

Abstract
The EU-supported EuroFlow Consortium aimed at innovation and standardization of immunophenotyping for diagnosis and classification of hematological malignancies by introducing 8-color flow cytometry with fully standardized laboratory procedures and antibody panels in order to achieve maximally comparable results among different laboratories. This required the selection of optimal combinations of compatible fluorochromes and the design and evaluation of adequate standard operating procedures (SOPs) for instrument setup, fluorescence compensation and sample preparation. Additionally, we developed software tools for the evaluation of individual antibody reagents and antibody panels. Each section describes what has been evaluated experimentally versus adopted based on existing data and experience. Multicentric evaluation demonstrated high levels of reproducibility based on strict implementation of the EuroFlow SOPs and antibody panels. Overall, the 6 years of extensive collaborative experiments and the analysis of hundreds of cell samples of patients and healthy controls in the EuroFlow centers have provided for the first time laboratory protocols and software tools for fully standardized 8-color flow cytometric immunophenotyping of normal and malignant leukocytes in bone marrow and blood; this has yielded highly comparable data sets, which can be integrated in a single database.
Keywords: flow cytometry, standardization, compensation, software, fluorochromes, immunophenotyping
Introduction
Immunophenotyping is currently one of the fundamental pillars for the diagnosis and classification of leukemia and lymphoma.1 In the last two decades multiparameter flow cytometry has become the preferred method to assess the immunophenotypic features of cells present in peripheral blood (PB), bone marrow (BM), lymph node (LN) biopsy specimens, cerebrospinal fluid (CSF) and other types of samples suspected of containing neoplastic hematopoietic cells.1, 2 During the first part of this period, the list of clinically useful antibodies (Abs) has progressively increased,3, 4, 5 leading to the definition of complex immunophenotypic profiles. In parallel, the number of antigens that can be assessed in a single measurement has increased dramatically owing to the availability of new multicolor digital instruments and a greater number of compatible fluorochromes.6, 7 This has facilitated more precise identification and phenotypic characterization of specific populations of tumor cells in samples over the background of the coexisting residual normal leukocyte subsets.8 However, the higher complexity of the immunophenotypic approaches and panels of reagents involved in such characterization demanded increasing expertise for correct interpretation of the data obtained. As a consequence, disturbing levels of subjectivity have been introduced, depending on the experience and knowledge of individual experts and the variable panels of reagents applied in different clinical diagnostic laboratories.
In order to decrease such variability and subjectivity, consensus recommendations and guidelines have been produced by several expert groups.3, 5, 9, 10, 11, 12, 13, 14 These documents have had a wide impact and they have been followed by many centers around the world, but they have been only partially successful for several reasons. First, they focus on lists of markers without specific recommendations about reagent clones, fluorochrome conjugates or optimally designed antibody combinations in the panel. Second, they fail to provide robust protocols for the selection of the most appropriate (i) combinations of fluorochromes and fluorochrome-conjugated reagents in a panel, (ii) sample preparation techniques, (iii) standard operating procedures (SOPs) to establish instrument settings prior to the measurements and (iv) the most adequate strategies for data analysis. Most importantly, the so far proposed sets of markers have never been prospectively evaluated.
In 2006 the EU-supported EuroFlow Consortium (EU-FP6, LSHB-CT-2006-018708) started a project aimed at the prospective design and evaluation of panels of antibodies for the diagnosis and classification of the most frequent subtypes of leukemias and lymphomas in which immunophenotyping has proven to be relevant. The major objectives were (i) to provide comprehensive multicolor combinations of fluorochrome-conjugated antibodies aimed at answering those medical questions for which multicolor flow cytometry immunophenotyping is indicated, (ii) to prospectively evaluate their performance in multiple diagnostic laboratories and (iii) to optimize the reagent panel whenever required. For this purpose, proven reproducibility in multiple diagnostic laboratories was mandatory. Therefore, the definition of optimal antibody panels also required an effort in the selection of the most appropriate combination of compatible fluorochromes, the design and evaluation of adequate SOPs for instrument setup, fluorescence compensation and sample preparation and elaboration of adequate software tools for the overall evaluation of the phenotypic profiles obtained.
In the first five sections of this paper, we provide detailed information about the selection of the most appropriate combination of fluorochromes for 8-color panels, the protocols recommended for instrument settings, fluorochrome compensation and sample preparation, together with the data analysis strategies adopted to evaluate the tested antibody reagents and panels. In the last section, results of multicentric evaluation of the level of reproducibility that can be achieved by implementation of all standardization efforts are provided. In each of the sections, we indicate what has been specifically evaluated versus adopted based on existing data.
SECTION 1. Fluorochrome selection for 8-color panels
J Flores-Montero1, T Kalina2, JJ Pérez3, S Böttcher4, VHJ van der Velden5, J Almeida1, L Lhermitte6, A Mendonça7, R de Tute8, M Cullen8, L Sedek9, E Mejstrikova2, JJM van Dongen5 and A Orfao1
1USAL, Salamanca, Spain; 2DPH/O, Prague, Czech Republic; 3HUS, Salamanca, Spain; 4UNIKIEL, Kiel, Germany; 5Erasmus MC, Rotterdam, The Netherlands; 6AP-HP, Paris, France; 7IPOLFG, Lisbon, Portugal; 8UNIVLEEDS, Leeds, UK and 9SUM, Zabrze, Poland
Background
Selection of the most appropriate combination of fluorochromes is a key step in designing a multicolor immunophenotypic panel.15 Usage of the new digital flow cytometers capable of simultaneously measuring multiple (for example, ⩾6) different fluorescence emissions has only recently become possible in diagnostic laboratories because of the increasing availability of compatible fluorochromes.16, 17, 18, 19, 20, 21 However, the varying spectral overlap of such fluorochromes has also led to a higher complexity of fluorescence compensation matrices.6, 22, 23
Fluorochrome selection largely depends on the intrinsic characteristics of each individual fluorescent compound, particularly its excitation and emission profile, its relative brightness, the spillover into other fluorescence detectors and its stability.24 The selection of the most adequate fluorochrome combination also depends on the specific optical configuration of the flow cytometer, that is, the number and type of laser lines it contains, the number of detectors available for each laser and the specific set of filters for each individual laser.7 Furthermore, the aim of an antibody panel, the type of samples to be stained (that is, PB, BM versus small cell samples) and the cells contained in it also contribute to the decision on the minimum number of reagents to be simultaneously assessed in individual tubes.25, 26 Finally, the availability of optimal clones of fluorochrome-conjugated antibodies also determines the selection of specific combinations of reagents in a panel.27, 28
On the basis of the innovative immunophenotyping strategy designed by the EuroFlow group in which new data merge and calculation tools are combined for improved diagnosis and classification of hematological malignancies, a minimum requirement of 8-color panels for cost-effective immunophenotyping was foreseen. Such panels should allow simultaneous usage of (i) backbone markers aimed at specific identification of the cell populations of interest and (ii) additional antibody markers devoted to a more detailed characterization of the said cell populations.29
In this section we review the selection of flow cytometer instruments, their optical configuration and the set of compatible fluorochromes, as performed during the construction and evaluation of the EuroFlow 8-color panels.
Selection of flow cytometry instruments and their optical configurations
At the time the EuroFlow project started in March 2006, four ⩾8-color flow cytometry instruments from two different manufacturers were available, with flexible and compatible optical configurations (Table 1), which could potentially be used in diagnostic laboratories. The four instruments were taken into consideration in selecting the combinations of fluorochromes to be used in the EuroFlow panels. All four instruments have a three laser-line configuration, with blue (488 nm), red (633 or 635 nm) and violet (405 or 407 nm) lasers.
Table 1. Typical default optical configuration and most common fluorochromes available for each detector of three lasers, ⩾8-color flow cytometry instruments available in March 2006.
| Channel |
FACSCanto II (BD Biosciences) |
FACSAria (BD Biosciences) |
LSR II (BD Biosciences) |
CyAn ADP (Dako/Beckman Coulter) |
Most commonly available fluorochromes | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Laser | DM | EF | Laser | DM | EF | Laser | DM | EF | Laser | DM | EF | ||
| 1 | 30 mW Violet (405 nm) | 450/50 | 10 mW Violet (407 nm) | 450/50 | 25 mW Violet (405 nm) | 450/50 | 100 mW Violet (405 nm) | 450/50 | PacB/HV450 | ||||
| 2 | 30 mW Violet (405 nm) | 502 | 510/50 | 10 mW Violet (407 nm) | 502 | 530/30 | 25 mW Violet (405 nm) | 502 | 525/50 | 100 mW Violet (405 nm) | 502 | 530/40 | AmCyan/PacO/HV500 |
| 3 | 20 mW Blue (488 nm) | 502 | 530/30 | 13 mW Blue (488 nm) | 502 | 530/30 | 20 mW Blue (488 nm) | 502 | 530/30 | 25 mW Blue (488 nm) | 502 | 530/40 | FITC/AF488 |
| 4 | 20 mW Blue (488 nm) | 556 | 585/42 | 13 mW Blue (488 nm) | 556 | 585/42 | 20 mW Blue (488 nm) | 556 | 575/26 | 25 mW Blue (488 nm) | 556 | 585/42 | PE |
| 5 | 13 mW Blue (488 nm) | 610 | 616/23 | 25 mW Blue (488 nm) | 613/20 | PE-TR | |||||||
| 6 | 20 mW Blue (488 nm) | 655 | 670LP | 13 mW Blue (488 nm) | 655 | 695/40 | 20 mW Blue (488 nm) | 655 | 695/40 | 25 mW Blue (488 nm) | 655 | 680/30 | PerCP/PerCPCy5.5 |
| 7 | 20 mW Blue (488 nm) | 735 | 780/60 | 13 mW Blue (488 nm) | 735 | 780/60 | 20 mW Blue (488 nm) | 735 | 780/60 | 25 mW Blue (488 nm) | 735 | 750LP | PECy7 |
| 8 | 17 mW Red (633 nm) | 660/20 | 11 mW Red (633 nm) | 660/20 | 35 mW Red (633 nm) | 660/20 | 60 mW Red (635 nm) | 665/20 | APC/AF647 | ||||
| 9 | 17 mW Red (633 nm) | 735 | 780/60 | 11 mW Red (633 nm) | 735 | 780/60 | 35 mW Red (633 nm) | 735 | 780/60 | 60 mW Red (635 nm) | 735 | 750LP | APCCy7/APCH7/AF700a |
Abbreviations: AF, alexa fluor; AmCyan, Anemonia Majano cyan fluorescent protein; APC, allophycocyanin; Cy5.5, cyanin5.5; Cy7, cyanin7; DM, dichroic mirror; EF, emission filter; FITC, fluorescein isothiocyanate; H7, hilite7; HV450, Horizon V450; HV500, Horizon V500; LP, long pass; PacB, pacific blue; PacO, pacific orange; PE, phycoerythrin; PerCP, peridinin–chlorophyll–protein; TR, Texas Red.
AF700 requires a 710/50 emission filter.
Selection of fluorochromes
A two-step approach was used by the EuroFlow group for selection of fluorochromes: (i) some fluorochromes were predefined without further specific testing based on previous experience, whereas (ii) others were evaluated prior to their selection. Accordingly, the first two positions for the blue laser line (emission at 488 nm) were pre-selected as fluorescein isothiocyanate (FITC) and phycoerythrin (PE) because of the extensive experience available with both fluorochromes, the large number of high-quality commercially available reagents and their compatibility with the optical configuration of all the four ⩾8-color instruments listed in Table 1. The same selection criteria were applied for Allophycocyanin (APC) as the first fluorochrome for the red laser line (emission at 633/635 nm). Similarly, either peridinin–chlorophyll–protein complex (PerCP) or PerCP–Cyanin5.5 (PerCPCy5.5) and PE–Cyanin7 (PECy7) were left as the most suitable fluorochrome choices for the third and fourth detectors of the blue laser line, respectively. In contrast, APC-Cyanin7 (APCCy7), Alexa Fluor 700 (AF700) and APC-Hilite7 (APCH7) were compared for the second detector of the red laser line, and Pacific Blue (PacB) versus Horizon V450 (HV450) and Pacific Orange (PacO) versus Anemonia Majano cyan fluorescent protein (AmCyan)17 versus Horizon V500 (HV500) were evaluated for the first and second detector of the violet laser line (emission at 405/407 nm), respectively.
For these evaluations several fluorochrome-conjugated antibody reagents were compared: PacB-conjugated CD2(TS1/8), CD3(UCHT1), CD4(RPA-T4), CD20(2H7), CD45(T29/33) and HLADR(L243) versus HV450-conjugated CD2(S5.2), CD3(UCHT1), CD4(RPA-T4), CD20(L27), CD45(HI30) and HLADR(L243); AmCyan-conjugated CD45(2D1) versus PacO-conjugated CD45(HI30) versus HV500-conjugated CD45(HI30); and APCCy7-conjugated CD4(RPA-T4) versus AF700-conjugated CD4(RPA-T4) versus APCH7-conjugated CD4(RPA-T4) antibody(clone) reagents. Antigen expression was evaluated as both mean fluorescence intensity (MFI) and stain index (SI; defined as the difference between the MFI of positive and negative cells divided by 2 s.d.'s of the MFI observed for the negative cell population).24 In all cases, staining of ⩾5 PB samples was used to evaluate the staining patterns of each pair/group of reagents to be compared. Sample preparation and instrument settings were performed in the eight different EuroFlow laboratories as described in Section 2 and Section 4 of this manuscript.
Comparison between the Pacific Blue (PacB) and Horizon V450 (HV450) fluorochromes
The PacB and HV450 fluorochromes showed very similar fluorescence profiles that adequately fit with the optical configuration of the first detector for the violet laser of the four flow cytometry instruments. Detailed comparison of the needs for compensation for the spillover into other detectors of the fluorescence emissions of these two fluorochromes showed that these were slightly higher (P>0.05; Mann–Whitney U test) for PacB versus HV450; nonetheless, both fluorochromes showed no spillover into any detector except for the second detector of the violet laser (Table 2).
Table 2. Mean values of compensation matrices (n=5) obtained at different time points in up to five different EuroFlow flow cytometer instruments for fluorochromes compared for the same fluorescence channel.
| Laser channel | |
|
Compensation requirements in other fluorescence channels |
|
|
|
||
|---|---|---|---|---|---|---|---|---|
| PacB | HV450 | PacO | AmCyan | HV500 | APCCy7 | AF700 | APCH7 | |
| Violet-1 | NA | NA | 2.2±0.3* | 11.5±1.5 | 7.5±1.6 | 0.2±0.3 | 0.1±0.1 | 0.1±0.3 |
| Violet-2 | 27.9±2.8 | 23.8±2.3 | NA | NA | NA | 0.3±0.4 | 0.1±0.1 | NR |
| Blue-1 | 0.1±0.1 | 0.1±0.1 | 0.8±0.4 | 17.1±2.6 | 2.8±1.2 | 0.4±0.4 | 0.2±0.1 | 0.2±0.4 |
| Blue-2 | NR | 0.1±0.1 | 0.4±0.2 | 1.4±0.2 | 0.5±0.2 | 0.2±0.2 | NR | 0.1±0.2 |
| Blue-3 | 0.1±0.1 | 0.1±0.1 | 0.5±0.2 | 0.4±0.1 | 0.3±0.2 | 1.2±1.0 | 3.6±0.7 | 0.6±1.2 |
| Blue-4 | NR | NR | 0.1±0.1 | NR | NR | 3.3±1.9 | 1.3±0.3 | 1.7±0.8 |
| Red-1 | NR | NR | 0.3±0.4 | NR | NR | 4.8±2.5 | 0.8±0.2 | 2.0±1.1 |
| Red-2 | 0.1±0.1 | NR | 0.1±0.2 | NR | NR | NA | NA | NA |
Abbreviations: AF700, alexa fluor 700; AmCyan, Anemonia Majano cyan fluorescent protein; APC, allophycocyanin; Cy7, cyanin7; H7, hilite7; HV450, Horizon V450; HV500, Horizon V500; NA, not applicable; NR, not required; PacB, pacific blue; PacO, pacific orange.
Results are expressed as percentage values±s.d.
*P<0.01 versus both AmCyan and HV500 (paired Student's T-test).
Regarding MFI and SI values, similar results with <10% differences were found when the same clone and manufacturer were compared. Conversely, when either the clones or the manufacturers were not the same, differences between reagents were higher (Table 3). We have chosen PacB for the EuroFlow panels, based on broader availability of PacB conjugates at the time of testing.
