

Abstract
The mast cell product, tryptase, has recently been implicated in fibrosis in the hypertensive heart. Tryptase has been shown to mediate non-cardiac fibroblast function via activation of protease activated receptor-2 and subsequent activation of the mitogen-activated protein kinase pathway, including extracellular signal-regulated kinase1/2. Therefore, we hypothesized that this pathway may be a mechanism leading to fibrosis in the hypertensive heart. Isolated adult cardiac fibroblasts were treated with tryptase, which induced activation of extracellular signal-regulated kinase1/2 via protease activated receptor-2. Blockade of protease activated receptor-2 with FSLLRY (10 μM) and inhibition of the extracellular signal-regulated kinase pathway with PD98059 (10 μM) prevented collagen synthesis in isolated cardiac fibroblasts stimulated with tryptase. p38 mitogen activated protein kinase and stress-activated protein/c-Jun N-terminal kinase were not activated by tryptase. Cardiac fibroblasts isolated from spontaneously hypertensive rats showed this same pattern of activation and treatment of spontaneously hypertensive rats with FSLLRY prevented fibrosis in these animals indicating the in vivo applicability of the cultured fibroblast findings. Also, tryptase induced a myofibroblastic phenotype indicated by elevations in α smooth muscle actin and ED-A fibronectin. Thus, the results from this study demonstrate the importance of tryptase for inducing a cardiac myofibroblastic phenotype, ultimately leading to the development of cardiac fibrosis through the activation of the extracellular signal-regulated kinase pathway. Specifically, tryptase causes cardiac fibroblasts to increase collagen synthesis via a mechanism involving activation of protease activated receptor-2 and subsequent induction of extracellular signal-regulated kinase signaling.
Keywords: Tryptase, Protease Activated Receptor, Mitogen-Activated Protein Kinase, heart, fibrosis, ED-A fibronectin, mast cell
INTRODUCTION
We recently presented evidence identifying the cardiac mast cell as central to the induction of fibrosis in the hypertensive rat heart.1 The mast cell product, tryptase, is associated with the pathogenesis of fibrosis in many tissues particularly that of the lung,2 kidney,3 and skin.4 We further identified that tryptase is elevated in the hypertensive heart and that tryptase stimulates collagen production and proliferation by isolated adult cardiac fibroblasts.1 However, the mechanism by which tryptase is able to elicit this response is unknown. Tryptase, as well as trypsin, are the major activators of protease activated receptor (PAR)-2, but do not activate PAR-1, -3, and -4.5 The PARs represent a novel group of G protein-coupled seven transmembranedomain receptors that are activated by serine proteases. These proteases cleave the N-terminal receptor sequence at 33SKGR↓SLIGRL42 sites, exposing the sequence of tethered ligand (SLIGRL for rat PAR-2). This tethered ligand binds to a site on the second extracellular loop of the receptor, triggering its autoactivation and subsequent signaling cascade events such as mitogen activated protein kinase (MAPK) phosphorelay, which includes extracellular signal-regulated kinase isoforms 1 and 2 (ERK1/2), p38 and stress activated protein kinase /c-Jun N-terminal kinases (SAPK/JNK).5 There are only a few descriptions of tryptase/PAR-2 signaling events in any type of fibroblast, with Frungieri et al. having reported that tryptase/PAR-2 signaling involves the activation of ERK1/2 in fetal foreskin fibroblasts.6
Accordingly, the present study sought to determine if tryptase activates cardiac fibroblasts via PAR-2, and identify whether specific MAPK pathways are involved. We found that tryptase acts via PAR-2 to induce ERK1/2, but not p38 or SAPK/JNK activation in isolated adult rat cardiac fibroblasts. Activation of this cascade led to transition to a myofibroblastic phenotype and increased secretion of collagen. This pathway was also found to be active in cardiac fibroblasts isolated from hypertensive rat hearts. This study is the first to establish PAR-2 activation in cardiac fibroblasts and the selective downstream activation of ERK1/2.
