region of cTnI in cNTnC•Ca2+•cTnI147-163 (both WT and L48Q). The total SASA of the hydrophobic patch residues was greater in cNTnĊCa2+˙cTnI147-163 simulations than in cNTnC.Ca2+ or Apo cNTnC in both WT and L48Q simulations (Table S2, supporting information), indicating the hydrophobic patch was more exposed in the presence of cTnI147-163. This is consistent with an NMR and X-ray studies showing that binding of the switch region of cTnI induces opening of cNTnC (31, 54). In addition, L48Q had greater SASA of the hydrophobic patch area in all structures from the simulations compared to WT (Table S2, supporting information), again showing the larger hydrophobic surface area in cNTnC(L48Q). Figure 9F shows the surface rendering of structures from cNTnC•Ca2+ (0 ns and 60 ns) and cNTnC(L48Q)•Ca2+ (60 ns) simulations. The selected hydrophobic patch residues are colored red in all structures with the rest of the protein colored in white. Overall, the increased mobility of helix B facilitated the exposure of the hydrophobic patch in cNTnC(L48Q) and disrupted the closed structure of cNTnC, which supports the idea of an increase in the binding affinity of the switch region of cTnI to cNTnC.
Effect of L48Q substitution on the Ca2+ coordinating residues at Ca2+ binding site II
cTnC is one member of the EF-hand superfamily of calcium binding proteins. The EF-hand is a highly conserved (both by sequence and structure) helix-loop-helix motif with Ca2+ binding loop containing 12 amino acids (55, 56). Among the 12 residues in the second Ca2+ binding loop (site II) of cNTnC, residues D65, D67, S69, D73, T71, and E76 at positions 1, 3, 5, 7, 9, 12 are involved in coordination to the calcium (57, 58). Since the interaction of these residues with each other and the Ca2+-binding pocket itself are critical regulators of the contractile process, we also analyzed the MD simulation trajectories of cNTnC(L48Q)•Ca2+•cTnI147-163 and cNTnC•Ca2+•cTnI147-163 to examine how the L48Q substitution influences Ca2+ binding at cNTnC site II. The distances between the center of mass of Ca2+ and the Ca2+-coordinating residues were calculated and averaged over time to evaluate the overall stability of the Ca2+ binding site (Figures 10 A, B). Variability was not significant at any of the Ca2+ coordinating residues, with the exception of S69, in the WT simulations. The L48Q simulations showed no significant perturbation at any residues, indicative of a very stable Ca2+ binding site.
FIGURE 10.

Ca2+ binding pocket at site II. A and B. Average distances between Ca2+ coordinating residues over 10 ns windows. C. Distances between Ca2+ and Ca2+ coordinating residues: D67, S69 and E76. D. Snapshots from L48Q (run1) and WT (run1) simulations showing the Ca2+ binding site compared with the starting structure (top), WT t=60 ns (middle), and L48Q t=60 ns (bottom) with Ca2+ coordinating residues shown in sticks and atoms from panel C shown in spheres.
Detailed coordinating information was obtained by investigating the changes in the distances between center of mass of Ca2+ and individual Ca2+-binding atoms, shown by examples in Figure 10C. Interestingly, in the 60 ns simulation snapshot of the WT cNTnC•Ca2+•cTnI147-163complex, D67-OD1, E76-OE2 and S69-OG (atoms shown as red spheres in Figure 13D) pointed away from the Ca2+ ion. However, for the cNTnC(L48Q)•Ca2+•cTnI147-163 complex, only D65-OD2 pointed away from the Ca2+ ion. Figure 10C summarizes the average distances between these atoms and Ca2+ throughout the simulations. When compared to the average distances between Ca2+ and these atoms from the original 40 structures in the 1MXL NMR ensemble and those in model 18 from 1MXL (data not shown), D67OD1 and E76OE2 were closer to Ca2+ for the L48Q variant than WT, suggesting possible improved interactions between Ca2+ and these coordinating residues in L48Q. This change within the calcium binding site in the L48Q variant observed by MD simulations is consistent with our observed experimental measurements of increased Ca2+ binding affinity for the L48Q cTnC variant (Figure 1).