Table 3. Mean fluorescence intensity (MFI) and stain index (SI) values obtained for different sets of reagents evaluated in normal PB samples (n=5).
| Marker | PacB | HV450 | P-valuea | |||||
|---|---|---|---|---|---|---|---|---|
| CD2 | Clone (manufacturer) | TS1/8 (BioLegend) | S5.2 (BD B) | |||||
| MFI±s.d. of CD2+ T and NK-cells | 5741±755.7 | 8259±1792.8 | 0.02 | |||||
| SIc | 37.0 | 41.4 | ||||||
| CD3 | Clone (manufacturer) | UCHT1 (BD Ph) | UCHT1 (BD B) | |
|
|
|
|
| MFI±s.d. of T-cells | 15 774±1503.7 | 17 246±814.2 | 0.08 |
APCCy7 |
AF700 |
APCH7 |
P-valueb |
|
| SId | 117.2 | 130.5 | ||||||
| CD4 | Clone (manufacturer) | RPA-T4 (BD Ph) | RPA-T4 (BD B) | RPA-T4 (BD B) | RPA-T4 (BD B) | RPA-T4 (BD B) | ||
| MFI±s.d. of CD4+ T-cells | 9474±710.2 | 9195±408.2 | 0.46 | 13 596±686.5 | 4307±174.1 | 9910±414.3 | <0.001 | |
| SIe | 61.8 | 66.6 | 42.6 | 41.4 | 35.0 | |||
| CD20 | Clone (manufacturer) | 2H7 (eBiosciences) | L27 (BD B) | |
|
|
|
|
| MFI±s.d. of B-cells | 30 073±3783.5 | 38 152±2857.4 | 0.005 |
PacO |
AmCyan |
HV500 |
P-valueb |
|
| SIf | 219.61 | 222.8 | ||||||
| CD45 | Clone (manufacturer) | T29/33 (Dako) | HI30 (BD B) | HI30 (Invitrogen) | 2D1 (BD B) | HI30 (BD B) | ||
| MFI±s.d. of lymphocytes | 30 742±824.8 | 53 709±2062.2 | <0.001 | 5521±150.6 | 30 681±2838.3 | 19 157±686.3 | <0.001 | |
| SIg | 3.9 | 5.2 | 3.8 | 4.8 | 3.8 | |||
| HLADR | Clone (manufacturer) | L243 (BioLegend) | L243 (BD B) | |||||
| MFI±s.d. of monocytes | 11 509±1721.4 | 16 874±1934.3 | 0.002 | |||||
| SIh | 47.7 | 64.8 |
Abbreviations: AF700, alexa fluor 700; AmCyan, Anemonia Majano cyan fluorescent protein; APCCy7, allophycocyanin–cyanin7; APCH7, allophycocyanin–hilite7; BD Ph, BD Pharmingen; BD B, BD Biosciences; HV450, Horizon V450; HV500, Horizon V500; PacB, pacific blue; PacO, pacific orange.
Paired Student's T-test.
P<0.001 for the following comparisons: APCH7 versus AF700, AF700 versus APCCy7, APCCy7 versus APCH7, PacO versus AmCyan, PacO versus HV500 and AmCyan versus HV500 (paired Student's T-test).
Positive reference population (PRP): CD2+ T- and NK-cells and negative reference population (NRP), CD2− lymphocytes.
PRP, T-cells; NRP, B- and NK-cells.
PRP, CD4+ T-cells; NRP, CD4− T-cells.
PRP, B-cells; NRP, T- and NK-cells.
PRP, lymphocytes; NRP, neutrophils.
PRP, monocytes; NRP, lymphocytes.
Comparison among the Anemonia Majano cyan fluorescent protein (AmCyan), Pacific Orange (PacO) and Horizon V500 (HV500) fluorochromes
Specific comparisons for the second detector of the violet laser line were made for the AmCyan, PacO and HV500 fluorochrome dyes. These fluorochromes showed clearly different fluorescence profiles. Accordingly, in terms of needs for fluorescence compensation, a higher spillover into other channels was observed for AmCyan, particularly in the first detector of the violet laser line (P<0.01 versus both PacO and HV500; paired Student's T-test) and in the first detector of the blue laser (P<0.01 versus both PacO and HV500; paired Student's T-test), where either PacB or HV450, and FITC, respectively, are typically measured. Table 2 summarizes the compensation matrix values obtained for these three dyes. In general, the MFI obtained for monoclonal Ab reagents conjugated with these fluorochromes directed against the same antigen was also higher for AmCyan, although different clones were compared and fluorescence differences may not be solely related to the fluorochrome (Table 3). AmCyan showed a higher resolution power, but the higher fluorescence intensity represented a disadvantage when a strong AmCyan signal for a marker was combined with a dim signal of FITC-conjugated reagents in the same cell populations, because of its relatively higher overlap with the first detector of the blue laser (data not shown). In turn, PacO showed low spillover into other channels (Table 2), together with clearly dimmer MFI values (Table 3); nonetheless, its resolution power, as reflected by the observed SI, was comparable to that of AmCyan (Table 3). HV500 showed an intermediate profile between AmCyan and PacO in terms of both needs for compensation and fluorescence intensity of positive cells (higher than PacO but lower than AmCyan), associated with a comparable resolution power (SI) between different cell populations (Table 3).
Comparisons among the Allophycocyanin–Cyanin7 (APCCy7), Alexa Fluor 700 (AF700) and Allophycocyanin–Hilite7 (APCH7) fluorochromes
Comparison of APCCy7, AF700 and APCH7 was performed in sequential steps. First, the performance of each individual fluorochrome was assessed. Accordingly, APCCy7 showed a relatively high intensity (Table 3), while its main disadvantage was the over-time instability, especially in the presence of formaldehyde-based fixatives. This instability resulted in a relatively high and variable degradation-associated ‘spillover' into the first channel of the red laser and the appearance in this channel of false-positive events (data not shown), in line with previous observations.24 More recently, such instability has also been related to a cell-dependent degradation phenomenon.30 In addition, APCCy7 showed great lot-to-lot differences in brightness and compensation needs (data not shown). AF700 showed little spillover into this latter channel (Table 2), but this dye required the use of a different mirror and filter –680 nm long pass (LP) and 710/50 nm band pass (BP), respectively– than those available by default in all four flow cytometers evaluated. In addition, the fluorescence intensity of AF700 translated into suboptimal discrimination of some antigens expressed at relatively low levels, particularly when they were expressed on cells that had a bright APC signal (decreased SI due to compensation-induced data spread; data not shown). Finally, the APCH7 dye, a more stable APC-based tandem dye with a long Stoke's shift, was tested. It showed a lower SI and MFI than its equivalent APCCy7-antibody conjugates (Table 3), but the major advantages of APCH7 conjugates included (i) improved stability and (ii) better compensation profile, while (iii) keeping the default optical configuration of the instrument unchanged. These results are illustrated by direct comparison of APCCy7, APCH7 and AF700 conjugates of the same CD4 monoclonal Ab clone from the same manufacturer after staining of normal PB samples (n=5) (Table 3).
Conclusion
Selection of appropriate fluorochromes to be combined was a key and pre-requisite step in developing the 8-color EuroFlow panels. On the basis of existing knowledge, experience and proven quality of evaluated reagents, several fluorochromes were pre-selected. For other fluorochrome positions, extensive comparisons were required. Finally, we selected the combination of PacB (or HV450), PacO (or HV500), FITC, PE, PerCPCy5.5, PECy7, APC and APCH7. However, it should be noted that some of these fluorochromes performed at the desirable conditions, but others (for example, APCH7) still leave room for improvement. Substitution of PacO by HV500 and PacB by HV450 might be feasible, provided that identical clones are used, that the new reagents are extensively compared to the reference reagents, and that new compensation matrices are applied, which are adequate for the selected fluorochromes.
SECTION 2. EuroFlow standard operating procedure (SOP) to establish standardized instrument settings at multiple sites
T Kalina1, JG te Marvelde2, VHJ van der Velden2, J Flores-Montero3, D Thűrner1, S Böttcher4, M Cullen5, L Lhermitte6, AS Bedin6, L Sedek7, A Mendonça8, O Hrusak1, JJM van Dongen2 and A Orfao3
1DPH/O, Prague, Czech Republic; 2Erasmus MC, Rotterdam, The Netherlands; 3IBMCC-CSIC-USAL, USAL, Salamanca, Spain; 4UNIKIEL, Kiel, Germany; 5UNIVLEEDS, Leeds, UK; 6AP-HP, Paris, France; 7SUM, Zabrze, Poland and 8IPOLFG, Lisbon, Portugal
Background
Flow cytometers are relatively flexible instruments that allow simultaneous measurement of the light scatter properties of different types of cells and the fluorescence emissions of distinct fluorophores attached to them.31 Because of their flexibility, adequate setting of instrument conditions, including fine tuning of the light scatter and fluorescence detectors, is required prior to a specific measurement, in order to establish the optimal window of analysis. An additional goal within the EuroFlow project was to define SOPs to establish standardized instrument settings that would allow reproducible (identical or at least highly comparable) measurements at different times in the same instrument or in different instruments at the same or at distinct sites through the application of predefined scatter and MFI values for specific reference particles. In general, with such SOPs, all particles that will be measured should fall in the previously defined window of analysis for the light scatter and each fluorescence detector.
The EuroFlow light scatter settings aim at reaching two goals: (i) all populations of interest (from small erythroblasts to eosinophils and plasma cells) fall centered within the scale limits and (ii) adequate scatter resolution between individual cell populations is obtained, for both cell surface and intracellular staining procedures. Lymphocytes were chosen as an internal biological reference population to control for adequate placement of instrument light scatter settings.
The EuroFlow setting of photomultiplier tube (PMT) voltages for a fluorescence detector is established at a voltage above the electronic noise in such a way that the least autofluorescent cell type to be measured is placed at the left side of the scale, as ‘negative' events clearly distinguishable from debris in the multidimensional space generated, dim fluorescent events can be discriminated from the negative, and no cell- or bead-associated fluorescence measurement reaches the upper limit of the scale.32 Each PMT is characterized by a response of accuracy to PMT voltage measured, as the robust coefficient of variation (rCV) of a dim particle. Optimal PMT voltage is set at the beginning of the plateau of a rCV versus PMT voltage curve.32 In this way, the electronic noise contribution to the signal is minimal whereas maximal dynamic range is left for the measurement of fluorescence. At the time of writing, Cytometer Setup and Tracking (CS&T) beads (BD Biosciences, San Jose, CA, USA) and Cyto-Cal Multifluor Plus Violet Intensity Calibrator (Thermo Scientific, Freemont, CA, USA) are being evaluated by EuroFlow as potentially suitable additional calibrators for long-term, multi-center studies.
In this section we summarize the most critical and relevant steps included in the EuroFlow SOPs developed for optimal placement of instrument settings.
Instruments and reagents
FACSCanto II (BD Biosciences) flow cytometers were used in seven centers and both an LSR II (BD Biosciences) and a CyAn ADP (Dako, Glostrup, Denmark/Beckman Coulter, Brea, CA, USA) were used in another center. All cytometers were equipped with three lasers emitting at 405/407, 488 and 633/635 nm. Optical filter configurations were identical, with the exceptions described in Table 1. Eight-peak Rainbow bead calibration particles (Spherotech, Lake Forest, IL, USA) were used throughout the study for initial PMT characterization and for setting target MFI values, as well as for daily checks; the same master lot of beads (RCP-30-5A master lot X02) was used throughout the study.
Placement of PMT voltages for fluorescence measurements
To place PMT voltages, the following sequential steps were used: the Rainbow 8-peak bead population showing the second dimmest fluorescence was gated and the rCV of that peak was calculated in each fluorescence channel for PMT voltages ranging from 300 to 999 mV at increments of 50 mV.33 The optimal voltage for each channel was first determined on one instrument (LSR II) and set at the beginning of the plateau phase of the curve generated. Using the PMT value obtained in this way, the brightest peak was gated and its fluorescence intensity recorded in all channels and then used as preliminary ‘Target MFIs' for all other instruments. Subsequently, verification of PMT settings was performed on each individual EuroFlow instrument. For verification of the lower boundary, PMT settings were checked on the rCV versus PMT voltage curve, as described above for the reference instrument. For the ‘Target MFI' to be accepted, PMT voltage on each instrument had to be at the plateau of the curve for all nine instruments. Additionally, all bright markers from the EuroFlow antibody panels29 were tested in the corresponding channels of all instruments; if the target MFI setting resulted in suboptimal PMT setting on any instrument, the target MFI values were adjusted accordingly till consensus target MFI values assuring optimal PMT settings for each instrument were reached.
Placement of instrument settings for light scatter measurements
Fine tuning of scatter settings was based on usage of normal human PB lymphocytes. For this purpose, 50 μl of PB samples obtained from healthy donors (after informed consent was given) and measured within the first 24 h after venipuncture were used at each site. Prior to measurement, non-nucleated red cells were lysed (10 min) using 2 ml of 10X FACS Lysing Solution (BD Biosciences) and diluted 1/10 (vol/vol) in distilled water (dH2O), according to the recommendations of the manufacturer. Then, the sample was centrifuged (5 min at 540 g), the cell pellet was washed with 2 ml of phosphate buffer saline (PBS; pH=7.4) containing 0.5% (w/v) bovine serum albumin (BSA; SIGMA-ALDRICH, St Louis, MO, USA) and 0.09% of sodium azide (NaN3; SIGMA-ALDRICH), centrifuged again under the same conditions and finally resuspended in 250 μl of PBS with 0.5% BSA+0.09% NaN3, and measured in the flow cytometer at a ‘low' flow rate mode within the first hour after sample preparation. PMT voltages were adjusted so that forward scatter (FSC)/sideward scatter (SSC)-gated lymphocyte singlets reached mean SSC and FSC values of 55 000±5000 and 13 000±2000, respectively.
EuroFlow instrument settings
Final PMT voltages for each fluorescence channel were set for each instrument to reach target MFI values using the brightest peak of Rainbow 8-peak beads of the same lot. Subsequent rainbow bead lots were assigned new target MFI values by cross-calibration using the previous lot for an instrument in a single laboratory (DPH/O, Prague, Czech Republic) (Table 4, see also www.euroflow.org for the updated target MFI of other Rainbow bead lots). In turn, light scatter settings were placed as described above. Inclusion of the FSC-H parameter will allow discrimination of doublets in a FSC-Area (FSC-A) versus FSC-Height (FSC-H) bivariate plot, contributing further to the accuracy of the results.34 The final instrument settings for both light scatter and fluorescence-associated PMT voltages are further referred as EuroFlow settings. The detailed EuroFlow SOP for instrument setup is available at the EuroFlow website (www.euroflow.org).
Table 4. Target mean fluorescence intensity (MFI) values obtained after optimal PMT adjustments for each fluorescence channel for the brightest peak of Rainbow 8-peak calibration beads in the LSR II instrument.
| Fluorochrome channel |
MFI values Rainbow lot no. |
||
|---|---|---|---|
| X02, Y02 | Z02 | EAB01 | |
| PacB | 195 572 | 194 818 | 215 352 |
| PacO | 231 265 | 216 293 | 217 908 |
| FITC | 59 574 | 58 372 | 65 283 |
| PE | 101 900 | 98 520 | 84 847 |
| PerCPCy5.5 | 216 064 | 223 940 | 228 818 |
| PECy7 | 27 462 | 27 185 | 29 865 |
| APC | 176 780 | 226 435 | 252 000 |
| APCH7 | 56 437 | 81 371 | 102 099 |
Abbreviations: APC, allophycocyanin; Cy7, cyanin7; FITC, fluorescein isothiocyanate; H7, hilite7; PacB, pacific blue; PacO, pacific orange; PE, phycoerythrin; PerCPCy5.5, peridinin–chlorophyll–protein–cyanin5.5.
Monitoring of instrument performance
Monitoring of instrument performance was done daily (at each cold start) after laser stabilization was allowed for 30 min. Rainbow 8-peak beads were acquired under EuroFlow settings (under ‘disabled compensation' conditions) and the MFI of the brightest peak in each fluorescence channel was compared with the corresponding target MFI value. The following criteria had to be reached for the instrument to pass the check: (i) MFI values within the target MFI±15%, and (ii) coefficient of variation (CV) of the brightest peak <4% for the blue and violet laser channels, but <6% for the red laser channels and the PECy7 channel. Whenever instrument performance failed, measures such as thorough cleaning, de-gassing flow cell and laser delay verification were taken. When the performance was not restored to pass the monitoring criteria, a service visit was requested. After a service visit, PMT settings were adjusted as described above and a new compensation experiment was performed as described in Section 3 of this manuscript.
MFI values of the brightest Rainbow bead peak were daily reported for each individual flow cytometer. As the scaling of axes is different on FACSCanto II and LSR II (262 144 channels) as compared to CyAn ADP (4096 channels), the Rainbow beads data file was first converted to FCS 2.0 format and then read with the CyAn ADP's Summit software (Dako) to calculate the corresponding numerical values with the same distribution over the scale.
Automated baseline settings and instrument monitoring
When FACSDiVa V6.0 software with the CS&T module and BD CS&T beads (BD Biosciences) were introduced in 2008, baseline PMT settings were placed according to the manufacturers' instructions for the FACSCanto II and the LSR II instruments. Subsequently, PMT voltage settings were adjusted manually in the CS&T module, to create EuroFlow baseline settings. Electronic noise (SDEN) and rCV of the dimmest CS&T bead values obtained with the two baseline settings were compared for eight instruments (data of one representative instrument is shown in Figure 1).