METHODS
Cardiac Fibroblast Isolation and Culture Studies
Cardiac fibroblasts were obtained from adult male Sprague Dawleys rats (8 weeks old, n=4), spontaneously hypertensive rats (SHR; 12 weeks old, n=4) and Wistar Kyoto rats (WKY; 12 weeks old, n=4). All studies conformed to the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of South Carolina Institution’s Animal Care and Use Committee. Anesthesia at the experimental end point was affected by sodium pentobarbital (50 mg/kg, IP). Briefly, hearts were minced and digested with Liberase 3 (Roche) and fibroblasts purified by selective attachment to plastic culture ware. Fibroblasts were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% neonatal bovine serum (NBS), and 5% fetal calf serum (FCS) with replacement of media every other day. All fibroblasts were used on the second passage to minimize changes in phenotype associated with culture.7 Prior to experimentation, one million fibroblasts were seeded on 100 mm plastic culture plates and allowed to adhere to the plate for 24 hours in DMEM with 10% NBS and 5% FCS. Next, cells were rinsed with Moscona’s Salt Solution and the media replaced with serum–free DMEM-F12 media for 24 hours. For the studies assessing collagen production, fibroblasts were either untreated, or treated with tryptase (1000 mU, Promega) dissolved in DMEM with 1.5% FBS for 24 or 72 hours. Additionally, tryptase treated fibroblast were pretreated for one hour with the PAR-2 peptide antagonist (FSLLRY, 10 μM, Peptides International) or the mitogen activated protein kinase kinase (MEK)1/2 inhibitor (PD98059, 10 μM, Cayman Chemical). PD98059 was used since MEK1/2 is immediately upstream of ERK1/2, thus, inhibiting its effects. For MAPK activation experiments, fibroblasts were incubated with tryptase, tryptase in the presence of FSLLRY or PD98059 for 10 minutes. At the completion of the experiments, fibroblasts and media were collected and snap frozen.
Collagen Production by Isolated Cardiac Fibroblasts
In response to experimental treatment, adult cardiac fibroblast production of collagen was determined by hydroxyproline analysis of media as described by Edwards et al.8 (Please see http://hyper.ahajournals.org)
Western Blot Analysis
Please see http://hyper.ahajournals.org
MAPK Activation
Please see http://hyper.ahajournals.org
Whole Animal Studies
Rats were housed under standard environmental conditions and maintained on commercial rat chow and tap water ad libitum. All studies conformed to the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of South Carolina Institution’s Animal Care and Use Committee. Anesthesia at the experimental end point was affected by sodium pentobarbital (50 mg/kg, IP). Twelve week-old SHR were randomly assigned to 2 groups: 1) untreated (n=6); and 2) those treated with the PAR-2 peptide antagonist, FSLLRY (10 μg/kg/day, Peptides International, IP; n=5), with the experimental endpoint being at 15 weeks of age. At twelve week of age, SHR are known to be hypertensive.9 This dosage of FSLLRY was based on previous work by Al-Ani et al.10 Untreated WKY rats served as age-matched controls. At the experimental end point, measurements of systolic, diastolic, and mean arterial pressures were made using a catheter attached to a pressure transducer, and inserted into the right carotid artery. Following sacrifice, the heart was removed and the atria and great vessels dissected away, with the left ventricle (LV) and right ventricle (RV) were separated and weighed. A transverse mid-section of the LV was placed in 4% paraformaldehyde for fixation. Lung weight was also obtained after the removal of the esophagus and trachea with the pleural surface blotted dry.
Analysis of Left Ventricular Collagen Volume Fraction
Paraffin-embedded blocks were prepared from midventricular sections of the LV as previously described.1 5 μm sections were stained with collagen specific picrosirius red (0.1% Sirius Red F3BA in picric acid). Photomicrographs of 20 random fields, avoiding perivascular collagen, were obtained using a Ziess Axiovert 200 fluorescent microscope with a 20X objective. Collagen volume fraction (CVF) was determined from these images using Image J and expressed as a percent of area. 11
Statistical Analysis
Data are expressed as mean ± SD or SEM as appropriate. The results were analyzed statistically by using the computer program SPSS software (Chicago, IL). Quantitative data were compared among the multiple groups by one-way ANOVA followed by Bonferroni post hoc testing. For two group comparisons, the Student’s t testing was used. Differences were considered to be significant when P<0.05.
RESULTS
Tryptase/PAR-2 Induces Selective MAPK Activation in Isolated Cardiac Fibroblasts
Knowing that tryptase induced collagen synthesis by isolated cardiac fibroblasts,1 we first sought to determine which specific MAPK pathways were activated by tryptase. There was a significant increase in the level of total ERK in isolated cardiac fibroblasts treated with tryptase. Nevertheless, the ratio of phosphorylated ERK (pERK) to total ERK significantly increased by 143% compared to control following stimulation with tryptase (P<0.01 vs control; Figure 1A). This ERK activation was induced by tryptase activation of PAR-2 since the PAR-2 antagonist FSLLRY prevented the increase in the ratio of pERK to total ERK (P<0.01 vs tryptase). Pretreatment with MEK1/2 inhibitor, PD98059, reduced phosphorylated ERK to values below the detectable limits of the ELISA, indicative of its effectiveness (<38.3 pg/ml, data not shown). For p38 and SAPK/JNK, there were no significant differences in the ratios of phosphorylated to total protein between the tryptase treated group and the controls (Figure 1B and C).