DISCUSSION
The combination of structural and functional information has been valuable for understanding the biochemical and biophysical mechanisms of proteins. Here we have used a highly integrative approach by combining fluorescence spectroscopy, micro-calorimetry, NMR methodologies and molecular dynamics simulations to determine the molecular consequences of an L48Q mutation on the conformation and dynamics of cTnC, as well as its interaction with cTnI upon Ca2+ binding.
It is important to elucidate the effects of cTnC modifications on its interactions with cTnI that may contribute to the change in cooperative myofilament activation. As such, knowledge of how the L48Q cTnC variant influences the cTnC-cTnI interaction will help in understanding molecular mechanisms of how altered cTn protein-protein interaction lead to changes in Ca2+ sensitivity of myofilament contraction (17). Our steady state fluorescence spectroscopy results indicate that interactions with cTnI, L48Q cTnC underwent greater conformational changes than the control indicated by the increased fluorescence signal in both the presence and absence of Ca2+. This increase in the L48Q cTnC-cTnI interaction suggests a strengthening of the Ca2+ signaling pathway between cTn subunits and tropomyosin (and actin) such that at any given subsaturating Ca2+ concentration, more myosin binding sites on thin filaments are available. This (for L48QcTnC) is demonstrated in myofibrils and demembranated cardiac muscle as a faster rate of myosin binding and force development at all sub-maximal (but not maximal) Ca2+ activations (17). Greater myosin binding is also demonstrated by a prolonged slow phase of relaxation in cardiac myofibrils (17), which is thought to reflect the rate of decay of the attached myosin population during relaxation (59). Finally, in terms of cooperative mechanisms of contractile force production, we have demonstrated that myosin binding to actin plays a larger role in cardiac vs. skeletal muscle thin filament activation (17, 60–62) and that the apparent cooperativity of activation (the slope, nH, of the force-pCa curve) is reduced by L48Q cTnC (17). The greater conformational change of L48Q cTnC upon binding Ca2+ and cTnI binding might lead to the exposure of the hydrophobic patch at lower concentrations of Ca2+ than for native cTnC. This may result in a lower dependence on allosteric activation of thin filaments by myosin in myofilaments containing L48Q cTnC.
To gain an understanding of the local structural perturbations caused by L48Q, we focused on the N-terminal domain of cTnC (cNTnC) and its interaction with Ca2+ and cTnI147-163. There were several reasons why we decided to shift to the truncated forms of cTnC and cTnI. The first reason is that as protein size increases it is more difficult to obtaining high-quality, high-resolution NMR data. More importantly, there are no significant differences in the structure of the N-domain of cTnC between cNTnC and cNTnC•Ca2+ with cTnC and cTnC•3Ca2+ (1) (3), respectively. Furthermore, the cTnC and cTnI fragments in the NMR structure of cNTnC•Ca2+•cTnI147-163 adopt the same conformation as in the X-ray structure of the core troponin complex (cTnC•3Ca2+•cTnI31-210•cTnT183-288) (31, 54). However, we cannot rule out the possibility that in addition to its local effect on the structure of cNTnC, L48Q may also have long-range effects on the structure or function of the C-terminal domain of cTnC. To explain the enhanced affinity of cTnI147-163 for cNTnC(L48Q) observed by NMR spectroscopy, we compared the amide chemical shifts of D73, E66, L29, G34, G68, and T71 from cNTnC(L48Q)•Ca2+ with the shifts of the closed state, cNTnC•Ca2+ and two open states, cNTnC•Ca2+•cTnI147-163 and cNTnC•Ca2+•bepridil. The average of chemical shifts suggest that cNTnC(L48Q)•Ca2+ is in a conformation somewhere between the closed and open states, closer to the closed state. Furthermore, cNTnC(L48Q) was found to dimerize more readily than cNTnC•Ca2+ but less than sNTnC•2Ca2+, which also supports the notion that L48Q stabilizes a slightly more open state of cNTnC. The detailed information about the interaction between cTnI147-163 region and cNTnC will help to unambiguously trace the overall interaction between cTnC-cTnI.