Figure 1.

Comparison of cytometer setting & tracking (CS&T) module and EuroFlow baseline settings obtained for the fluorescein isothiocyanate (FITC) channel (blue laser line) in one representative instrument. CS&T (mid-fluorescence peak in a) and EuroFlow (Rainbow beads brightest, eighth peak in b) baseline settings are compared in c (gray and red vertical lines, respectively) for the robust coefficient of variation (CV) and robust electronic noise (SDEN). Note that although EuroFlow settings used lower PMT voltages, the robust CV values (orange line) and robust SDEN values (green line) are still in their plateau phases.
Instrument monitoring with the CS&T module was performed in parallel to the EuroFlow instrument performance-monitoring SOP on three different instruments (two FACSCanto II and one LSR II), for a 3-month period. To evaluate instrument performance, we calculated the CV of MFI values obtained for the brightest peak of 8-peak Rainbow particles.
Reproducibility of fluorescence intensity measurements with EuroFlow settings
The level of standardization of the EuroFlow settings was evaluated at two different time points, before standardization evaluation experiments were performed as described in Section 6. Results of such evaluation showed nearly identical MFI values for individual PMTs when their voltage was set to match the target MFI fluorescence channels listed in Table 4. In all eight instruments, the CV for the MFI values obtained for the brightest peak of Rainbow beads was systematically lower than 5.5% (Table 5).
Table 5. Variation of mean fluorescence intensity (MFI) values obtained for the brightest bead population of the Rainbow 8-peak beads in individual instruments placed in eight different EuroFlow centers (seven FACSCanto II and one LSR II flow cytometers).
| Fluorochrome-associated PMT detector | Target MFI | Mean MFIa of individual measurements (n=12) | CV |
|---|---|---|---|
| PMT 1—PacB | 195 572 | 193 109 | 5.40% |
| PMT 2—PacO | 231 265 | 225 152 | 4.63% |
| PMT 3—FITC | 59 574 | 59 003 | 2.08% |
| PMT 4—PE | 101 900 | 100 763 | 2.38% |
| PMT 5—PerCPCy5.5 | 216 064 | 215 596 | 2.11% |
| PMT 6—PECy7 | 27 462 | 27 639 | 3.13% |
| PMT 7—APC | 176 780 | 176 190 | 1.68% |
| PMT 8—APCH7 | 56 437 | 56 610 | 2.16% |
Abbreviations: APC, allophycocyanin; CV, coefficient of variation; Cy7, cyanin7; FITC, fluorescein isothiocyanate; H7, hilite7; PacB, pacific blue; PacO, pacific orange; PE, phycoerythrin; PerCPCy5.5, peridinin–chlorophyll–protein–cyanin5.5; PMT, photomultiplier tube.
Results are expressed as arbitrary MFI channel values scaled from 0 to 262 144.
Long-term evaluation of the MFI signal fluctuation with fixed PMT voltages revealed that in each of the eight instruments evaluated, changes of up to ±15% of the mean target MFI might transiently occur, whereas significant maintenance or hardware issues were highlighted by not meeting the above-described monitoring criteria, with deviations in these values (Figure 2).
Figure 2.

Overtime stability of Rainbow 8-peak bead mean fluorescence intensity (MFI) profile, illustrating the results obtained for three fluorescence channels: pacific blue (PacB) channel of the violet laser (blue dots); phycoerythrin (PE) channel of the red laser (yellow dots); and fluorescein isothiocyanate (FITC) channel of the green laser (green dots), for the same flow cytometer instrument upon long-term monitoring of MFI measurements for the brightest peak of the Rainbow 8-peak beads. As shown, faulty violet laser was recognized as a source for the decreased MFI values falling below 15% of the target MFI (boxes A and C). Acceptable±15% range for each channel are depicted by gray lines and a colored background. After a service visit and laser alignment, MFI values above 15% of the target MFI were detected (box B); thus, photomultiplier tube (PMT) voltages were adjusted at this time point manually (closed circles). Please note that by placement of instrument settings as per the cytometer setting & tracking (CS&T) module the PMT could be adjusted to correct for the violet laser failure (open circles) until the laser failed completely and was replaced.
Electronic noise level with EuroFlow settings
The SDEN level obtained with individual flow cytometers using EuroFlow settings was highly comparable to that obtained through the CS&T module (Figure 1), except for the PerCPCy5.5 channel (Table 6). Thus, it could be concluded that the EuroFlow approach for PMT settings yields high-quality data with no impairment of the quality of the results obtained, due to higher electronic noise over individual CS&T module baseline. On average, the EuroFlow approach set PMT voltages at lower levels (Table 6), which allows for slightly larger dynamic ranges for measurements on the detectors. Of note, the significantly higher SDEN value obtained for the PerCPCy5.5 channel was still well-fitted in the plateau phase of the voltage versus SDEN curve.
Table 6. PMT voltages and electronic noise (SDEN) obtained with the EuroFlow settings versus the CS&T module.
| Fluorochrome-associated PMT detector |
PMT voltage |
SDEN |
P-valuea | ||
|---|---|---|---|---|---|
| CS&T module settings | EuroFlow settings | CS&T module settings | EuroFlow settings | ||
| PMT 1—PacB | 431 (357–490) | 412 (360–460) | 24.1 (20–29.8) | 24 (20.6–29.1) | 0.92 |
| PMT 2—PacO | 509 (414–633) | 466 (395–581) | 25.2 (21.3–28.1) | 24.5 (20.2–27.3) | 0.08 |
| PMT 3—FITC | 483 (399–555) | 438 (375–518) | 28.2 (25.4–31.2) | 28.9 (26.2–29.7) | 0.98 |
| PMT 4—PE | 462 (411–501) | 395 (370–445) | 30.9 (18.1–33.6) | 31.1 (18.3–32.4) | 0.46 |
| PMT 5—PerCPCy5.5 | 543 (456–610) | 522 (440–591) | 28.1 (18.1–31.3) | 29.1 (18.2–32.9) | 0.03 |
| PMT 6—PECy7 | 624 (589–757) | 552 (539–707) | 29 (22.1–32.6) | 29.5 (20.7–31.8) | 0.49 |
| PMT 7—APC | 614 (543–687) | 576 (501–629) | 26 (16.8–28.9) | 25.9 (12.8–28.9) | 0.95 |
| PMT 8—APCH7 | 489 (435–662) | 524 (481–687) | 25.1 (17.5–36) | 26 (14.1–36.6) | 0.50 |
Abbreviations: APC, allophycocyanin; CS&T, Cytometer Setting & Tracking; Cy7, cyanin7; FITC, fluorescein isothiocyanate; H7, hilite7; PacB, pacific blue; PacO, pacific orange; PE, phycoerythrin; PerCPCy5.5, peridinin–chlorophyll–protein–cyanin5.5; PMT, photomultiplier tube. aTwo-tailed Student's T-test.
Results are expressed as mean (min–max) values.
Conclusion
The EuroFlow SOP was designed to establish and daily monitor standard instrument settings for a common bright signal placed at the same level in different flow cytometer instruments. Overall, our results show optimal performance at different sites and instruments (even from different manufacturers), with early alarms for changes in hardware components that may impact the results. At the same time, the EuroFlow SOP avoids performing full calibration of the instrument (including compensation) on a daily basis.
SECTION 3. Design and evaluation of EuroFlow standard operating procedure for establishing optimal compensation settings
T Kalina1, JG te Marvelde2, VHJ van der Velden2, J Flores-Montero3, D Thűrner1, S Böttcher4, M Cullen5, L Lhermitte6, L Sedek7, A Mendonça8, O Hrusak1, JJM van Dongen2 and A Orfao3
1DPH/O, Prague, Czech Republic; 2Erasmus MC, Rotterdam, The Netherlands; 3USAL, Salamanca, Spain; 4UNIKIEL, Kiel, Germany; 5UNIVLEEDS, Leeds, UK; 6AP-HP, Paris, France; 7SUM, Zabrze, Poland and 8IPOLFG, Lisbon, Portugal
Background
Most fluorochromes used in multicolor flow cytometry have relatively broad fluorescence emission spectra.7, 35 Therefore, measurement of their fluorescence emissions is typically not restricted to a single fluorescence channel but the emissions are also measured in detectors other than the primary channel for a particular fluorochrome (secondary fluorescence channels).7 Spectral overlap of light into secondary channels might lead to false-positive signals. However, the proportion of light spillover from the total fluorescence emission is constant for each fluorochrome, implying that this spillover can be mathematically calculated and subtracted.7 The term ‘fluorescence compensation' is typically used to describe this calculation and subtraction process. In general, the specific compensation values required depend on the spectral characteristics of the dyes, the optical bandpass filters and dichroic mirrors mounted in the flow cytometer, the intensity of the measured signal and the specific voltage used for the PMT where it is detected.7 In digital flow cytometers, fluorescence compensation is applied after data acquisition.36 Accurate calculation of the compensation values for a set of fluorochromes across multiple detectors is provided by the compensation tools available in conventional flow cytometry software once applied to data derived from the flow cytometric measurement of one or more sets of single fluorochrome-stained standards/controls.36 A full compensation matrix is calculated by the software based on each standard/control, and then it is applied to the measured data. A prerequisite to establish appropriate compensation settings is that the spectral characteristics of light emissions collected in individual channels for the standards/controls exactly match those of the dye(s) used in the experiment. Despite this, special attention should be paid to the fact that several currently used dyes are compound tandem dyes, where one fluorochrome serves as an acceptor of laser light energy and transfers this energy to the second dye of the tandem by fluorescence resonance energy transfer (FRET).7 Tandem dyes greatly enhance the Stoke's shift of the compound fluorochrome, but their manufacturing process may lead to non-uniform spectral characteristics of the tandems.7 Thus, tandem dyes (that is, PECy7, APCH7) present with variable spillover light to the donor dye channel depending on the proximity and amount of FRET acceptor dyes used;7 this frequently translates into the need for specific compensation controls/standards and settings for individual 8-color combinations containing different reagents conjugated to the same tandem dye.7 A second prerequisite for optimal compensation settings is that standards/controls must contain bright signals, so that the distance between the positive and negative subsets of events used to calculate fluorescence compensation values is as high as the maximum distance in the experimental samples to be measured. In practice, single reagent-stained cells or mouse immunoglobulin (Ig)-capture beads are used as compensation standards.37 It should be noted that compensation settings must be defined only after the PMT voltage is set for the experiment, because of its impact on fluorescence intensity and spillover into secondary channels.37
In this section we describe the procedures used to design and evaluate the compensation matrix required for routine use of the EuroFlow panels proposed for the different 8-color combinations of fluorochrome-conjugated antibodies, defined in the EuroFlow 8-color panels.29
Fluorescence compensation standards and controls
Specific subsets of PB leukocytes stained with fluorochrome-conjugated antibody reagents in single antibody-stained tubes (SAbST) were used as standards (Table 7) to establish the fluorescence compensation matrices to be applied to flow cytometric data measured using the 8-color EuroFlow panels for the diagnosis and classification of leukemias and lymphomas. SAbST were prepared as described in Section 4 for multiple single-stained aliquots of a normal PB sample showing negative to very bright expression of the stained reagents. In addition, reagent-specific SAbSTs for molecules not present on normal PB cells (for example, CD117 PECy7) were created using Ig-capture beads (CompBead, BD Biosciences) as specific standards for these specific reagents in the panel. Furthermore, normal and patient samples stained with the preliminary and final versions of the EuroFlow panels were used to confirm the utility of the calculated compensation matrices. The specific set of reagents used for fluorescence compensation purposes varied depending on the selected fluorochrome-conjugated antibodies at each round of evaluation of the EuroFlow panels, as described in van Dongen et al.29 Table 7 displays the set of markers used for the final version of the EuroFlow panels.29
Table 7. List of fluorochrome-conjugated antibodies used to set up fluorescence compensation matrices at individual centers.
|
Generic fluorochromes and tandem fluorochromes | |||||
|---|---|---|---|---|---|
|
Generic fluorochromes |
Tandem fluorochromes |
||||
| Generic targets | Positive target (bead or cell) populationa | PECy7 targets | Positive target (bead or cell) populationa | APCH7 targets | Positive target (bead or cell) populationa |
| CD20-PacB | B-cells | CD2-PECy7 | CD2+ T/NK-cells | CD3-APCH7 | T-cells |
| CD45-PacO | Lymphocytes | CD8-PECy7 | CD8hi T-cells | CD4-APCH7 | CD4+ T-cells |
| CD8-FITC | CD8hi T-cells | CD10-PECy7b | CompBead | CD8-APCH7 | CD8hi T-cells |
| CD8-PEc | CD8hi T-cells | CD16-PECy7 | NK-cells | CD9-APCH7b | CompBead |
| CD5-PerCPCy5.5d | CD5+ T-cells | CD19-PECy7 | B-cells | CD10-APCH7b | CompBead |
| CD8-APCc | CD8hi T-cells | CD45RA-PECy7 | CD45RA+ T-cells | CD14-APCH7e | Monocytes |
| CD45RO-PECy7 | CD45RO+ T-cells | CD19-APCH7 | B-cells | ||
| CD56-PECy7 | NK- and CD56+ T-cells | CD24-APCH7 | B-cells | ||
| CD117-PECy7b | CompBead | CD38-APCH7 | CD38hi Lymphocytes | ||
| HLADR-PECy7 | B- and HLADRhi T-cells | CD43-APCH7 | T-cells | ||
| CD49d-APCH7 | T-cells | ||||
| CD71-APCH7b | CompBead | ||||
| CD81-APCH7 | B-cells | ||||
| anti-λ−APCH7b | CompBead | ||||
Abbreviations: APC, allophycocyanin; Cy7, cyanin7; FITC, fluorescein isothiocyanate; H7, hilite7; PacB, pacific blue; PacO, pacific orange; PE, phycoerythrin; PerCPCy5.5, peridinin–chlorophyll–protein–cyanin5.5.
Unless otherwise indicated, the negative reference population used for each reagent was the lymphocytes from the ‘unstained' control tube. For more information about the specific clones used, please see van Dongen et al.29
‘Negative' CompBead used as negative reference population.
The CD8-PE and CD8-APC antibodies are not part of the EuroFlow antibody panels and might be used from any reliable source.
This tandem dye requires generic compensation;
Artificially CD14− monocyte population created by ‘appending' 5000 events from the unstained tube to this single antibody-stained tubes (SAbST) acquisition.
Fluorescence compensation setup
Compensation standards and controls were acquired with FACSDiVa software or Summit software using the software compensation tools. The setup containing the PMT voltage for each fluorescence channel and the compensation matrix calculated by the software was saved as ‘EuroFlow' Setup into the FACSDiVa Setup Catalog, or as ‘EuroFlow Protocol' in Summit. Templates were prepared for experiments and tubes labeled with the reagents' names beforehand, linked to the EuroFlow settings. Thus, reagent-specific compensation was applied accurately to the matching reagent labels, even when the compensation matrix was recalculated. In every center, compensation setup experiments were performed by default once a month. Whenever instrument monitoring failed and PMT voltages were reset to match target MFI values, the compensation setup experiment was repeated.
Comparison of fluorescence compensation matrices obtained at different days and at distinct centers
Compensation setup experiments showed that generic compensation matrices could be used for all antibody reagents in the EuroFlow panels conjugated with the PacB, PacO, FITC, PE and APC fluorochromes, as well as for the PerCPCy5.5 tandem fluorochrome (data not shown). In contrast, different values were required for both the PECy7 and APCH7 tandem fluorochromes, depending on the specific reagent conjugates used (Supplementary Table 1).
To evaluate and compare the fluorescence compensation settings established at different times in each center, compensation matrices were evaluated from 14 listmode data files in FCS 3.0 format, measured in seven centers (two per center); each of the two compensation matrices used per center had been established after a new compensation experiment (Table 8).