Figure 1.

Ratio of total to phosphorylated (A) ERK1/2, (B) p38, and (C) SAPK/JNK in isolated cardiac fibroblasts. Adult cardiac fibroblasts were untreated (control), treated with tryptase or treated with tryptase +FSLLRY (PAR-2 antagonist) for 10 minutes. All values are mean ±SEM, *P<0.05 vs Control; † P<0.05 vs Tryptase.
Isolated Cardiac Fibroblast PAR-2 Levels
The presence of PAR-2 was examined on isolated adult cardiac fibroblasts. PAR-2 was detected by western blot on untreated fibroblasts (Figure 2). PAR-2 levels increased above that of untreated cells following 72 hours, but not 24 hours of tryptase treatment. This was prevented by blockade of PAR-2.
Figure 2.

(A) Representative image of a western blot detecting PAR-2 on isolated adult cardiac fibroblasts. Ratio of PAR-2 to GAPDH on isolated cardiac fibroblasts following stimulation with tryptase for (B) 24 hours, and (C) 72 hours.
Collagen Production by Tryptase Treated Isolated Cardiac Fibroblasts
Isolated adult cardiac fibroblasts were treated with either tryptase, tryptase together with the PAR-2 antagonist FSLLRY, or tryptase with the MEK 1/2 inhibitor PD98059 for 24 and 72 hours and hydroxyproline concentration in the media measured as a marker for collagen (Figure 3). Tryptase significantly increased collagen production at both 24 and 72 hours time-points. PAR-2 blockade prevented collagen production at both time-points. The MEK1/2 inhibitor, PD98059, also prevented collagen production at both 24 and 72 hours.
Figure 3.
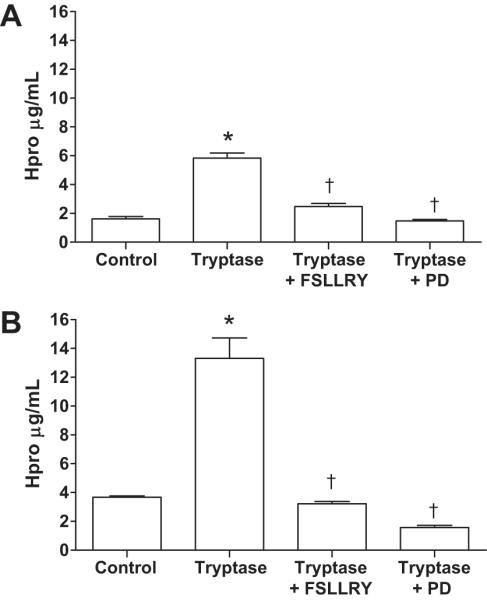
Hydroxyproline concentration in media from isolated adult cardiac fibroblast treated for 24 hr (A) and 72 hr (B). Adult cardiac fibroblast were treated with tryptase, tryptase + FSLLRY (PAR-2 antagonist), or Tryptase + PD (PD98059, MEK1/2 inhibitor). All values are mean ±SEM, *P<0.05 vs Control; † P<0.05 vs tryptase.
Isolated Cardiac Fibroblast α-Smooth Muscle Actin and ED-A Fibronectin Levels
Tryptase treatment induced a trend toward increased α-SMA compared to control, but this increase did not reach significance (P=0.067; Figure 4A and B). Pretreatment with FSLLRY or PD98059 prevented this trend. Tryptase treatment significantly increased ED-A fibronectin levels in adult cardiac fibroblasts (P<0.001 vs control). Pretreatment with FSLLRY or PD98059 prevented the induction of the myofibroblast marker, ED-A fibronectin, in adult cardiac fibroblast (P<0.001 for both groups vs Tryptase, Figure 4 C and D).
Figure 4.