The MD simulations demonstrate how the L48Q mutation causes the opening of cNTnC, with the helix B swinging away from the hydrophobic core of cNTnC and remaining in that position till throughout multiple 70 ns simulations. Most previous studies have followd simulations for only a short timespan (0–10ns). Recently, the McCammon group published a study (48) in which molecular dynamics was performed over 100 ns to assess the stability of the helices A and B, and the dynamics of wild-type cNTnC. Our work builds on this and extends it to study the dynamic behavior of cNTnC (L48Q) in apo, Ca2+ saturated and cTnI147-163 bound states in multiple long (70ns) MD simulations. . Our MD simulations predicted a decrease of approximately 10° in the AB interhelical angle of cNTnC(L48Q)•Ca2+. This magnitude of change is consistent with a recent study that used couple of different computational methods in combination with NMR data to predict that the AB angle of cNTnC(L48Q)•Ca2+ would go from ~130 to ~120° (50). This structural impact is most likely the result of changing the hydrophobic Leu to the hydrophilic Gln at position 48 at the end of the B-helix. L48 makes key hydrophobic contacts with the side-chains of residues on the A-helix, such as F20, A23, and F27 (Figure S3, supporting information). Overall, the results presented herein suggest the L48Q mutation modulates Ca2+ sensitivity of cTnC and myofilament contraction by disrupting the structure of cNTnC, destabilizing the closed conformation of cNTnC. This decreases the energetic barrier of opening, enhancing both Ca2+ and cTnI binding (Figure 11).
FIGURE 11.
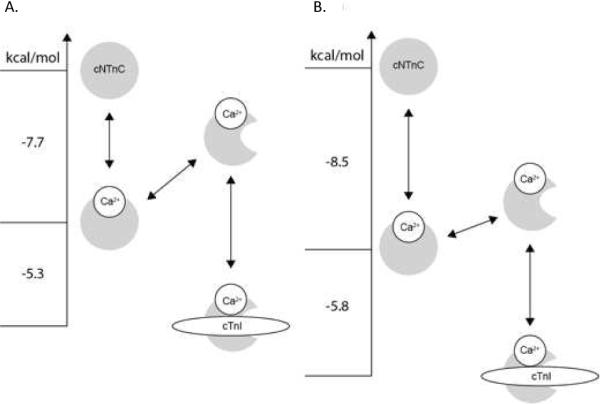
Energy level diagram highlighting the Ca2+-sensitizing mechanism of the L48Q mutation of cTnC. A. In apo cNTnC, cNTnC is closed, once Ca2+-binds to cNTnC, cNTnC oscillates between open and closed states, but the domain remains in predominantly the closed state. cTnI147-163 binds to the open state of cNTnC to stabilize its open conformation (Figure is adapted from (47, 64, 65)). B. The L48Q mutation shifts the closed-to-open equilibrium towards the open state enhancing both the Ca2+ and cTnI147-163 affinities. The free energy for A. is from McKay et al. (47), and for B. the NMR data in this work are used to calculate the free energy of binding. The relationship: ΔG°=−RTlnKD was used to calculate the free energies.