Table 8. Fluorescence compensation matrix values obtained from listmode data files (n=14) generated in 7 centers at two different time points for a total of 7 different flow cytometry instrumentsa.
|
Secondary fluorescence channel |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PacB | PacO | FITC | PE | PerCPCy5.5 | PECy7 | APC | APCH7 | |||
| Primary fluorescence channel | PacB | MIN | 24.3 | 0.0 | 0.0 | 0.0 | ||||
| MEDIAN | NA | 27.7 | 0.0 | NR | 0.0 | 0.0 | NR | NR | ||
| MAX | 31.0 | 0.2 | 0.6 | 0.1 | ||||||
| PacO | MIN | 1.9 | 0.2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| MEDIAN | 2.4 | NA | 0.4 | 0.2 | 0.3 | 0.0 | 0.0 | 0.0 | ||
| MAX | 2.9 | 0.5 | 0.3 | 0.5 | 0.1 | 0.3 | 0.5 | |||
| FITC | MIN | 0.0 | 4.8 | 10.0 | 3.0 | 0.2 | 0.0 | 0.0 | ||
| MEDIAN | 0.0 | 5.6 | NA | 12.0 | 3.5 | 0.3 | 0.0 | 0.0 | ||
| MAX | 0.1 | 6.4 | 16.0 | 4.0 | 0.5 | 0.2 | 0.2 | |||
| PE | MIN | 0.0 | 0.0 | 0.2 | 30.1 | 2.2 | 0.0 | |||
| MEDIAN | 0.0 | 0.1 | 1.3 | NA | 32.9 | 2.5 | 0.1 | NR | ||
| MAX | 0.1 | 0.3 | 1.7 | 38.9 | 2.8 | 0.1 | ||||
| PerCPCy5.5 | MIN | 0.0 | 0.0 | 0.0 | 0.0 | 12.5 | 1.6 | 1.0 | ||
| MEDIAN | 0.0 | 0.0 | 0.0 | 0.0 | NA | 16.5 | 2.4 | 5.5 | ||
| MAX | 0.9 | 0.9 | 0.2 | 0.1 | 18.8 | 3.7 | 8.0 | |||
| PECy7 | MIN | 0.0 | 0.0 | 0.0 | 0.2 | 0.6 | 0.0 | 3.2 | ||
| MEDIAN | 0.0 | 0.0 | 0.1 | 0.7 | 2.9 | NA | 0.0 | 6.8 | ||
| MAX | 0.4 | 0.6 | 0.5 | 13.1 | 5.7 | 0.9 | 9.1 | |||
| APC | MIN | 1.0 | 0.1 | 8.5 | ||||||
| MEDIAN | NR | NR | NR | NR | 1.2 | 0.1 | NA | 9.6 | ||
| MAX | 1.4 | 0.2 | 11.6 | |||||||
| APCH7 | MIN | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.3 | 1.3 | ||
| MEDIAN | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.5 | 1.8 | NA | ||
| MAX | 0.2 | 0.1 | 0.2 | 0.1 | 0.2 | 2.0 | 3.9 | |||
Abbreviations: APC, allophycocyanin; Cy7, cyanin7; FITC, fluorescein isothiocyanate; H7, hilite7; NA, not applicable; NR, compensation was never required; PacB, pacific blue; PacO, pacific orange, PE, phycoerythrin; PerCPCy5.5, peridinin–chlorophyll–protein–cyanin5.5.
Results are expressed as median percentage values and range. Median values are highlighted in bold.
Overall, compensation matrices were shown to be similar in all seven instruments evaluated (Table 8) and their variability among instruments was similar to that observed with time within each of the laboratories for individual instruments (P>0.05, paired Student's T-test). Although compensation requirements depend on the specific PMT voltage settings, overall, high spillover was detected for the PacB into the PacO channel and for PE into the PerCPCy5.5 channel. Furthermore, intermediate spillover was found between PerCPCy5.5 and PECy7, between FITC and PE, PECy7 and APCH7, and between APC and APCH7 detectors (Table 8). Compensation experiments performed 1 month apart yielded very similar compensation values (P>0.05; paired Student's T-test).
Conclusion
Fluorescence compensation setup procedures were designed to establish fluorescence compensation matrices for every individual 8-color combination of fluorochrome-conjugated reagents in the 8-color EuroFlow panels.29 The complexity of the procedure was higher than desired due to the need for different compensation values for reagents conjugated with the PECy7 and APCH7 fluorochrome tandems. Fortunately, the frequency of compensation could be set to a time interval of 1 month, during which only minor deviations from target MFI values were recorded on well-performing instruments, as assessed by routine (daily) monitoring of the standard instrument settings (see Section 2). Notably, highly stable compensation matrices were obtained at different times among all different EuroFlow laboratories with the proposed fluorescence compensation setup SOP. This suggests that in the future, software solutions for automated establishment of compensation matrices to experiments performed with adjusted PMT voltages to target MFI values may potentially be developed and implemented.
SECTION 4. Sample preparation and staining
VHJ van der Velden1, J Flores-Montero2, JG te Marvelde1, S Böttcher3, L Lhermitte4, AS Bedin4, J Almeida2, JJ Pérez5, M Cullen6, P Lucio7, E Mejstrikova8, T Szczepański9, T Kalina8, A Orfao2 and JJM van Dongen1
1Erasmus MC, Rotterdam, The Netherlands; 2USAL, Salamanca, Spain; 3UNIKIEL, Kiel, Germany; 4AP-HP, Paris, France; 5HUS, Salamanca, Spain; 6UNIVLEEDS, Leeds, UK; 7IPOLFG, Lisbon, Portugal; 8DPH/O, Prague, Czech Republic and 9SUM, Zabrze, Poland
Background
At present multiple protocols and reagents are available for staining leukocytes.5, 26, 38, 39, 40, 41, 42 Most protocols include a staining step, one or more washing steps and an erythrocyte lysing step (whenever non-nucleated red cells are present in the sample), but for enumeration of leukocytes the washing step is frequently omitted.5 Erythrocytes can be lysed using ammonium chloride or other commercially available reagents, for example, FACS Lysing Solution, QuickLysis (Cytognos SL, Salamanca, Spain) and VersaLyse (Beckman Coulter).5 For staining of intracellular proteins (for example, cytoplasmic (Cy)CD3, CyMPO, nuclear (Nu)TdT) the leukocytes need to be fixed and permeabilized as well.38, 42 For this purpose, several reagents, such as BD Perm/Wash buffer (BD Biosciences), Fix&Perm (AN DER GRUB Bio Research GmbH, Vienna, Austria), IntraStain (Dako) and IntraPrep (Beckman Coulter), are commercially available. Cell samples other than BM and PB, such as LN biopsies, CSF, pleural effusion fluid and vitreous humor, may need extra steps prior to the staining procedure.43 For example, CSF samples need to be collected in tubes with special medium in order to prevent substantial cell loss26 and LN biopsies need to be cut into small pieces and homogenized.41
The choice of procedure and reagents applied to stain leukocytes depends on the aim of the experiment, but generally the best procedure should fulfill the following criteria: (a) low CVs on FSC and SSC; (b) large differences in mean channel values for FSC and SSC between major leukocyte populations; (c) minimal cell loss; (d) preservation of fluorochrome brightness; (e) no impact on the stability of tandem fluorochromes; (f) low background staining; (g) minimal inter-laboratory variation; and (h) easy and fast performance. Taking this into account, the EuroFlow Consortium has evaluated several procedures for the staining of samples suspected of containing neoplastic hematopoietic cells.
Cell samples
The EuroFlow antibody panels29 are designed for diagnosis and classification of all major hematological malignancies. Although most EuroFlow antibody panels are primarily designed for evaluation of BM and/or PB samples, other samples, for example, pleural effusions and fine-needle aspirates, can be used as well. The preferred patient materials for these panels are discussed elsewhere.29
Erythrocyte lysing and staining procedures evaluated
Overall, four different erythrocyte lysing solutions (ammonium chloride, FACS Lysing Solution, QuickLysis and VersaLyse) were evaluated to assess which best fulfilled the above-listed criteria. Reagents were evaluated in all eight EuroFlow centers on PB samples obtained from 30 healthy donors, who gave their informed consent to participate in the study. Three different tubes were stained for each lysing solution: (1) CD4-PacB, CD8-AmCyan, CD45-FITC, CD19-PE and CD14-APC (all from BD Biosciences); (2) CD4-PerCPCy5.5, CD19-PECy7 and CD8-APCH7 (all from BD Biosciences) and (3) CD19-PECy7 (from Beckman Coulter). Briefly, 50 μl of PB was incubated (15 min in darkness) with the antibodies in a final volume of 100 μl. Subsequently, the lysing solution was added to the tube according to the instructions of the manufacturers and incubated for 10 min at room temperature in darkness. After centrifugation (5 min at 540 g), the supernatant was discarded and the cell pellet resuspended in 2 ml PBS+0.5% BSA. After another centrifugation step (5 min at 540 g), the supernatant was discarded and the cell pellet resuspended in 250 μl PBS+0.5% BSA. For tube 1, 50 μl of PerfectCOUNT beads (Cytognos SL) was added immediately prior to the acquisition in the flow cytometer. All samples were acquired in a flow cytometer at four different time points (0, 1, 3 and 24 h after staining) and data about 100 000 events per tube were recorded and stored. Stained samples were stored at 4 °C till acquisition at the 1-, 3- and 24-h time points.
Data recorded for tube 1 included: (a) qualitative comparison of the separation obtained among major leukocyte populations; (b) mean FSC and SSC channel and CVs detected for eosinophils, neutrophils, monocytes and total lymphocytes; (c) absolute number of eosinophils, neutrophils, monocytes, CD19+ B-cells, CD4+ T-cells and CD8hi T-cells; (d) MFI and CV values observed for CD45 (for each cell population) and for CD19, CD4, CD8 and CD14 for CD19+ B-cells, CD4+ T-cells, CD8hi T-cells and CD14hi monocytes, respectively. Data recorded for the other two monoclonal Ab combinations (tubes 2 and 3) included MFI and CVs of positive cells in the specific channel, MFI and CVs of negative cells in the same channel, and, for the tandem fluorochromes, the fluorescence signals (MFI values) in all other channels than the primary fluorochrome-specific one.
Overall, three different staining procedures were evaluated: stain-lyse-wash (SLW), stain-lyse-wash-fix (SLWF) and stain-lyse-no wash (SLNW). The SLW procedure is described above; for the SLWF procedure the final cell pellet was resuspended in PBS containing 0.5% paraformaldehyde instead of PBS+0.5% BSA. For the SLNW procedure, sample preparation ended after incubation (10 min) with the lysing solution without any further washing step.
Qualitative comparison of the scatter characteristics of the major PB cell populations for the four erythrocyte lysing solutions evaluated showed that FACS Lysing Solution and ammonium chloride yielded the best discrimination among them, independently of the staining procedure used. Furthermore, comparison between the three staining procedures tested showed that CVs for both FSC and SSC were lower and more homogeneous with the SLNW method, except when the FACS Lysing Solution was used, which improved the FSC and SSC CVs with the washing step (Figure 3).
Figure 3.

Illustrating example of the differences in the light scatter characteristics of the major subsets of peripheral blood leukocytes observed for the distinct lysing solutions and staining protocols. Please note the significant reduction in the light scatter CV for the different leukocyte populations observed with FACS Lysing Solution and a SLW protocol (red square). Events shown in the upper-left corner of each dot plot correspond to PerfectCOUNT beads (Cytognos SL) introduced for the evaluation of cell loss. SLW, stain-lyse-wash; SLWF, stain-lyse-wash-fix; SLNW, stain-lyse-no wash.
In general, the SLNW resulted in the highest cell numbers, whereas specific loss of lymphocytes (Figure 4a) and lymphocyte subsets (Figure 4b) was observed with the SLW and SLWF procedures. However, cell loss was significantly lower when FACS Lysing Solution was used (versus all other lysing reagents) (Figure 4).
Figure 4.

Comparison of the absolute cell counts of major leukocyte populations (a) and lymphocyte subsets (b) obtained with the four different lysing solutions (FACS Lysing Solution, Ammonium Chloride, QuickLysis and VersaLyse Lysing Solution) evaluated in combination with the three different staining procedures (SLNW, SLW, SLWF) tested. Results are shown as mean values (open circles) and 95% confidence intervals (vertical lines). FACS Lyse, FACS Lysing Solution; NH4Cl, ammonium chloride; VersaLyse, VersaLyse Lysing Solution. SLW, stain-lyse-wash; SLWF, stain-lyse-wash-fix; SLNW, stain-lyse-no wash.
Subsequently, we evaluated the effect of the different lysing solutions and staining procedures on the fluorescence intensities. Both the washing step and the final fixation step induced some decrease in the MFI of all antibodies evaluated. Overall, FACS Lysing Solution generally resulted in the highest MFI values (Figure 5). There were no clear differences in MFI values or spillover of fluorescence emissions into secondary channels (MFI of ‘non-specific' channels) between the four different lysing solutions tested.
Figure 5.

Comparison of the mean fluorescence intensity (MFI) values of six fluorochrome-conjugated antibodies obtained with the four different lysing solutions evaluated in combination with the three different staining procedures (SLNW, SLW, SLWF) tested. CD45-fluorescein isothiocyanate (FITC) was evaluated on total peripheral blood (PB) lymphocytes, CD14-allophycocyanin (APC) was evaluated on PB monocytes, CD4-peridinin chlorophyll protein cyanin5.5 (PerCPCy5.5) was evaluated on PB CD4+ T-lymphocytes, CD8-APC hilite7 (H7) on PB CD8hi T-lymphocytes and the two CD19-phycoerythrin cyanin 7 (PECy7) reagents were both evaluated on PB CD19+ B-lymphocytes. Results are shown as mean values (open circles) and 95% confidence intervals (vertical lines). FACS Lyse, FACS Lysing Solution; NH4Cl, ammonium chloride; VersaLyse, VersaLyse Lysing Solution. SLW, stain-lyse-wash; SLWF, stain-lyse-wash-fix; SLNW, stain-lyse-no wash.
Based on the data derived from the performance of the four different lysing reagents and the different sample preparation protocols, it was decided to use a stain-lyse-wash procedure with FACS Lysing Solution for all cell surface membrane (Sm) labelings. The detailed protocols recommended are shown in Table 9. As displayed there, due to the presence of Igs in plasma, membrane stainings for Ig chains (for example, Igκ, Igλ and Igμ) required washing steps prior to antibody incubation. Based on experience, practical feasibility and additional testing (data not shown), it was agreed to include NaN3 (at a concentration of 0.09%) in all washing solutions and to ensure that all immunostainings including SmIgs were preceded by two washing steps with 10 ml PBS+0.5% BSA (Table 9). The latter procedure resulted in maximal SmIg staining intensities (data not shown).
Table 9. Detailed EuroFlow Standard Operating Procedures (SOPs) for sample preparation and staining.