(A) Representative western blot for α-smooth muscle actin in isolated adult cardiac fibroblasts. Adult cardiac fibroblasts were treated with either tryptase, tryptase + FSLLRY (PAR-2 antagonist), or tryptase + PD (PD98059, MEK1/2 inhibitor) for 24 hours. (B) Graphic representation of α-smooth muscle actin in adult cardiac fibroblasts treated with tryptase, tryptase+ FSLLRY or tryptase + PD. All values are mean ± SEM, *P<0.05 vs Tryptase. (C) Representative western blot for ED-A fibronectin in isolated adult cardiac fibroblasts treated as previous stated. (D) Graphic representation of ED-A fibronectin in isolated adult cardiac fibroblast treated as previously stated. All values are mean ± SEM, *P<0.05 vs Control; †P<0.05 vs Tryptase.
MAPK Activation in SHR Fibroblasts
To determine if the pattern of MAPK pathway activation was the same in fibroblasts from hypertensive hearts, fibroblasts were isolated from SHR and the three MAPK pathways examined. SHR fibroblasts had significant increases in the ratio of phosphorylated to total ERK compared to WKY fibroblasts (0.218 ±0.055 and 0.027±0.004, P=0.0013, respectively; Figure 5A). There were no differences in the ratio of phosphorylated to total protein of p38 and SAPK/JNK in the SHR compared to the WKY fibroblast (Figure 5B and C).
Figure 5.

Ratio of total to phosphorylated (A) ERK1/2, (B) p38, and (C) SAPK/JNK in cardiac fibroblasts isolated from SHR and WKY. All values are mean ±SEM, *P<0.05 vs WKY.
Whole Animals Studies
Having identified PAR-2 as a mechanism by which tryptase activates cardiac fibroblasts; we then sought to determine if this receptor did in fact play a role in fibrosis in vivo in the hypertensive heart. Table 1 contains the average values for the biometric measurements for WKY, SHR, and SHR+FSLLRY animals. While the LV weights and the LV to body weight ratio of the SHR and SHR + FSLLRY animals were significantly increased from that of the WKY, these two groups were not significantly different from each other. RV weights were not significantly different between any of the groups. SHR and SHR+FSLLRY had significant increases in systolic blood pressure, diastolic blood pressure, and mean arterial pressure compared to the WKY animals, but were not significantly different from each other. Collagen volume fraction was significantly increased in the SHR group compared with the WKY (Figure 6). Collagen volume fraction was normalized in the SHR+FSLLRY (P<0.05 compared to SHR).
Table 1.
Biometric Data.
| Groups | N | BW (g) |
LV (mg) |
LV/BW (mg/g) |
RV (g) |
RV/BW (mg/g) |
Lungs (mg) |
SBP (mmHg) |
DBP (mmHg) |
MAP (mmHg) |
|---|---|---|---|---|---|---|---|---|---|---|
| WKY | 6 | 322 ± 9 | 721.5 ± 35.4 | 2.24 ± 0.11 | 203.8 ± 21.8 | 0.63 ± 0.05 | 1149.5 ± 96.5 | 90 ± 11 | 62 ± 11 | 71 ± 11 |
| SHR | 6 | 310 ± 10 | 802.2 ± 42.2* | 2.59 ± 0.14* | 180.5 ± 9.8 | 0.58 ± 0.05 | 1164 ± 59.9 | 191 ± 23* | 153 ± 15* | 166 ± 17* |
| SHR+FSLLRY | 5 | 301 ± 7* | 792.4 ± 21.2* | 2.63 ± 0.06* | 179.2 ± 19.7 | 0.59 ± 0.05 | 1175 ± 59.5 | 173 ± 23* | 135 ± 17* | 148 ± 20* |
All values are mean ± standard deviation. BW, body weight; LV, left ventricle; RV, right ventricle; SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure.
= P<0.05 v WKY.
Figure 6.

(A) Representative images of picrosirius red-stained LV sections from WKY, SHR and SHR + FSLLRY. (B) Graphical representation of LV collagen volume fraction. All values are mean ± SEM, *P<0.05 vs WKY; †P<0.05 vs SHR.
DISCUSSION
We recently found that cardiac mast cells can induce fibrosis in the hypertensive heart, and that tryptase levels were increased in these hearts.1 We further demonstrated that tryptase was able to induce proliferation and collagen production by isolated adult cardiac fibroblasts. The stimulatory action of tryptase on fibroblast function is thought to be a PAR-2 mediated event;5 however, this had not been investigated in cardiac fibroblasts. The major finding of this study is that tryptase induces collagen production in cardiac fibroblasts via the activation of PAR-2 and subsequent selective induction of MAPKs.