In terms of the Ca2+-sensitivity of cTnC, our solution spectrofluorimetry measurements demonstrate that the Ca2+ binding affinity (Ka) is increased by the L48Q variant for both cTnC in isolation and the cTn complex, as previously reported (15). This increase is due primarily to a slower rate of Ca2+ dissociation (koff), as measured for isolated cTnC, whole cTn and reconstituted thin filaments (15, 17). S69 was the least stable Ca2+ coordinating residue in the WT cNTnC MD simulations. In contrast, D67 and E76 (in addition to S69) in site II were more tightly coordinated with Ca2+ in the L48Q variant relative to WT cNTnC (Figures 10C, D). Similar results have been reported for MD simulations of other EF-hand Ca2+ binding proteins, parvalbumin and its variants (63), although the simulations were too short (300 ps) to make confident conclusions. L48 is located at the end of helix B (Figure 8A), and is too distant to directly affect Ca2+ binding at site II. Instead L48Q causes local changes that are propagated to the calcium site. Interestingly, the Cα RMSD of helices C and D calculated from all MD simulations are 1.59 ± 0.06 Å for L48Q (6 runs) and 1.88 ± 0.07 Å for WT (5 runs), showing that these helices are more stable in L48Q. The exposure of the hydrophobic surface requires the helices B and C to move away from the helix N, A, and D bundle, and L48Q catalyzes the lifting movement of helix B (Movie 1&2 Supporting Information).
Although many physiological and animal model studies have revealed the functional changes between native and disease-associated cTnC mutations that may underlie the pathogenesis of heart disease, there is little information available about the structural consequences of the change in the interaction of cTnI with cTnC caused by these substitutions. Our results provide the structural, dynamic and functional effects of the L48Q mutation of cTnC on cTnC-cTnI interactions and emphasize the importance of the conformational change in the regulatory domain of cNTnC in cardiac muscle regulation. Future studies may build on this information to determine if there is commonality of these properties for mutations associated with HCM that enhance Ca2+ binding to cTn.
Supplementary Material
ACKNOWLEDGEMENTS
IMR and BDS acknowledge helpful discussions on NMR relaxation with Professor Leo Spyracopoulos. DW and MR acknowledge all work done on Core facility instruments in the Analytical Biopharmacy Core (UW), help from Dr. John Sumidaits, the Center for Intracellular Delivery Biologics and the Washington State Life Sciences Discovery Fund. Computer time through the DOE Office of Biological Research was provided by the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under contract No.DE-AC02-05CH11231. IMR is the recipient of a Graduate Studentship from the Alberta Heritage Foundation for Medical Research. DW is the recipient of the pre-doctoral fellowship from the American Heart Association. MR is an Established Investigator of the American Heart Association. MEM acknowledges support by a grant from the Department of Defense through the National Defense Science and Engineering Graduate Fellowship Program for the MD studies.
This work was funded by grants from the U.S. National Institutes of Health (HL65497 to MR and GM50789 to VD), the Canadian Institutes of Health Research (BDS) and the Heart and Stroke Foundation of Canada (BDS)
Abbreviations used
- cTnC
intact cardiac troponin C
- cNTnC
N-domain of cTnC
- cTn
cardiac whole troponin
- cTnC(L48Q)
intact cTnC variant with the Leu48Gln mutation
- cTnI
cardiac troponin I
- cTnI147-163
cTnI peptide corresponding to residues 147-163
- IANBD
N-(2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diozole
- HSQC