| A. Common initial procedure when the EuroFlow antibody panel includes SmIg staining |
| If the EuroFlow antibody panel is going to be applied to a sample that includes SmIg staining, follow these initial steps; otherwise go directly to the backbone, surface or intracellular staining protocols (sections B, C, D, respectively): |
| 1. Pipette 300 μl of sample into a 10-ml tube (see Note 1). Note 1: For small samples (i.e. CSF, vitreous aspirates) spin down the total volume (5 min at 540 g), discard the supernatant (see point 5) and resuspend in 300 μl of PBS+0.5% of bovine serum albumin (BSA)+0.09% sodium azide (NaN3). 2. Add 10 ml filtered PBS+0.5% BSA+0.09% NaN3 3. Mix well 4. Centrifuge for 5 min at 540 g 5. Discard the supernatant using a Pasteur pipette or vacuum system without disturbing the cell pellet 6. Add 10 ml PBS+0.5% of BSA+0.09% NaN3 to the cell pellet 7. Mix well 8. Centrifuge for 5 min at 540 g 9. Discard the supernatant using a Pasteur pipette or vacuum system without disturbing the cell pellet 10. Resuspend the cell pellet in 200 μl of PBS+0.5% BSA+0.09% NaN3 11. Continue with conventional EuroFlow SOPs for staining of cell surface or cell surface plus intracellular markers as described below in procedures B, C and D, respectively |
| B. Staining of backbone markers |
| 1. Calculate the total volume of surface membrane backbone antibodies based on the number of tubes in the panel (see Note 2). Note 2: Intracellular backbone markers should not be added here. 2. Pipette these antibodies in one tube (backbone tube) 3. Calculate the total volume of sample to be stained, also based on the number of tubes in the panel and a volume of 50 μl per tube 4. Pipette this sample volume into the backbone tube 5. Mix well 6. Pipette equal amounts of the sample/backbone mix into the various tubes included in the applied EuroFlow panel (see Note 3). Note 3: Both the volume pipetted into each tube and the overall number of tubes depends on the specific EuroFlow panel that is applied. 7. Continue with the steps described below in procedure C |
| C. Staining of surface markers only (see Note 4): |
| Note 4: PCD tube 2 is processed identically to PCD tube 1 as described in section D if CD138-PacO is used. |
| 1. Add the appropriate volume of antibodies directed against cell surface markers (except for the backbone markers), as recommended for each specific EuroFlow panel 2. If necessary, use PBS+0.5% BSA+0.09% NaN3 to reach a final volume of 100 μl per tube (see information on the EuroFlow panels) 3. Mix well 4. Incubate for 15 min at room temperature (RT) protected from light 5. Add 2 ml of 1x FACS Lysing Solution (10x FACS Lysing Solution diluted 1/10 vol/vol in distilled water (dH2O)) 6. Mix well 7. Incubate for 10 min at RT protected from light 8. Centrifuge for 5 min at 540 g 9. Discard the supernatant using a Pasteur pipette or vacuum system without disturbing the cell pellet, leaving approximately 50 μl residual volume in each tube 10. Add 2 ml of PBS+0.5% BSA+0.09% NaN3 to the cell pellet 11. Mix well 12. Centrifuge for 5 min at 540 g 13. Discard the supernatant using a Pasteur pipette or vacuum system without disturbing the cell pellet, leaving approximately 50 μl residual volume in each tube 14. Resuspend the cell pellet in 200 μl PBS+0.5% BSA+0.09% NaN3 15. Acquire the cells after staining or (if not immediately acquired) store at 4 °C maximally for 3 h until measured in the flow cytometer |
| D. Combined staining of intracellular and surface membrane markers (see Note 5): |
| Note 5: Tube 4 of the AML/MDS panel should be stained/processed further as described in Procedure E |
| 1. Add the appropriate volumes of antibodies for cell surface markers, as recommended for each specific EuroFlow panel 2. If necessary, use PBS+0.5% BSA+0.09% NaN3 to reach a volume of 100 μl per tube (see information on the EuroFlow panels) 3. Mix well 4. Incubate for 15 min at RT protected from light 5. Add 2 ml of PBS+0.5% BSA+0.09% NaN3 to the cell pellet 6. Mix well 7. Centrifuge for 5 min at 540 g 8. Discard the supernatant using a Pasteur pipette or vacuum system without disturbing the cell pellet, leaving approximately 50 μl residual volume in each tube 9. Resuspend the cell pellet by mixing gently 10. Add 100 μl of Reagent A (fixative; Fix&Perm, An der Grub, Vienna, Austria) 11. Incubate for 15 min at RT protected from light 12. Add 2 ml of PBS+0.5% BSA+0.09% NaN3 to the cell pellet 13. Mix well 14. Centrifuge for 5 min at 540 g 15. Discard the supernatant using a Pasteur pipette or vacuum system without disturbing the cell pellet, leaving approximately 50 μl residual volume in each tube 16. Resuspend the cell pellet by mixing gently 17. Add 100 μl of Reagent B (permeabilizing solution; Fix&Perm) 18. Mix well 19. Add the appropriate volume of the intracellular antibodies (see EuroFlow panels) 20. Mix well 21. Incubate for 15 min at RT protected from light 22. Add 2 ml of PBS+0.5% BSA+0.09% NaN3 to the cell pellet 23. Mix well 24. Centrifuge for 5 min at 540 g 25. Discard the supernatant using a Pasteur pipette or vacuum system without disturbing the cell pellet, leaving approximately 50 μl residual volume in each tube 26. Resuspend the cell pellet in 200 μl PBS+0.5% BSA+0.09% NaN3 27. Acquire the cells after staining or (if not immediately acquired) store at 4 °C maximally for 3 h until measured in the flow cytometer. |
| E. Nuclear (Nu)TdT staining (Tube 4 AML/MDS EuroFlow panel): |
|
1. Continued from procedure C step 13
2. Add the appropriate amount of the TdT antibody to the cell pellet
3. Mix well
4. Incubate for 15 min at RT protected from light
5. Add 2 ml of PBS+BSA 0.5%+0.09% NaN3 to the cell pellet
6. Mix well
7. Centrifuge for 5 min at 540 g
8. Resuspend the cell pellet in 200 μl PBS+BSA 0.5%+0.09% NaN3
9. Acquire the cells after staining or (if not immediately acquired) store at 4 °C maximally for 3 h until measured in the flow cytometer. |
| Overview of protocol Sections for the various EuroFlow antibody panels and corresponding tubes. |
| Antibody panel | Tube(s) |
Protocol procedure |
||||
|---|---|---|---|---|---|---|
| A | B | C | D | E | ||
| ALOT | 1 | X | ||||
| BCP-ALL | 1,4 | X | X | X | ||
| 2,3 | X | X | X | |||
| T-ALL | 1–4 | X | X | |||
| AML/MDS | 1–3, 5–7 | X | X | |||
| 4 | X | X | X | |||
| LST | 1 | X | X | |||
| SST | 1 | X | X | |||
| PCD | 1–2 | X | X | |||
| B-CLPD | 1–4 | X | X | X | ||
| T-CLPD | 1,2,4,6 | X | X | |||
| 3,5 | X | X | ||||
| NK-CLPD | 1,2 | X | X | |||
| 3 | X | X | ||||
Abbreviations: ALOT, acute leukemia orientation tube; AML/MDS, acute myeloid leukemia and myelodysplastic syndrome; BCP-ALL, B-cell precursor acute lymphoblastic leukemia; CLPD, chronic lymphoproliferative disorder; LST, lymphoid screening tube; PCD, plasma cell disorders; SST, small sample tube; T-ALL, T-cell acute lymphoblastic leukemia.
Intracellular stainings
For the staining of intracellular antigens, special procedures are needed to permeabilize and fix the cells.38, 42 On the basis of the extensive experience of the EuroFlow laboratories, the Fix&Perm reagents were selected for this purpose; no additional comparison with other commercially available reagents was performed. The detailed protocols are shown in Table 9.
Although the Fix&Perm reagents work well for NuTdT staining, it was decided that within the acute myeloid leukemia (AML)/myelodysplastic syndrome (MDS) protocol, staining of NuTdT will be done using FACS Lysing Solution, based on the performance previously reported,38 because all tubes can then be treated in a similar way and additional effects on the light scatter characteristics of leukocytes (which could potentially hamper their use as common parameters to every stained aliquot) are avoided. This was not applied to staining of NuTdT in the BCP-ALL and T-ALL panels,29 because in such cases additional stainings for other intracellular markers were required (that is, CyIgμ, CyTCRβ and CyCD3), for which Fix&Perm reagents already was shown to be of utility.38, 42
To ensure similar staining intensities of the backbone markers in all tubes (for both membrane and intracellular stainings), all antibodies were titrated for a total volume (antibodies and sample) of 100 μl in every tube. If this volume was not reached, PBS+0.5% BSA+0.09% NaN3 was added to increase the volume to 100 μl. In some EuroFlow tubes, the total volume exceeded 100 μl. This was accepted as long as the total volume remained below 115 μl, as such minor deviations had no impact on the staining intensities of the backbone markers (data not shown).
Processing of cell samples with low nucleated cell counts
As described above, the sample preparation protocols and the different lysing solutions tested here were evaluated for the staining of whole BM and PB samples. However, in some patients the cell count may be rather low. This occurs, for example, in a substantial number of pediatric MDS patients and certainly will occur in samples obtained during therapy. We therefore evaluated whether it was possible to perform bulk lysis of erythrocytes with ammonium chloride prior to the EuroFlow protocol, to increase considerably the concentration of nucleated cells in the sample. Initially, within the AML/MDS panel,29 slight differences were observed for CD16, CD11b and CD15, but after titration of antibodies, fluorescence emissions were highly comparable between both procedures (Figure 6). Therefore, bulk lysis may be used prior to antibody staining when nucleated cell concentration needs to be increased, such as for the AML/MDS EuroFlow panel.29 As low cell counts less likely occur in other hematological diseases at diagnosis, prior bulk lysis was not specifically tested for these protocols.
Figure 6.

Parameter band plot of all individual parameters evaluated in a bone marrow sample from an MDS patient treated according to the EuroFlow protocol with (light colors) or without (dark colors) prior bulk lysis. Colored circles represent median scatter and fluorescence intensity (MFI) values obtained for the lymphocytes (dark green/light green), monocytes (red/orange) and neutrophils (dark blue/light blue).
Sample acquisition in the flow cytometer
As the time between staining of the samples and data acquisition in the flow cytometer may have an impact on the MFI of individual markers (particularly of those detected by reagents containing tandem fluorochromes), we acquired the samples immediately after staining, as well as 1, 3 and 24 h after sample preparation was completed. Our results show that MFI generally decreased over time, particularly when lysing solutions that did not contain fixative (that is, ammonium chloride) were used (Figure 7a). The most stable results were obtained with FACS Lysing Solution combined with either the SLNW or the SLW procedures (Figure 7b). Data became somewhat more variable when acquired 3 h and particularly 24 h after staining (Figures 7a and b).
Figure 7.

Effect of time between completion of staining and data acquisition in the flow cytometer (0 h, 1 h, 3 h and 24 h) and the sample preparation protocol on the mean fluorescence intensity (MFI) of CD19-phycoerythrin cyanin 7 (PECy7), CD4-peridinin chlorophyll protein cyanin5.5 (PerCPCy5.5) and CD8-allophycocyanin hilite7 (APCH7) on peripheral blood (PB) B-cells, CD4+ T-cells and CD8hi T-cells, using ammonium chloride (a) or FACS Lysing Solution (b) as lysing reagents. Three different sample preparation protocols were evaluated: SLNW; SLW and SLWF. Results are shown as mean values (open circles) and 95% confidence intervals (vertical lines). FACS Lyse, FACS Lysing Solution; NH4Cl, ammonium chloride. SLW, stain-lyse-wash; SLWF, stain-lyse-wash-fix; SLNW, stain-lyse-no wash.
On the basis of the results reported above, it was agreed that all samples should preferably be acquired within 1 h after completing the staining procedure. If not measured immediately, they should be stored at 4 °C in the darkness. Samples should be acquired on flow cytometers that have been set up according to the EuroFlow SOPs as described in Sections 2 and 3. For the EuroFlow screening and orientation tubes (acute leukemia orientation tube (ALOT), lymphoid screening tube (LST), small sample tube (SST) and plasma cell dyscrasia (PCD)),29 a minimum of 50 000 cells (typically 100 000) should be acquired in order to reach sufficient sensitivity for recognition of abnormal populations.
Conclusion
The EuroFlow protocols for sample preparation and staining were designed based on previous experience and experimental data available in the literature together with the results of specific experiments performed by the EuroFlow Consortium. Based on the combined results, the EuroFlow Consortium favors the use of a SLW procedure with FACS Lysing Solution for cell surface antigens, where measurements are performed shortly (<1 h) after sample preparation is completed. Special situations were envisaged for the staining of SmIgs, intracellular markers and samples with low nucleated cell counts, where introduction of additional washing steps, a fixation/permeabilization step and bulk lysis prior to staining, respectively, are recommended. The EuroFlow sample preparation and staining protocols described here are designed to be used together with EuroFlow SOPs for instrument setup (Section 2) and fluorescence compensation (Section 3) for the selected fluorochromes (Section 1). The proposed sample preparation and staining protocols perfectly fit with the EuroFlow antibody panels designed for the diagnosis and classification of hematological malignancies29 when using the most common types of samples, such as PB and BM. Specific issues related to other types of samples that have peculiar features and require unique sample preparation protocols (for example, CSF) are addressed in the EuroFlow antibody panel report.29
SECTION 5. EuroFlow strategies and tools for data analysis
M Martín-Ayuso1, ES Costa2, CE Pedreira3, Q Lécrevisse4, J Hernández1, L Lhermitte5, S Böttcher6, JJM van Dongen7 and A Orfao4
1Cytognos SL, Salamanca, Spain; 2Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil; 3Engineering Graduate Program, Electrical Engineering Program (COPPE PEE) and Faculty of Medicine (FM), Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil; 4USAL, Salamanca, Spain; 5AP-HP, Paris, France; 6UNIKIEL, Kiel, Germany and 7Erasmus MC, Rotterdam, The Netherlands
Background
Even though we have seen considerable improvements of clinical flow cytometry over the last years, the multicolor capabilities of currently available flow cytometers are still far behind the requested needs in routine clinical diagnostic laboratories. For example, the current immunophenotypic diagnosis of distinct WHO categories of hematological malignancies frequently requires the assessment of ∼30 different markers on neoplastic cells, which cannot be routinely studied on the same cell, owing to technical limitations.44, 45, 46, 47 In order to overcome these technical limitations, multiple aliquots of a sample are stained with different combinations of markers.47 In this approach, a few markers aim at the reproducible definition of the cell population(s) of interest; the so-called backbone markers are repeatedly used in every aliquot of the same sample and combined with other sets of markers, which together aim at the detailed immunophenotypic characterization of the cell population(s) of interest.47
Despite their clear benefits, these advances in multiparameter flow cytometry have led to a significantly increased complexity of data analysis and data interpretation because of the higher number of parameters simultaneously assessed in greater numbers of individual cells, and the expanded number of variables that might have an impact on the quality of the results.44, 45, 46, 47 Moreover, these technical improvements have not been paralleled (or followed) by innovations of data analysis and interpretation tools in the software packages routinely used in hematology laboratories. This lack of innovation has further contributed to the increased complexity of immunophenotyping of hematological malignancies.44, 45 In recent years, the EuroFlow Consortium has proposed several new data analysis tools48, 49, 50 aimed at decreasing such complexity through the development of new and more objective data analysis and interpretation strategies.48, 49, 50, 51 These novel tools have been progressively incorporated into the Infinicyt software (Cytognos SL) developed by the EuroFlow Consortium.
In this section we describe the new data analysis strategy proposed by the EuroFlow Consortium to be used in combination with the EuroFlow antibody panels and the EuroFlow SOPs for multiparameter immunophenotypic diagnosis and classification of hematological disorders.
Merge of flow cytometry data files and calculation of 'missing values‘
The EuroFlow antibody panels are composed of multiple 8-color combinations of antibodies that contain three or four fluorochrome-conjugated antibodies as common backbone markers, essential for gating the cells of interest in every aliquot of a sample stained with a specific EuroFlow antibody panel.29 The Merge function (first step in Figure 8) was used to fuse different data files corresponding to distinct aliquots of the same sample, each stained with a unique combination of reagents from the EuroFlow antibody panels. This results in a new single merged data file that contains all information measured in the same sample.52, 53 Such data file consists of a data matrix (Figure 9) in which the information (the measured parameters) for each different cellular event evaluated is aligned in one column per tube, which includes light scatter- and fluorescence emission-associated parameters, placed in different rows of the data matrix. The data matrix contains filled and unfilled boxes, corresponding to parameters that were directly measured and parameters not evaluated directly (missing values) for an individual event in a given aliquot of the sample, respectively48 (Figure 9).
Figure 8.

Flow chart diagram illustrating the sequential steps used during data analysis for the evaluation of the performance of the EuroFlow antibody panels.
Figure 9.
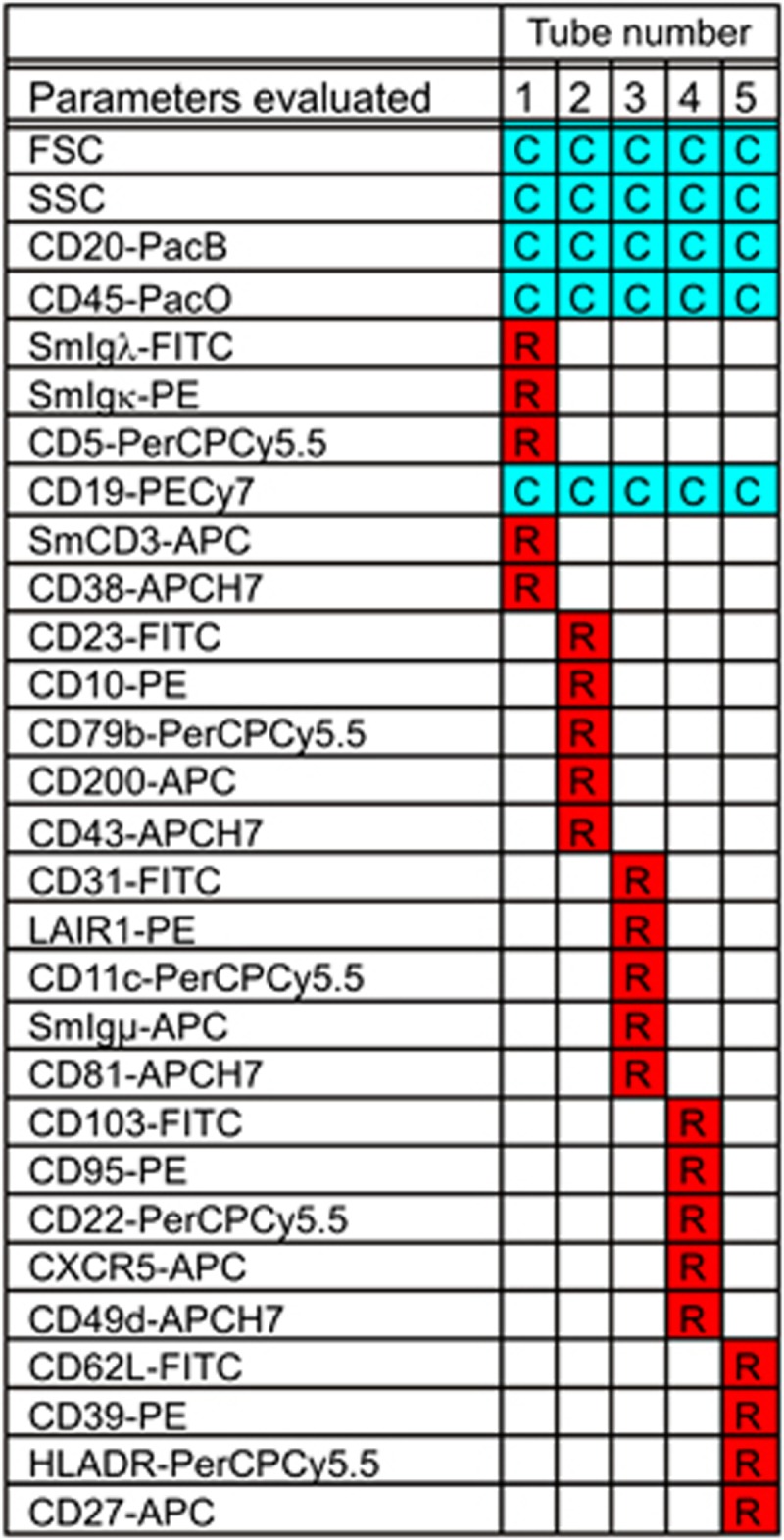
Data matrix obtained from the EuroFlow B-CLPD (B-cell chronic lymphoproliferative disorders) antibody panel, showing merging of five original data files into a single data file containing data about 29 parameters (2 scatter parameters and 27 markers). Columns correspond to the different B-CLPD tubes (sample aliquots) measured and rows correspond to the different parameters evaluated. ‘C' means ‘common' marker defined as measured in all aliquots; ‘R' means ‘real' data measured in any of the tubes. Blank spaces represent the parameter information that was not measured on an individual aliquot of the sample.