heteronuclear single-quantum coherence
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- MD
molecular dynamics
- WT
wild-type
- PDB
Protein Data Bank
- Cα-RMSD
root-mean-square deviation of Cα atom coordinates from the starting structure
- COMdist
distances between the center of mass of two objects
- ATMdist
distances between the center of mass of two atoms
- SASA
solvent accessible surface area
Footnotes
SUPPORTING INFORMATION AVAILABLEMD Movies and Figures S1–S4 and Tables S1–S2 as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Sia SK, Li MX, Spyracopoulos L, Gagne SM, Liu W, Putkey JA, Sykes BD. Structure of cardiac muscle troponin C unexpectedly reveals a closed regulatory domain. Journal of Biological Chemistry. 1997;272:18216–18221. doi: 10.1074/jbc.272.29.18216. [DOI] [PubMed] [Google Scholar]
- 2.Gagne SM, Tsuda S, Li MX, Smillie LB, Sykes BD. Structures of the Troponin-C Regulatory Domains in the Apo and Calcium-Saturated States. Nat. Struct. Biol. 1995;2:784–789. doi: 10.1038/nsb0995-784. [DOI] [PubMed] [Google Scholar]
- 3.Spyracopoulos L, Li MX, Sia SK, Gagne SM, Chandra M, Solaro RJ, Sykes BD. Calcium-induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry. 1997;36:12138–12146. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- 4.Putkey JA, Sweeney HL, Campbell ST. Site-Directed Mutation of the Trigger Calcium-Binding Sites in Cardiac Troponin-C. J. Biol. Chem. 1989;264:12370–12378. [PubMed] [Google Scholar]
- 5.Baryshnikova OK, Robertson IM, Mercier P, Sykes BD. The dilated cardiomyopathy G159D mutation in cardiac troponin C weakens the anchoring interaction with troponin I. Biochemistry. 2008;47:10950–10960. doi: 10.1021/bi801165c. [DOI] [PubMed] [Google Scholar]
- 6.Ramakrishnan S, Hitchcock-DeGregori SE. Structural and functional significance of aspartic acid 89 of the troponin C central helix in Ca2+ signaling. Biochemistry. 1996;35:15515–15521. doi: 10.1021/bi961788u. [DOI] [PubMed] [Google Scholar]
- 7.Biesiadecki BJ, Kobayashi T, Walker JS, John Solaro R, de Tombe PP. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ Res. 2007;100:1486–1493. doi: 10.1161/01.RES.0000267744.92677.7f. [DOI] [PubMed] [Google Scholar]
- 8.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol. Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 9.Rarick HM, Tu XH, Solaro RJ, Martin AF. The C terminus of cardiac troponin I is essential for full inhibitory activity and Ca2+ sensitivity of rat myofibrils. J. Biol. Chem. 1997;272:26887–26892. doi: 10.1074/jbc.272.43.26887. [DOI] [PubMed] [Google Scholar]
- 10.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann B, Schmidt-Traub H, Perrot A, Osterziel KJ, Gessner R. First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Hum. Mutat. 2001;17:524. doi: 10.1002/humu.1143. [DOI] [PubMed] [Google Scholar]
- 12.Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circ Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomes AV, Potter JD. Molecular and cellular aspects of troponin cardiomyopathies. Ann. N. Y. Acad. Sci. 2004;1015:214–224. doi: 10.1196/annals.1302.018. [DOI] [PubMed] [Google Scholar]
- 14.Tikunova SB, Liu B, Swindle N, Little SC, Gomes AV, Swartz DR, Davis JP. Effect of calcium-sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry. 2010;49:1975–1984. doi: 10.1021/bi901867s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tikunova SB, Davis JP. Designing calcium-sensitizing mutations in the regulatory domain of cardiac troponin C. J Biol Chem. 2004;279:35341–35352. doi: 10.1074/jbc.M405413200. [DOI] [PubMed] [Google Scholar]
- 16.Parvatiyar MS, Pinto JR, Liang J, Potter JD. Predicting cardiomyopathic phenotypes by altering Ca2+ affinity of cardiac troponin C. J. Biol. Chem. 2010;285:27785–27797. doi: 10.1074/jbc.M110.112326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kreutziger KL, Piroddi N, McMichael JT, Tesi C, Poggesi C, Regnier M. Calcium binding kinetics of troponin C strongly modulate cooperative activation and tension kinetics in cardiac muscle. J. Mol. Cell. Cardiol. 2011;50:165–174. doi: 10.1016/j.yjmcc.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong WJ, Rosenfeld SS, Wang CK, Gordon AM, Cheung HC. Kinetic studies of calcium binding to the regulatory site of troponin C from cardiac muscle. J. Biol. Chem. 1996;271:688–694. doi: 10.1074/jbc.271.2.688. [DOI] [PubMed] [Google Scholar]
- 19.Pearlstone JR, Chandra M, Sorenson MM, Smillie LB. Biological function and site IICa2+−induced opening of the regulatory domain of skeletal troponin C are impaired by invariant site I or II Glu mutations. J. Biol. Chem. 2000;275:35106–35115. doi: 10.1074/jbc.M001000200. [DOI] [PubMed] [Google Scholar]
- 20.Li MX, Corson DC, Sykes BD. Structure determination by NMR. Isotope labeling. Methods Mol. Biol. 2002;173:255–265. doi: 10.1385/1-59259-184-1:255. [DOI] [PubMed] [Google Scholar]
- 21.Martyn DA, Regnier M, Xu D, Gordon AM. Ca2+ − and cross-bridge-dependent changes in N- and C-terminal structure of troponin C in rat cardiac muscle. Biophys. J. 2001;80:360–370. doi: 10.1016/S0006-3495(01)76020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon AM, Qian Y, Luo Z, Wang CK, Mondares RL, Martyn DA. Characterization of troponin-C interactions in skinned barnacle muscle: comparison with troponin-C from rabbit striated muscle. J Muscle Res Cell Motil. 1997;18:643–653. doi: 10.1023/a:1018631806182. [DOI] [PubMed] [Google Scholar]
- 23.Dong WJ, Robinson JM, Stagg S, Xing J, Cheung HC. Ca2+−induced conformational transition in the inhibitory and regulatory regions of cardiac troponin I. J. Biol. Chem. 2003;278:8686–8692. doi: 10.1074/jbc.M212886200. [DOI] [PubMed] [Google Scholar]
- 24.Patton C, Thompson S, Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 25.George SE, Su Z, Fan D, Wang S, Johnson JD. The Fourth EF-Hand of Calmodulin and Its Helix-Loop-Helix Components:□ Impact on Calcium Binding and Enzyme Activation†. Biochemistry. 1996;35:8307–8313. doi: 10.1021/bi960495y. [DOI] [PubMed] [Google Scholar]
- 26.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 27.Johnson BA, Blevins RA. Nmr View - a Computer-Program for the Visualization and Analysis of Nmr Data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 28.Spyracopoulos L. A suite of Mathematica notebooks for the analysis of protein main chain N-15 NMR relaxation data. Journal of Biomolecular Nmr. 2006;36:215–224. doi: 10.1007/s10858-006-9083-0. [DOI] [PubMed] [Google Scholar]
- 29.d'Auvergne EJ, Gooley PR. The use of model selection in the model-free analysis of protein dynamics. J. Biomol. NMR. 2003;25:25–39. doi: 10.1023/a:1021902006114. [DOI] [PubMed] [Google Scholar]
- 30.Palmer AG, Rance M, Wright PE. Intramolecular Motions of a Zinc Finger DNA-Binding Domain from Xfin Characterized by Proton-Detected Natural Abundance C-12 Heteronuclear Nmr-Spectroscopy. J. Am. Chem. Soc. 1991;113:4371–4380. [Google Scholar]
- 31.Li MX, Spyracopoulos L, Sykes BD. Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac tropoin-C. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 32.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Beck DAC, McCully ME, Alonso DOV, Daggett V. in lucem molecular mechanics ilmm) University of Washington; Seattle: 2000-2012. [Google Scholar]
- 34.Levitt M, Hirshberg M, Sharon R, Daggett V. Potential-energy function and parameters for simulations of the molecular-dynamics of proteins and nucleic-acids in solution. Comput. Phys. Commun. 1995;91:215–231. [Google Scholar]