A calculation function was used to fill in the ‘missing values' in the above-mentioned data matrix corresponding to the merged data file. For this purpose, the common backbone parameters were used for identification of the neoplastic and/or normal cell populations of interest present in each of all tubes coming from a single sample (second step in Figure 8). Afterwards, a new data file was created, which only contained information about those parameters measured for individual events contained in the gated cell population.48 Then, the ‘missing values' in the data matrix corresponding to the gated cell population were calculated (third step in Figure 8). For this latter purpose, for each event to be calculated inside the selected population, the software searches for the ‘nearest neighbor'54, 55 event in each of the other aliquots of the sample, based only on its unique position in the multidimensional space created by all common backbone parameters (Figure 10). Therefore, the ‘nearest neighbor' of each event to be calculated in a merged data file measured in one sample aliquot will be the event showing the shortest distance from it in the n-dimensional space generated by the same common parameters in another sample aliquot from the merged data file. Finally, the software applies the values obtained for the ‘nearest neighbor' for all those parameters measured for the event in the latter sample aliquot but not measured in the former sample aliquot. This calculation process is done for each individual event in the merged data file till the data set is completed. At the end of the calculation process, the new data file contains both the data that were actually measured in the flow cytometer for each event and the calculated data for those parameters not measured in the same group of events in the other aliquots.48 The calculation process requires optimal definition with maximum biological heterogeneity within the cell population to become apparent with the common parameters (for example, backbone parameters) for the cell population to be calculated; thus, backbone marker selection is crucial. In order to obtain a high accuracy of the calculation process, each event in the cell population of interest is required for its definition in the EuroFlow antibody panels, based on five or six backbone parameters (two scatter parameters and three to four fluorescence markers).
Figure 10.

Schematic representation of the data calculation process with the Infinicyt software based on the ‘nearest neighbor' principle. First, one event from a cell population (B-cells highlighted in red) in a is identified in a first data file (tube 2 of B-cell chronic lymphoproliferative disorders (B-CLPD) panel) based on the backbone markers; then the event corresponding to the nearest neighbor of this event is identified in the second data file (right; tube 5 of B-CLPD panel) as that event occupying the same (closest) position in a multidimensional space formed by the same backbone parameters (b). Third, through the data calculation process the values for those parameters that were only measured for the later event in the second data file (d) but not for the former event in the first data file are assigned to the said event in the first data file and vice versa (c). Finally, the calculation process is completed for all other events in the cell population of interest (red events). Through this approach, all events in the merged and calculated data file have information about each of the parameters measured in both tubes (e).
As previously described in this paper (see Section 4), EuroFlow antibody panels include both surface and intracellular stainings. Therefore, variations in the FSC/SSC values or in the fluorescence levels of the backbone markers may occur because of the different sample preparation procedures (Table 9). In order to allow the calculation process when cells are treated with different staining protocols, a harmonization procedure was developed56 and applied to those cell populations of interest, for all parameters measured in common in the different sample aliquots, which are prepared differently (Figure 11). Such harmonization process consists of the translation of a data matrix defined in a tube by a given set of parameters for a given cell population into a data matrix defined by the same parameters for the same cell population measured in another tube under different conditions (for example, surface versus surface plus intracellular stainings). Of note, this harmonization tool did not affect the calculation process, as similar results were obtained when we compared the calculated values in files that contained information about a sample for which some aliquots/tubes were submitted to intracellular staining procedures and others were treated for Sm staining only.
Figure 11.

Illustrating example of the impact of different sample preparation protocols on the immunophenotypic and light scatter features of lymphocytes from a normal peripheral blood (PB) sample (a) and blast cells from B-cell precursor acute lymphoblastic leukemia (BCP-ALL) (n=9; c) and how the harmonization process reduces such impact (b and d, respectively). In a and b, FSC versus SSC representation of duplicates of a sample stained with two different protocols (permeabilized versus non-permeabilized lymphocytes) is shown without (a) and with (b) data harmonization applied, respectively; in both a and b, green and violet populations correspond to non-permeabilized and permeabilized aliquots, respectively. In c and d, different BCP-ALL blast cell populations from nine different BCP-ALL patients each stained in five different aliquots with the BCP-ALL EuroFlow panel are displayed. Each population is represented as median values in a principal component (PC) 1 versus PC2 analysis diagram (automatic population separator (APS)1 view based on the discrimination obtained for the following parameters: FSC, SSC, CD19, CD34 and CD45), where paired duplicated samples are colored identically. In c samples contain both permeabilized and non-permeabilized aliquots within the panel and the harmonization process was applied for five patient samples (duplicates colored dark yellow, light green, dark violet, red and cyan) for which duplicates show a very close position in the APS1 view; conversely for the other pairs of duplicates (light yellow, dark green, violet, dark blue show greater differences between paired samples). In d, one group of duplicates was processed by permeabilizing all aliquots within the panel, while in the other group each sample contained permeabilized and non-permeabilized sample aliquots, with data harmonization being applied to the latter group; note that now all pairs of sample duplicates overlap, confirming that with data harmonization blast cell populations processed differently (permeabilized versus non-permeabilized) are highly comparable to those who underwent a uniform sample preparation protocol.
As an end result of the calculation procedure, all individual events from each of the original data files corresponding to different aliquots of the same sample contain information about each reagent/parameter included in the whole antibody panel. The overall number of parameters for which values can be assigned to each individual cellular event included in the new data file are virtually unlimited, and equals that of the number of parameters measured in the whole set of merged data files for a given number of stained aliquots of a sample. This allows visualization of previously ‘impossible' bivariate dot plots for individual events (for example, staining patterns for two reagents conjugated with the same fluorochrome) 48 (Figure 10).
Generation of reference data files
A reference data file is a data file constructed by merging two or more data files, each corresponding to a cell population measured in different samples with the same panel of reagents (fourth step in Figure 8). Hereby, the reference data file contains information about all parameters (measured or calculated) for each individual event of the targeted cell population.50 Reference data files may contain information about normal or neoplastic cell populations, which may be homogeneous or heterogeneous with regard to different parameters evaluated. The generation of the reference data files aims at building libraries of reference cases to be compared between each other or with a new case that has been stained with the same panel of reagents (fifth step in Figure 8). On the basis of the existence of different patterns of protein expression in normal versus neoplastic cells, as well as among different WHO disease entities, a library can be built, which contains all normal and aberrant patterns that represent each of the different normal and pathological cell populations studied with the different EuroFlow antibody panels. Such a library can be used for (1) further evaluation of the utility and performance of antibody panels and (2) pattern-guided prospective classification of new cases diagnosed in different individual laboratories, which use the same EuroFlow antibody panels and laboratory procedures.50
Evaluation of the EuroFlow antibody panels based on comparisons of groups of reference data files
The EuroFlow 8-color antibody panels for the diagnosis and classification of hematological malignancies are designed to answer specific clinical questions, which can be grouped into two general categories: (1) Is a given hematopoietic cell population normal or reactive/regenerating or abnormal/neoplastic? (2) When an abnormal/neoplastic cell population is identified, which WHO disease category does it belong to? In order to evaluate the utility and performance of the EuroFlow antibody panels, different groups of reference files that had been stained with the same antibody panels have been constructed. To answer the first question, reference data files from a normal/reactive cell population were compared with their neoplastic counterpart from one or multiple WHO disease entities in a multivariate 1 × 1 set of comparisons approach. To answer the second question, reference data files corresponding to the neoplastic cell population from multiple cases of a single WHO disease entity were compared against single or multiple reference data files corresponding to one or more WHO disease entities.
For such comparisons, multiple approaches such as principal component analysis (PCA) can be used with the corresponding multiple-dimensions (that is, bi- or tridimensional) graphical representations of, for example, Principal Component (PC) X versus PC Y, and PC X versus PC Y versus PC Z, respectively, using the Automatic Population Separator (APS) graphical representation of the Infinicyt software (Figure 12).
Figure 12.

Example of principal component (PC1 versus PC2) analysis (PCA; automatic population separator (APS)1 views) for comparison of a new sample—red circles (median values) and dots—with a library of cases (median values/case represented as circles) from three different reference groups, each being colored differently (green, cyan and violet circles). In the upper panels the unknown case is compared to each pair of reference groups and it only overlaps systematically with the dark blue cases (a–c). In the lower panels (d–f), the new sample is separately compared with each individual reference group, showing again a high degree of overlap with the dark blue reference cases (f). Contour lines in each panel correspond to one (inner line) and two (outer line) s.d.'s of the mean value of the corresponding group of reference cases.
On the basis of this APS representation, information about the separation between the two groups of reference data files is obtained through definition of median and/or mean±s.d. borders (Figure 12) together with information about the most informative (versus redundant) parameters.57 It also allows re-evaluation of a panel after excluding one or multiple markers to objectively evaluate the contribution of each marker. A similar approach can then be used to prospectively compare one new case against two different groups of reference data files. Through such comparison, information is obtained about whether new cases belong to one of the reference groups or whether they differ from the reference groups, for those markers which are relevant in such comparison.
Through such comparisons one can also easily and objectively identify the phenotypic differences and similarities between the cell populations compared in the different reference groups and the markers that account for them. In fact, it allows direct (multivariate) comparisons of one or more cell populations from a given sample with other (for example, reference) cell populations from a pool of ⩾2 different samples (Figure 12). In a certain way, this mimics what an expert follows in his mind when he compares the immunophenotypic profiles obtained with a given antibody panel in a sample with the profiles obtained for the same combinations of antibodies in another sample (or group of samples) composed of normal, reactive, activated, aberrant or malignant cells. For example, the APS comparison of normal with malignant B-cell precursors allows identification of the best combination of markers to distinguish between them and thereby define the most common aberrant phenotypes. In addition, APS comparison between malignant B-cells from patients with different B-cell chronic lymphoproliferative disorders (B-CLPD) defined according to the WHO 2008 classification1 allows identification of the most informative parameters for their differential diagnosis. Noteworthily, fully objective information is obtained through this approach about the specific contribution of each marker in the panel.49
Conclusion
During the last 6 years, the EuroFlow Consortium has built new approaches for data analysis, which provide a new objective strategy for evaluation of the performance and utility of individual markers and antibody panels. The newly proposed strategy for data analysis of samples stained with the EuroFlow antibody panels includes a set of sequential steps (Figure 8): merge of data files corresponding to individual samples stained with the EuroFlow antibody panels, calculation of missing values for the cell populations of interest, merge of data files for the reference groups and evaluation of antibody panels through multiple comparisons between different sets of reference cases stained with the same panel of reagents. This new analytical strategy also provides a pattern-guided approach for the immunophenotypic classification of normal and malignant cell populations.49, 50 The tools required to use the new analytical approach have been implemented into the Infinicyt software by the EuroFlow Consortium, which allows their usage in routine practice by any other group around the world. Herewith a full set of flow cytometry data analysis tools is provided to the flow cytometry field to help expert-based interpretation of highly complex multiparameter data sets. In combination with the reference data files generated, the new software tools also provide a robust and reliable method for data comparison between different diagnostic laboratories on a sample-by-sample basis. The robustness and reliability of this approach is also based on the use of antibody panels with sufficient and adequate backbone markers that have been selected for careful identification of the subset of cells of interest, which is essential for accurate calculation of missing data and their graphical representation. Accordingly, use of the EuroFlow antibody panels and the EuroFlow SOPs for sample preparation allows for (1) building databases with reference groups of well-defined WHO categories; (2) classification of a new case; (3) assuring internal and external quality control by any other user if the same tools and reference groups are used. Therefore, the new software tools contribute significantly to the standardization of flow cytometry data analysis.
SECTION 6. Results of multicenter measurements
T Kalina1, J Flores-Montero2, VHJ van der Velden3, S Böttcher4, M Cullen5, L Lhermitte6, L Sedek7, A Mendonça8, HK Wind3, JG te Marvelde3, E Mejstrikova1, O Hrusak1, JJM van Dongen3 and A Orfao2
1DPH/O, Prague, Czech Republic; 2USAL, Salamanca, Spain; 3Erasmus MC, Rotterdam, The Netherlands; 4UNIKIEL, Kiel, Germany; 5UNIVLEEDS, Leeds, UK; 6AP-HP, Paris, France; 7SUM, Zabrze, Poland and 8IPOLFG, Lisbon, Portugal
Background
In order to design and apply the EuroFlow antibody panels for the immunophenotypic diagnosis and classification of leukemias and lymphomas, SOPs were developed and evaluated as described in the previous sections. However, multicentric implementation of such antibody panels29 and SOPs would require further evaluation of the protocols at the multicentric level. For this purpose two different series of experiments could be envisaged: (1) staining of comparable samples with the same SOPs and antibody panels at multiple sites, and (2) staining of the same sample at different sites with the EuroFlow antibody panels and SOPs.
Results of the multi-step procedure to standardize EuroFlow setup of all instruments were evaluated on a set of PB samples following the two approaches. The variation observed in multi-center experiments may be caused by multiple factors. Among them, the most relevant ones include (1) the pattern of expression of the molecule investigated (that is, tight peaks for CD4 expression versus non-homogeneous expression of CD27 in T-cells); (2) stability of the fluorochrome and its emission spectra (that is, stable FITC emitting in green versus relatively less stable APCH7 and PECy7 tandem fluorochromes with emission in far red resulting in variation due to photon-counting statistics); and (3) affinity and thus titration profile of antibodies (that is, for some antibody clones pipetting errors may lead to changes in staining levels).
The present experiment was chosen to allow analysis of distinct populations defined by positive markers in every fluorescence channel and it was designed to mimic the performance of the antibody panels for the EuroFlow ‘small sample tube'.29 A complex, 8-color tube was chosen as testing tube to include correct compensation in the end-point of the test. All measurements were subjected to the previously described EuroFlow SOPs, including analysis of merged data files using the Infinicyt software. The main question of the presented experiment was whether biological differences between distinct cell subsets will be resolved well when all setup procedures described so far are used in eight different EuroFlow laboratories and when the merged data are analyzed by the same software tools.
Standardized instrument settings and SOP evaluation experiments
The PB of one donor was stabilized using TransFix reagent (Cytomark, Buckingham, UK) and distributed in 1-ml aliquots to the eight EuroFlow centers; in addition, PB samples were obtained (after informed consent) from 30 different healthy volunteers—that is, one PB sample distributed to all eight centers and 30 different PB samples analyzed at eight centers (three to four samples per center). Instrument setup, compensation and sample preparation were performed exactly as described in Sections 2, 3 and 4, respectively. Reagents used for staining were modified from one of the 8-color EuroFlow panels (that is, SST)29 as follows: CD20-PacB (eBiosciences, San Diego, CA, USA), CD45-PacO (Invitrogen, Carlsbad, CA, USA), CD8-FITC, CD27-PE (ExBio, Prague, Czech Republic), CD4-PerCPCy5.5, CD14-APC and CD3-APCH7 (all from BD Biosciences) and CD19-PECy7 (Beckman Coulter). After acquisition in the flow cytometers, data were exported as FCS 3.0 data files. At each center, the following cell subsets were gated: SSClo/CD45hi total lymphocytes, CD14+/CD45hi monocytes, CD20hi/CD19+ B-lymphocytes, and CD3+/CD27+ memory T-lymphocytes with both CD3+/CD4+ T-cells and CD3+/CD8hi T-cells. Then the MFI values obtained for individual markers were calculated and reported (Table 10 and Figure 13a). Subsequently, both MFI values and the original listmode data files were sent to one center (DPH/O, Prague, Czech Republic) for central analysis. Then, the CV of the MFI values obtained for each subset in each channel was calculated. In addition, listmode data files were merged with Infinicyt software (version 1.3), monocytes were gated as CD45hi/CD14+ cellular events and total lymphocytes were gated as FSClo/SSClo/CD45hi events and their subsets further defined as listed in Table 10. Next, the merged file was displayed in an APS view (PC1 versus PC2), where each subset was color-coded, and the median of each subset was depicted as a color-coded circle as illustrated in Figures 13b and c.