- 35.Levitt M, Hirshberg M, Sharon R, Laidig KE, Daggett V. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J. Phys. Chem. B. 1997;101:5051–5061. [Google Scholar]
- 36.Kell GS. Precise Representation of Volume Properties of Water at 1 Atmosphere. J. Chem. Eng. Data. 1967;12:66–69. [Google Scholar]
- 37.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 38.Gether U, Lin S, Ghanouni P, Ballesteros JA, Weinstein H, Kobilka BK. Agonists induce conformational changes in transmembrane domains III and VI of the beta2 adrenoceptor. EMBO J. 1997;16:6737–6747. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gether U, Lin S, Kobilka BK. Fluorescent labeling of purified beta 2 adrenergic receptor. Evidence for ligand-specific conformational changes. J Biol Chem. 1995;270:28268–28275. doi: 10.1074/jbc.270.47.28268. [DOI] [PubMed] [Google Scholar]
- 40.Dong WJ, Cheung HC. Calcium-induced conformational change in cardiac troponin C studied by fluorescence probes attached to Cys-84. Biochim Biophys Acta. 1996;1295:139–146. doi: 10.1016/0167-4838(96)00028-3. [DOI] [PubMed] [Google Scholar]
- 41.Hoffman RMB, Li MX, Sykes BD. The bindin of W7, an inhibitor of striated muscle contraction, to cardiac troponin C. Biochemistry. 2005;44:15750–15759. doi: 10.1021/bi051583y. [DOI] [PubMed] [Google Scholar]
- 42.Li MX, Gagne SM, Spyracopoulos L, Kloks CP, Audette G, Chandra M, Solaro RJ, Smillie LB, Sykes BD. NMR studies of Ca2+ binding to the regulatory domains of cardiac and E41A skeletal muscle troponin C reveal the importance of site I to energetics of the induced structural changes. Biochemistry. 1997;36:12519–12525. doi: 10.1021/bi971222l. [DOI] [PubMed] [Google Scholar]
- 43.Tripet B, Van Eyk JE, Hodges RS. Mapping of a second actintropomyosin and a second troponin C binding site within the C terminus of troponin I, and their importance in the Ca2+-dependent regulation of muscle contraction. J. Mol. Biol. 1997;271:728–750. doi: 10.1006/jmbi.1997.1200. [DOI] [PubMed] [Google Scholar]
- 44.Li MX, Saude EJ, Wang X, Pearlstone JR, Smillie LB, Sykes BD. Kinetic studies of calcium and cardiac troponin I peptide binding to human cardiac troponin C using NMR spectroscopy. Eur. Biophys. J. Biophy. 2002;31:245–256. doi: 10.1007/s00249-002-0227-1. [DOI] [PubMed] [Google Scholar]
- 45.Paakkonen K, Sorsa T, Drakenberg T, Pollesello P, Tilgmann C, Permi P, Heikkinen S, Kilpelainen I, Annila A. Conformations of the regulatory domain of cardiac troponin C examined by residual dipolar couplings. Eur. J. Biochem. 2000;267:6665–6672. doi: 10.1046/j.1432-1327.2000.01763.x. [DOI] [PubMed] [Google Scholar]
- 46.Eichmueller C, Skrynnikov NR. Observation of mu s time-scale protein dynamics in the presence of Ln(3+)stop ions: application to the N-terminal domain of cardiac troponin C. J. Biomol. NMR. 2007;37:79–95. doi: 10.1007/s10858-006-9105-y. [DOI] [PubMed] [Google Scholar]
- 47.McKay RT, Saltibus LF, Li MX, Sykes BD. Energetics of the induced structural change in a Ca2+ regulatory protein: Ca2+ and troponin I peptide binding to the E41A mutant of the N-domain of skeletal troponin C. Biochemistry. 2000;39:12731–12738. doi: 10.1021/bi001240u. [DOI] [PubMed] [Google Scholar]
- 48.Lindert S, Kekenes-Huskey PM, Huber G, Pierce L, McCammon JA. Dynamics and Calcium Association to the N-Terminal Regulatory Domain of Human Cardiac Troponin C: A Multiscale Computational Study. J Phys Chem B. 2012 doi: 10.1021/jp212173f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Love ML, Putkey JA, Cohen C. Bepridil opens the regulatory N-terminal lobe of cardiac troponin C. P Natl Acad Sci USA. 2000;97:5140–5145. doi: 10.1073/pnas.090098997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robertson IM, Boyko RF, Sykes BD. Visualizing the principal component of (1)H,(15)N-HSQC NMR spectral changes that reflect protein structural or functional properties: application to troponin C. J. Biomol. NMR. 2011;51:115–122. doi: 10.1007/s10858-011-9546-9. [DOI] [PubMed] [Google Scholar]
- 51.Spyracopoulos L, Gagne SM, Sykes BD. In: Proceedings of the International School of Structural Biology and Magnetic Resonance. Jardetzky O, Lefevre JF, editors. Plenum Press; New York: 2001. pp. 37–44. 37-44. [Google Scholar]
- 52.Baryshnikova OK, Sykes BD. Backbone dynamics of SDF-1 alpha determined by NMR: Interpretation in the presence of monomer-dimer equilibrium. Protein Sci. 2006;15:2568–2578. doi: 10.1110/ps.062255806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spyracopoulos L, Gagne SM, Li MX, Sykes BD. Dynamics and thermodynamics of the regulatory domain of human cardiac troponin C in the apo- and calcium-saturated states. Biochemistry. 1998;37:18032–18044. doi: 10.1021/bi9816960. [DOI] [PubMed] [Google Scholar]
- 54.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 55.Lewit-Bentley A, Rety S. EF-hand calcium-binding proteins. Curr Opin Struct Biol. 2000;10:637–643. doi: 10.1016/s0959-440x(00)00142-1. [DOI] [PubMed] [Google Scholar]
- 56.Marsden BJ, Shaw GS, Sykes BD. Calcium binding proteins. Elucidating the contributions to calcium affinity from an analysis of species variants and peptide fragments. Biochem Cell Biol. 1990;68:587–601. doi: 10.1139/o90-084. [DOI] [PubMed] [Google Scholar]
- 57.Herzberg O, James MN. Structure of the calcium regulatory muscle protein troponin-C at 2.8 A resolution. Nature. 1985;313:653–659. doi: 10.1038/313653a0. [DOI] [PubMed] [Google Scholar]
- 58.Wilkinson JM, Grand RJ. The amino acid sequence of troponin I from rabbit skeletal muscle. Biochem J. 1975;149:493–496. [PMC free article] [PubMed] [Google Scholar]
- 59.Belus A, Piroddi N, Tesi C. Mechanism of cross-bridge detachment in isometric force relaxation of skeletal and cardiac myofibrils. J Muscle Res Cell M. 2003;24:261–267. [PubMed] [Google Scholar]
- 60.Regnier M, Rivera AJ, Wang CK, Bates MA, Chase PB, Gordon AM. Thin filament near-neighbour regulatory unit interactions affect rabbit skeletal muscle steady-state force-Ca(2+) relations. J Physiol. 2002;540:485–497. doi: 10.1113/jphysiol.2001.013179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gordon AM, Rivera AJ, Wang CK, Regnier M. Cooperative activation of skeletal and cardiac muscle. Adv Exp Med Biol. 2003;538:371–378. doi: 10.1007/978-1-4419-9029-7_34. discussion 378–379. [DOI] [PubMed] [Google Scholar]
- 62.Gillis TE, Martyn DA, Rivera AJ, Regnier M. Investigation of thin filament near-neighbour regulatory unit interactions during force development in skinned cardiac and skeletal muscle. J Physiol. 2007;580:561–576. doi: 10.1113/jphysiol.2007.128975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elkins KM, Gatzeva-Topalova PZ, Nelson DJ. Molecular dynamics study of Ca(2+) binding loop variants of parvalbumin with modifications at the “gateway” position. Protein Eng. 2001;14:115–126. doi: 10.1093/protein/14.2.115. [DOI] [PubMed] [Google Scholar]
- 64.Paakkonen K, Sorsa T, Drakenberg T, Pollesello P, Tilgmann C, Permi P, Heikkinen S, Kilpelainen I, Annila A. Conformations of the regulatory domain of cardiac troponin C examined by residual dipolar couplings. Eur. J. Biochem. 2000;267:6665–6672. doi: 10.1046/j.1432-1327.2000.01763.x. [DOI] [PubMed] [Google Scholar]
- 65.Sorsa T, Pollesello P, Solaro RJ. The contractile apparatus as a target for drugs against heart failure: Interaction of levosimendan, a calcium sensitiser, with cardiac troponin c. Mol. Cell. Biochem. 2004;266:87–107. doi: 10.1023/b:mcbi.0000049141.37823.19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.