Table 10. Overall results of synchronized experiments expressed in terms of variability obtained in the 8 EuroFlow laboratories for the measurement of antigen expression profiles in normal PB monocytes and lymphocytes (n=30 different samples), stained, prepared and measured in all 8 centers in parallel versus a stabilized sample obtained in a single center, distributed and then stained, prepared and measured locally at each center.
| Channel | PacB | PacO | FITC | PE | PerCPCy5.5 | PECy7 | APC | APCH7 |
|---|---|---|---|---|---|---|---|---|
| Target MFI (Rainbow beads) | 195 572 | 231 265 | 59 574 | 101 900 | 216 064 | 27 462 | 176 780 | 56 437 |
| Mean actual MFI (Rainbow beads) | 193 109 | 225 152 | 59 003 | 100 763 | 215 596 | 27 639 | 176 190 | 56 610 |
| CV of Rainbow MFI | 5.4% | 4.6% | 2.1% | 2.4% | 2.1% | 3.1% | 1.7% | 2.2% |
| Antibody conjugate evaluated | CD20 | CD45 | CD8 | CD27 | CD4 | CD19 | CD14 | CD3 |
| Gating parameters and cell subset | CD20hi/CD19+ B-cells | CD45hi total lymphocytes | CD3+/CD8hi T-cells | CD3+/CD27+ memory T-cells | CD3+/CD4+ T-cells | CD20hi/CD19+ B-cells | CD45hi/CD14+ monocytes | CD3+ T-cells |
| MFI CV for the cell subset (n=8 for 1 stabilized sample) | 15.2% | 13.9% | 11.4% | 32.9% | 24.7% | 11.1% | 43.8% | 38.7% |
| MFI CV of the cell subset (n=30 samples) | 16.9% | 15.5% | 16.9% | 28.0% | 28.4% | 15.4% | 22.7% | 48.4% |
Abbreviations: APC, allophycocyanin; Cy7, cyanin7; CV, coefficient of variation; FITC, fluorescein isothiocyanate; MFI, mean fluorescence; H7, hilite7; PacB, pacific blue; PacO, pacific orange; PB, peripheral blood; PE, phycoerythrin; PerCPCy5.5, peridinin–chlorophyll–protein–cyanin5.5.
Figure 13.

Results of synchronized EuroFlow experiments performed on different centers and instruments. (a) Box plot representations of mean fluorescence intensity (MFI) values observed for all antigens evaluated in the eight gated subsets of peripheral blood (PB) monocytes and lymphocytes from 30 healthy donor PB samples. Results corresponding to a total of 30 merged data files are displayed. (b) Principal component (PC)1 versus PC2 view (automatic population separator (APS) 1 view) of individual cellular events of the cell populations depicted in a; the median values of each gated subset (circles) are color-coded as follows: B-cells, red; CD4+/CD27+ memory T-cells, light blue; CD4+/CD27− T-cells, dark blue; CD8hi/CD27+ memory T-cells, dark green; CD8hi/CD27− T-cells, light green; CD3+/CD4− and CD8− T-cells, violet; NK-cells, yellow and monocytes, orange. (c) APS1 view of a single stabilized peripheral blood sample measured in 8 different EuroFlow laboratories for illustration of intra-donor variability (color coding is the same as in b). In a, results are displayed as box plots, where the line in the middle represents median values, the upper and lower limits of the box represent the 75th and 25th percentiles, respectively, and the upper and lower ends of vertical lines represent the 95% confidence interval. In b, each population is represented as a circle surrounded by dots corresponding to median values of median expression for all immunophenotypic parameters measured and to individual cells, respectively.
Comparison of data obtained at each of the centers showed that instrument-related differences caused a CV of target MFI values of <5.5% (see Section 2 and Table 10). When a stabilized PB sample obtained at one center was stained, measured and analyzed manually at each of the eight centers, CVs for the MFI values of each cell population evaluated were systematically <44%. Similarly, a maximal CV of 44% for CD3-APCH7 on T-cells was observed for normal PB samples obtained, stained, measured and analyzed at each individual center. Notably, CVs below 17% were obtained for 4/8 fluorochrome-conjugated markers assessed in specific cell subsets. Merging all listmode data files, followed by gating on the different subsets of lymphocytes and monocytes showed that we were able to clearly distinguish clusters of PB events corresponding to the same cell subsets from samples drawn from different donors, stained at different centers and measured on different instruments (Figures 13a and b). This illustrates that biological differences are not hidden or affected by the technical variability. To test the feasibility of merged data analysis across flow cytometry platforms, we acquired the same tube (except for CD14-APC) on LSR II and CyAn ADP instruments. Both the conventional analysis of dot plots (Supplementary Figure 1A) and graphical analysis of the APS view (Supplementary Figure 1B) showed separation of major lymphocyte subsets.
Conclusion
Our collective experiments showed that standardized instrument settings, compensation procedures, staining protocols and data analysis in multi-institutional collaboration programs are feasible. Data variation resulting from hardware differences (optical elements might have different quality; in some channels different filters are used on LSR II and FACSCanto II instruments) or variation from other sources is negligible when compared to biological differences between cell types. However, initial training of local operators in the applied SOPs is strongly recommended.
Discussion
In constructing the EuroFlow antibody panels for the diagnosis and classification of leukemias and lymphomas using ⩾8-color flow cytometry, among all technical issues, selection of the most adequate and feasible combination of fluorochromes to be used in the available multicolor flow cytometers was a pre-requisite. Usage of an increasingly high number of fluorochromes is associated with an exponential increase in the amount of information obtained from a single combination of fluorochrome-conjugated antibodies. However, such multicolor/multi-marker approaches are associated with an increasing complexity and the need to select the most appropriate/optimal combinations of individual reagents, including compatible fluorochromes for which the required high-quality antibodies are commercially available. As stated above, several fluorochromes were pre-selected because of the extensive experience and proven utility of a high number of good-quality antibody conjugates and their match with the default optical configuration of the available instruments. In contrast, for other fluorochrome positions, extensive experimental comparisons among different fluorochrome conjugates were required. Finally, the combination of PacB, PacO, FITC, PE, PerCPCy5.5, PECy7, APC and APCH7 was selected. However, while the majority of these fluorochromes performed satisfactorily well, others still require improvement (for example, APCH7). Preliminary testing of several new alternative fluorochromes (e.g., HV450, HV500 and brilliant violet fluorochromes) show promising results, implying that they might be suited as replacements. However, for some fluorochrome positions, alternative fluorochromes are not available (or became available very recently) or they are just conjugated with a restricted number of CD markers, which limits their current applicability but also points to the need for further improvements.
The technical EuroFlow approach was designed to establish and monitor standard instrument settings to a common bright signal placed at the same level in different flow cytometer instruments. This implies the possibility that some variation might occur in the measurement of dim/negative signals owing to small differences between individual instruments. However, it assures that individual flow cytometers work above their detector's background noise. Slight differences in laser power output, sharpness of the optical filters' edges and other hardware-associated variables might account for such small deviations.32, 33 In fact, we detected higher MFI for unstained cells in the violet laser fluorescence channels at one occasion in one instrument. The violet laser of this instrument had to be replaced by the manufacturer owing to a low laser power delivered to the flow cell. In 2008 the CS&T module was introduced in the 6.0 version of the FACSDiVa software.58 Please note that this module is made for automated instrument characterization and automated calculation of optimal voltage settings for single instruments and does not deal with standardized multicenter setting approaches. With the CS&T module, PMT voltages are set at a value that is 10 times the standard deviation of electronic noise. Small differences were found between CS&T and EuroFlow settings owing to the different criteria for optimal setting of PMT voltages. Whereas CS&T favors higher sensitivity for dim signals, the EuroFlow settings are tailored to measure both dim signals and signals generated by molecules with very high levels of expression (for example, CD38 expression on normal plasma cells). In practice, both methods (the EuroFlow settings and the CS&T module) were associated with optimal PMT settings and allow for an early detection of instrument failure (for example, laser failure). Unfortunately, automated adjustments at daily monitoring by the CS&T module systematically require a full new compensation experiment, even if the variation obtained for the new adjustment is as low as ±1 mV with no real impact on the compensation matrix. Daily performance of a full compensation experiment is expensive, time-consuming and consequently inefficient in diagnostic laboratories. In addition, it is not supported by the minor changes observed in the compensation matrix. An additional advantage of the EuroFlow approach is its flexibility and its applicability for 8-color flow cytometers from different manufacturers. Based on the long-term stability of MFI measurements, once-fixed PMT voltages were used. We adopted acceptance criteria for deviations of up to 15% from the target MFI values, for instrument settings to pass during daily monitoring. A user-friendly software tool and graphics were built into the Infinicyt software for a quick color-code assessment of any deviation from the accepted criteria for optimal instrument settings. The stringency of such criteria should be driven by the purpose of standardization. In immunophenotyping of hematological malignancies, the biological intra- and inter-sample differences are quite high and they are not hidden or affected by changes in fluorescence intensity values of up to 30% (EuroFlow data; not shown). As described in Section 5 with new automated software-driven analytical approaches that simultaneously take into account all markers and their intensities at the same time, the relative relevance of small MFI changes in individual markers is significantly diminished. In addition, we also show that these criteria can be easily met by different instruments at different sites.
Usage of an optimal fluorescence compensation matrix is currently considered as a requirement for optimal identification of single- versus double-positive cells in multicolor flow cytometry immunophenotyping.37 The complexity of the procedure designed to set up the optimal fluorescence compensation matrix depends on the specific multicolor antibody panels. As could be predicted, single compound dyes were represented by one ‘generic' SAbST (one representative marker stained in a specific cell population), while tandem fluorochromes were represented by one tube for each specific fluorochrome conjugate antibody. The only exception to this rule was the PerCPCy5.5 tandem fluorochrome. However, it should be noted that, in contrast to PECy7 and APCH7, PerCPCy5.5 is a tandem fluorochrome where both compounds of the tandem show maximum emission into the same bandpass filter; this could explain why no fluorochrome-specific compensation is needed. The similar spillover values for different PerCPCy5.5 reagents were confirmed in a small-scale experiment (data not shown).
Fluorescence compensation experiments consisting of a full set of compensation controls (n=30 tubes) represent a challenge for time-stressed laboratories as well as a burden for laboratory budget. Thus, the frequency of compensation could be set to a time interval of 1 month, during which only minor deviations from target MFI were recorded on well-performing instruments. However, gradual 405-nm laser power failures often resulted in significant signal shifts that required new instrument setup and compensation experiments, more frequently than initially planned. Careful selection of reagents with sufficient life-span, especially with regard to tandem fluorochromes, and protection of light-sensitive reagents is crucial for acquisition of high-quality data in the once-per-month compensation scheme. Based on the comparison of the fluorescence compensation matrices obtained over time for the same instrument, we concluded that it is not necessary to repeat the compensation experiment whenever both the reagents and the signal collection on the instrument are stable. However, the stability of tandem fluorochromes is not reliably constant for all manufacturers and it depends on the storage and handling conditions. Our 1-month compensation approach was feasible as judged by evaluation of ⩾2000 merged PB, BM, LN, CSF and vitreous humor samples of multiple disease categories acquired over the past 6 years (data not shown). In turn, a software solution that would allow automated and rapid establishment of fluorescence compensation settings to experiments, after PMT voltages had been adjusted to ‘Target MFI', could be of great help for clinical flow cytometry laboratories. Interestingly, our multicenter results indicate that such an approach is apparently feasible, owing to the highly stable compensation settings observed in our study at both the inter- and intra-laboratory level.
The EuroFlow SOPs for sample preparation was developed because of its ability to provide robust and reliable data that meet all the criteria indicated above. In combination with the standardized EuroFlow SOPs to define instrument settings and fluorescence compensation, it allows generation of highly comparable and reproducible data for a single instrument and between different instruments within the same laboratory and between different laboratories. Such highly reproducible data are required not only for the comparison of data obtained within the different EuroFlow laboratories, but also for the construction of a database with immunophenotypic data from large numbers of patients suffering from the various subtypes of relevant hematological malignancies, which can be potentially used as a reference by any laboratory worldwide.
Until now, standardization of flow cytometry in hematological diagnostic processes remains a challenge and it is rarely achieved in a multi-institutional setting. There have been some attempts to standardize the analysis of minimal residual disease in multiple study groups9, 59, 60, 61 that restricted the standardization on the analytical stage by exchanging the listmode data files. Kraan et al.62 have presented general rules for cytometer setup in clinical settings using analog flow-cytometric systems with up to four colors, whereas Shankey et al.63 presented complete standardization of 4-color ZAP-70 investigation in three institutions. Our present study goes further beyond the so-far-reported multi-center studies and aims at standardization of the data to the level at which listmode files measured in all centers can be meta-analyzed by software tools specifically designed for this purpose. The whole process of cytometer settings, compensation settings, fluorochrome selection and antibody panel selection was re-evaluated and fully controlled. The need for such extensive standardization arises from the possibilities that are brought by three-laser ⩾8-color digital flow cytometers to measure increasingly detailed subsets in complex cellular samples. Cell definitions using >4 colors are thought to enhance the accuracy of rare cell detection such as used for minimal residual disease studies. Analysis of surface/cytoplasmic expression patterns on large cohorts of samples by computational tools is possible only when the input data are supplied in a fully standardized format. Sharing of knowledge and diagnosing rare diseases will be made possible by manual or computer-assisted analysis of data files acquired in multi-institutional cooperation. Here again, appropriate interpretation of the data is possible only when standardized instrument settings and controls are used, the quality of the data is ensured, and the performance of the antibody panels is evaluated and taken into account during analysis.
We conclude that the 6 years of extensive collaborative experiments and the analysis of hundreds of patients' samples in the EuroFlow centers have indeed provided innovative protocols, software tools and antibody panels for fully standardized diagnosis and classification of hematological malignancies.
Acknowledgments
We are grateful to Dr Jean-Luc Sanne of the European Commission for his support and monitoring of the EuroFlow project. We thank Marieke Comans-Bitter for graphic design of the figures and for her continuous support in the management of the EuroFlow Consortium, and Bibi van Bodegom, Caroline Linker, and Monique van Rossum for their secretarial support of the consortium activities. We are grateful to Ria Bloemberg and Gellof van Steenis for support in the financial management of the project funds. The research activities of the EuroFlow Consortium were supported by the European Commission (grant STREP EU-FP6, LSHB-CT-2006-018708, entitled ‘Flow cytometry for fast and sensitive diagnosis and follow-up of hematological malignancies') and the following national grants: Spanish Network of Cancer Research Centers (ISCIII RTICC-RD06/0020/0035-FEDER), FIS 08/90881 from the ‘Fondo de Investigación Sanitaria', Ministerio de Ciencia e Innovación (Madrid, Spain), SA016-A-09 from the Consejería de Educación, Junta de Castilla y León, Valladolid, Spain, and PIB2010BZ-00565 from the Dirección General de Cooperación Internacional y Relaciones Institucionales, Secretaría de Estado de Investigación, Ministerio de Ciencia e Innovación (Madrid, Spain). T Kalina, E Mejstrikova and O Hrusak were supported by the Czech Ministry of Education Grant No. MSM0021620813, Czech Ministry of Health grant NT/12425-4 and TK is supported as an ISAC Scholar by The International Society for Advancement of Cytometry.
The EuroFlow Consortium is an independent scientific consortium which aims at innovation and standardization of diagnostic flow cytometry. All acquired knowledge and experience will be shared with the scientific and diagnostic community after protection of the relevant Intellectual Property, for example by filing of patents. The involved patents are collectively owned by the EuroFlow Consortium and the revenues of the patents are exclusively used for EuroFlow Consortium activities, such as for covering (in part) the costs of the Consortium meetings, the EuroFlow Educational Workshops and the purchase of custom-made reagents for collective experiments. Cytognos is for-profit company developing the Infinicyt software.
Footnotes
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
Supplementary Material
References
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues4th edn.International Agency for Research on Cancer: Lyon; 2008. 439 pp. [Google Scholar]
- Davis BH, Holden JT, Bene MC, Borowitz MJ, Braylan RC, Cornfield D, et al. Bethesda International Consensus recommendations on the flow cytometric immunophenotypic analysis of hematolymphoid neoplasia: medical indications. Cytometry B Clin Cytom. 2007;72 (Suppl 1:S5–S13. doi: 10.1002/cyto.b.20365. [DOI] [PubMed] [Google Scholar]
- Wood BL, Arroz M, Barnett D, DiGiuseppe J, Greig B, Kussick SJ, et al. Bethesda International Consensus recommendations on the immunophenotypic analysis of hematolymphoid neoplasia by flow cytometry: optimal reagents and reporting for the flow cytometric diagnosis of hematopoietic neoplasia. Cytometry B Clin Cytom. 2007;72 (Suppl 1:S14–S22. doi: 10.1002/cyto.b.20363. [DOI] [PubMed] [Google Scholar]
- Stewart CC, Behm FG, Carey JL, Cornbleet J, Duque RE, Hudnall SD, et al. U.S.-Canadian Consensus recommendations on the immunophenotypic analysis of hematologic neoplasia by flow cytometry: selection of antibody combinations. Cytometry. 1997;30:231–235. doi: 10.1002/(sici)1097-0320(19971015)30:5<231::aid-cyto3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Stetler-Stevenson M, Ahmad E, Barnett D, Braylan R, DiGiuseppe J, Marti G, et al. Clinical Flow Cytometric Analysis of Neoplastic Hematolymphoid Cells. Approved guideline. 2nd edn. CLSI document H43-A2 ed. Clinical and Laboratory Standards Institute: Wayne, PA, 2007.
- Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4:648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- Baumgarth N, Roederer M. A practical approach to multicolor flow cytometry for immunophenotyping. J Immunol Methods. 2000;243:77–97. doi: 10.1016/s0022-1759(00)00229-5. [DOI] [PubMed] [Google Scholar]
- Craig FE, Foon KA. Flow cytometric immunophenotyping for hematologic neoplasms. Blood. 2008;111:3941–3967. doi: 10.1182/blood-2007-11-120535. [DOI] [PubMed] [Google Scholar]
- van de Loosdrecht AA, Alhan C, Bene MC, Della Porta MG, Drager AM, Feuillard J, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica. 2009;94:1124–1134. doi: 10.3324/haematol.2009.005801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawstron AC, Orfao A, Beksac M, Bezdickova L, Brooimans RA, Bumbea H, et al. Report of the European Myeloma Network on multiparametric flow cytometry in multiple myeloma and related disorders. Haematologica. 2008;93:431–438. doi: 10.3324/haematol.11080. [DOI] [PubMed] [Google Scholar]
- Ruiz-Arguelles A, Rivadeneyra-Espinoza L, Duque RE, Orfao A. Report on the second Latin American consensus conference for flow cytometric immunophenotyping of hematological malignancies. Cytometry B Clin Cytom. 2006;70:39–44. doi: 10.1002/cyto.b.20083. [DOI] [PubMed] [Google Scholar]
- Owens MA, Vall HG, Hurley AA, Wormsley SB. Validation and quality control of immunophenotyping in clinical flow cytometry. J Immunol Methods. 2000;243:33–50. doi: 10.1016/s0022-1759(00)00226-x. [DOI] [PubMed] [Google Scholar]
- Davis BH, Foucar K, Szczarkowski W, Ball E, Witzig T, Foon KA, et al. U.S.-Canadian Consensus recommendations on the immunophenotypic analysis of hematologic neoplasia by flow cytometry: medical indications. Cytometry. 1997;30:249–263. [PubMed] [Google Scholar]
- Bene MC, Nebe T, Bettelheim P, Buldini B, Bumbea H, Kern W, et al. Immunophenotyping of acute leukemia and lymphoproliferative disorders: a consensus proposal of the European LeukemiaNet Work Package 10. Leukemia. 2011;25:567–574. doi: 10.1038/leu.2010.312. [DOI] [PubMed] [Google Scholar]
- Mahnke YD, Roederer M. Optimizing a multicolor immunophenotyping assay. Clin Lab Med. 2007;27:469–485. doi: 10.1016/j.cll.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roederer M, Kantor AB, Parks DR, Herzenbergzzz LA. Cy7PE and Cy7APC: bright new probes for immunofluorescence. Cytometry. 1996;24:191–197. doi: 10.1002/(SICI)1097-0320(19960701)24:3<191::AID-CYTO1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, et al. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- Berlier JE, Rothe A, Buller G, Bradford J, Gray DR, Filanoski BJ, et al. Quantitative comparison of long-wavelength Alexa Fluor dyes to Cy dyes: fluorescence of the dyes and their bioconjugates. J Histochem Cytochem. 2003;51:1699–1712. doi: 10.1177/002215540305101214. [DOI] [PubMed] [Google Scholar]
- Telford W, Kapoor V, Jackson J, Burgess W, Buller G, Hawley T, et al. Violet laser diodes in flow cytometry: an update. Cytometry A. 2006;69:1153–1160. doi: 10.1002/cyto.a.20340. [DOI] [PubMed] [Google Scholar]
- Abrams B, Diwu Z, Guryev O, Aleshkov S, Hingorani R, Edinger M, et al. 3-Carboxy-6-chloro-7-hydroxycoumarin: a highly fluorescent, water-soluble violet-excitable dye for cell analysis. Anal Biochem. 2009;386:262–269. doi: 10.1016/j.ab.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Maecker H, Trotter J.Selecting Reagents for Multicolor Flow Cytometry. Application Note. BD Biosciences: San Jose, CA, 2009. [PubMed]
- Stewart CC, Stewart SJ. Four color compensation. Cytometry. 1999;38:161–175. doi: 10.1002/(sici)1097-0320(19990815)38:4<161::aid-cyto3>3.3.co;2-1. [DOI] [PubMed] [Google Scholar]
- Roederer M. Spectral compensation for flow cytometry: visualization artifacts, limitations, and caveats. Cytometry. 2001;45:194–205. doi: 10.1002/1097-0320(20011101)45:3<194::aid-cyto1163>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Frey T, Nomura LE, Trotter J. Selecting fluorochrome conjugates for maximum sensitivity. Cytometry A. 2004;62:169–173. doi: 10.1002/cyto.a.20092. [DOI] [PubMed] [Google Scholar]
- Wood B. 9-color and 10-color flow cytometry in the clinical laboratory. Arch Pathol Lab Med. 2006;130:680–690. doi: 10.5858/2006-130-680-CACFCI. [DOI] [PubMed] [Google Scholar]
- Kraan J, Gratama JW, Haioun C, Orfao A, Plonquet A, Porwit A, et al. Flow cytometric immunophenotyping of cerebrospinal fluid Curr Protoc Cytom 2008. Chapter 6: Unit 6 25. [DOI] [PMC free article] [PubMed]
- Basso G, Buldini B, De Zen L, Orfao A. New methodologic approaches for immunophenotyping acute leukemias. Haematologica. 2001;86:675–692. [PubMed] [Google Scholar]
- McLaughlin BE, Baumgarth N, Bigos M, Roederer M, De Rosa SC, Altman JD, et al. Nine-color flow cytometry for accurate measurement of T cell subsets and cytokine responses. Part I: Panel design by an empiric approach. Cytometry A. 2008;73:400–410. doi: 10.1002/cyto.a.20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen JJM, Lhermitte L, Böttcher S, Almeida J, van der Velden VHJ, Flores-Montero J, et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes Leukemia 2012. doi: 10.1038/leu.2012.120 [DOI] [PMC free article] [PubMed]
- Le Roy C, Varin-Blank N, Ajchenbaum-Cymbalista F, Letestu R. Flow cytometry APC-tandem dyes are degraded through a cell-dependent mechanism. Cytometry A. 2009;75:882–890. doi: 10.1002/cyto.a.20774. [DOI] [PubMed] [Google Scholar]
- Hulett HR, Bonner WA, Sweet RG, Herzenberg LA. Development and application of a rapid cell sorter. Clin Chem. 1973;19:813–816. [PubMed] [Google Scholar]
- Maecker HT, Trotter J. Flow cytometry controls, instrument setup, and the determination of positivity. Cytometry A. 2006;69:1037–1042. doi: 10.1002/cyto.a.20333. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, Ambrozak D, Nguyen R, Chattopadhyay P, Roederer M. Quality assurance for polychromatic flow cytometry. Nat Protoc. 2006;1:1522–1530. doi: 10.1038/nprot.2006.250. [DOI] [PubMed] [Google Scholar]
- Hughes OR, Stewart R, Dimmick I, Jones EA. A critical appraisal of factors affecting the accuracy of results obtained when using flow cytometry in stem cell investigations: where do you put your gates. Cytometry A. 2009;75:803–810. doi: 10.1002/cyto.a.20764. [DOI] [PubMed] [Google Scholar]
- Shapiro HM.Excitation and emission spectra of common dyes Curr Protoc Cytom 2004. Chapter 1: Unit 1 19. [DOI] [PubMed]
- Tung JW, Parks DR, Moore WA, Herzenberg LA. New approaches to fluorescence compensation and visualization of FACS data. Clin Immunol. 2004;110:277–283. doi: 10.1016/j.clim.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Roederer M.Compensation in flow cytometry Curr Protoc Cytom 200211Chapter 1: Unit 1 14. [DOI] [PubMed] [Google Scholar]
- Groeneveld K, te Marvelde JG, van den Beemd MW, Hooijkaas H, van Dongen JJ. Flow cytometric detection of intracellular antigens for immunophenotyping of normal and malignant leukocytes. Leukemia. 1996;10:1383–1389. [PubMed] [Google Scholar]
- Macey MG, McCarthy DA, Milne T, Cavenagh JD, Newland AC. Comparative study of five commercial reagents for preparing normal and leukaemic lymphocytes for immunophenotypic analysis by flow cytometry. Cytometry. 1999;38:153–160. doi: 10.1002/(sici)1097-0320(19990815)38:4<153::aid-cyto2>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Stewart CC, Stewart SJ.Immunophenotyping Curr Protoc Cytom 2001. Chapter 6: Unit 6 2. [DOI] [PubMed]
- Holmes K, Lantz LM, Fowlkes BJ, Schmid I, Giorgi JV.Preparation of cells and reagents for flow cytometry Curr Protoc Immunol 2001. Chapter 5: Unit 5 3. [DOI] [PubMed]
- Kappelmayer J, Gratama JW, Karaszi E, Menendez P, Ciudad J, Rivas R, et al. Flow cytometric detection of intracellular myeloperoxidase, CD3 and CD79a. Interaction between monoclonal antibody clones, fluorochromes and sample preparation protocols. J Immunol Methods. 2000;242:53–65. doi: 10.1016/s0022-1759(00)00220-9. [DOI] [PubMed] [Google Scholar]
- Quijano S, Lopez A, Sancho JM, Panizo C, Deben G, Castilla C, et al. Identification of leptomeningeal disease in aggressive B-cell non-Hodgkin's lymphoma: improved sensitivity of flow cytometry. J Clin Oncol. 2009;27:1462–1469. doi: 10.1200/JCO.2008.17.7089. [DOI] [PubMed] [Google Scholar]
- Owens MA, Loken MR.Flow Cytometry Principles for Clinical Laboratory Practice: Quality Assurance for Quantitative Immunophenotyping Wiley-Liss: New York; Chichester; 1995. xiii, 224 pp. [Google Scholar]
- Caraux A, Klein B, Paiva B, Bret C, Schmitz A, Fuhler GM, et al. Circulating human B and plasma cells. Age-associated changes in counts and detailed characterization of circulating normal CD138- and CD138+ plasma cells. Haematologica. 2010;95:1016–1020. doi: 10.3324/haematol.2009.018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braylan RC, Orfao A, Borowitz MJ, Davis BH. Optimal number of reagents required to evaluate hematolymphoid neoplasias: results of an international consensus meeting. Cytometry. 2001;46:23–27. doi: 10.1002/1097-0320(20010215)46:1<23::aid-cyto1033>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Sanchez ML, Almeida J, Vidriales B, Lopez-Berges MC, Garcia-Marcos MA, Moro MJ, et al. Incidence of phenotypic aberrations in a series of 467 patients with B chronic lymphoproliferative disorders: basis for the design of specific four-color stainings to be used for minimal residual disease investigation. Leukemia. 2002;16:1460–1469. doi: 10.1038/sj.leu.2402584. [DOI] [PubMed] [Google Scholar]
- Pedreira CE, Costa ES, Barrena S, Lecrevisse Q, Almeida J, van Dongen JJ, et al. Generation of flow cytometry data files with a potentially infinite number of dimensions. Cytometry A. 2008;73:834–846. doi: 10.1002/cyto.a.20608. [DOI] [PubMed] [Google Scholar]
- Pedreira CE, Costa ES, Almeida J, Fernandez C, Quijano S, Flores J, et al. A probabilistic approach for the evaluation of minimal residual disease by multiparameter flow cytometry in leukemic B-cell chronic lymphoproliferative disorders. Cytometry A. 2008;73A:1141–1150. doi: 10.1002/cyto.a.20638. [DOI] [PubMed] [Google Scholar]
- Costa ES, Pedreira CE, Barrena S, Lecrevisse Q, Flores J, Quijano S, et al. Automated pattern-guided principal component analysis vs expert-based immunophenotypic classification of B-cell chronic lymphoproliferative disorders: a step forward in the standardization of clinical immunophenotyping. Leukemia. 2010;24:1927–1933. doi: 10.1038/leu.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa ES, Arroyo ME, Pedreira CE, Garcia-Marcos MA, Tabernero MD, Almeida J, et al. A new automated flow cytometry data analysis approach for the diagnostic screening of neoplastic B-cell disorders in peripheral blood samples with absolute lymphocytosis. Leukemia. 2006;20:1221–1230. doi: 10.1038/sj.leu.2404241. [DOI] [PubMed] [Google Scholar]
- Robinson JP, Durack G, Kelley S. An innovation in flow cytometry data collection and analysis producing a correlated multiple sample analysis in a single file. Cytometry. 1991;12:82–90. doi: 10.1002/cyto.990120112. [DOI] [PubMed] [Google Scholar]
- Robinson JP, Ragheb K, Lawler G, Kelley S, Durack G. Rapid multivariate analysis and display of cross-reacting antibodies on human leukocytes. Cytometry. 1992;13:75–82. doi: 10.1002/cyto.990130112. [DOI] [PubMed] [Google Scholar]
- Cover TM, Hart PE. Nearest neighbour pattern clasification. IEEE Trans Inf Theory. 1967;IT-13:21–27. [Google Scholar]
- Duda RO, Hart PE, Stork DG, RO Duda.PC, scene aIn: Duda RO, Hart PE, Stork DG, (eds)Pattern Classification2nd ednWiley: New York; Chichester; 2001. 654 pp. [Google Scholar]
- Costa ES, Peres RT, Almeida J, Lecrevisse Q, Arroyo ME, Teodosio C, et al. Harmonization of light scatter and fluorescence flow cytometry profiles obtained after staining peripheral blood leucocytes for cell surface-only versus intracellular antigens with the Fix & Perm reagent. Cytometry B Clin Cytom. 2010;78:11–20. doi: 10.1002/cyto.b.20486. [DOI] [PubMed] [Google Scholar]
- Lugli E, Roederer M, Cossarizza A. Data analysis in flow cytometry: the future just started. Cytometry A. 2010;77:705–713. doi: 10.1002/cyto.a.20901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stall A.Qr and Br in BD FACSDiva v6 Software: Parameters for Characterizing Detector Performance. Application Note 23-10516-00. BD Biosciences: San Diego, CA, 2008.
- Bjorklund E, Matinlauri I, Tierens A, Axelsson S, Forestier E, Jacobsson S, et al. Quality control of flow cytometry data analysis for evaluation of minimal residual disease in bone marrow from acute leukemia patients during treatment. J Pediatr Hematol Oncol. 2009;31:406–415. doi: 10.1097/MPH.0b013e3181a1c0e8. [DOI] [PubMed] [Google Scholar]
- Dworzak MN, Gaipa G, Ratei R, Veltroni M, Schumich A, Maglia O, et al. Standardization of flow cytometric minimal residual disease evaluation in acute lymphoblastic leukemia: multicentric assessment is feasible. Cytometry B Clin Cytom. 2008;74:331–340. doi: 10.1002/cyto.b.20430. [DOI] [PubMed] [Google Scholar]
- Irving J, Jesson J, Virgo P, Case M, Minto L, Eyre L, et al. Establishment and validation of a standard protocol for the detection of minimal residual disease in B lineage childhood acute lymphoblastic leukemia by flow cytometry in a multi-center setting. Haematologica. 2009;94:870–874. doi: 10.3324/haematol.2008.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraan J, Gratama JW, Keeney M, D'Hautcourt JL. Setting up and calibration of a flow cytometer for multicolor immunophenotyping. J Biol Regul Homeost Agents. 2003;17:223–233. [PubMed] [Google Scholar]
- Shankey TV, Forman M, Scibelli P, Cobb J, Smith CM, Mills R, et al. An optimized whole blood method for flow cytometric measurement of ZAP-70 protein expression in chronic lymphocytic leukemia. Cytometry B Clin Cytom. 2006;70:259–269. doi: 10.1002/cyto.b.20135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.