PAR -1, -2, -3, and -4 are found in a variety of cells including many cell types throughout the cardiovascular system. PAR-1 has been shown to be expressed by cardiac fibroblasts and able to induce proliferation through the activation of ERK 1/2.12 However, while tryptase activates PAR-2, it is not a ligand for PAR-1, -3, and -4. To date, little is known about PAR-3 and PAR-4 activation in cardiac fibroblasts. This is the first study to examine PAR-2 activation in the cardiac fibroblast.
PAR-2 has been described in cardiac tissue; in fact, the expression of PAR-2 is present as early as E17 in mice and is constitutively expressed in the adult murine heart.13 Bohn et al. also reported PAR-2 transcript expression to be present in the adult human heart.14 While the location of this receptor was previously thought to be mainly on cardiomyocytes, we established herein that isolated adult cardiac fibroblasts also have PAR-2. We show that PAR-2 levels in isolated adult cardiac fibroblasts increase in the presence of tryptase; however, this increase may be due to proliferation of the cardiac fibroblasts caused by tryptase.1
Frungieri et al. reported that tryptase/PAR-2 activation, leading to phosphorylation of the MAPK ERK1/2, increased the fibrotic response in skin fibroblasts.6 However, it was unknown if tryptase activation of PAR-2 activates this same pathway in cardiac fibroblasts. The potential role of MAPKs in mediating tryptase-induced fibrosis in the heart had also previously been unexplored. MAPK signaling includes the activation of the serine/threonine kinases, ERK1/2, p38 isoforms, and SAPK/JNK, all of which have extensive homology to one another. Growth and differentiation factors that induce early activation of ERK do so by the sequential phosphorylation of the protein kinases Raf-1, MAP kinase kinase (MEK) 1 and 2 and ERK1/2 in most cells.15 We now show for the first time that there is an increase in ERK1/2 phosphorylation in isolated adult cardiac fibroblasts following treatment with tryptase. PAR-2 antagonism prevented ERK 1/2 phosphorylation and also prevented collagen production by cardiac fibroblasts. Furthermore, the MEK 1/2 inhibitor, PD98059 prevented collagen synthesis by cardiac fibroblasts stimulated with tryptase. This suggests that tryptase binding to PAR-2 receptor causes signaling events through ERK1/2 to elicit the fibrotic response, while p38 and SAPK/JNK do not appear to be involved (Figure 7).
Figure 7.
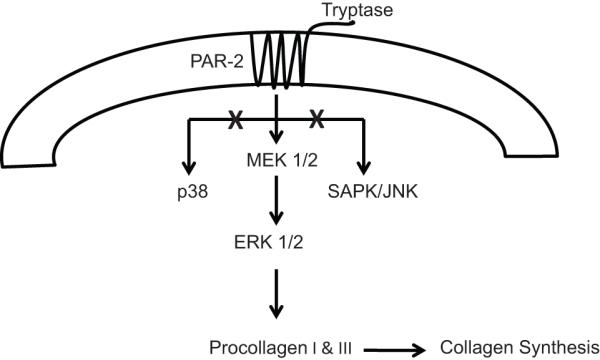
Schematic of Tryptase/PAR-2 Signaling. Tryptase activates PAR-2 in isolated adult cardiac fibroblast via ERK 1/2 to cause subsequent collagen production and fibrosis in the heart.
ERK1/2 has been shown to regulate procollagen gene expression in cardiac fibroblasts. ERK1/2 translocates to the nucleus to act directly on the α1(I) procollagen gene. The transcription factors of Egr, Ets, and AP-1 families are downstream of ERK1/2.16-18 Specifically, the α1(I) procollagen gene contains both AP-1 and ETS binding sites in the 5′UTR and first intron, and therefore these sequences may represent downstream targets of ERK1/2.19-21 Consequently, the α1 (I) collagen has been shown to be a biomarker for fibrosis in the hypertensive hearts of humans and rats.22-24 In order to determine if the same pattern of MAPK activation occurred in cardiac fibroblasts in the hypertensive heart, we isolated these cells from SHR and compared MAPK activation to that in fibroblasts from normotensive WKY hearts. We found that SHR cardiac fibroblasts had increased phosphorylation of ERK1/2 with no upregulation of p38 and SAPK/JNK, similar to what we observed in the isolated cultured fibroblasts treated with tryptase. This is consistent with our previous findings that tryptase is increased in SHR hearts.1
While together our data shows that tryptase stimulation of PAR-2 on isolated cardiac fibroblasts induces collagen synthesis, we wanted to know if PAR-2 was in fact important in hypertensive hearts in vivo. Therefore, we treated SHR with the PAR-2 antagonist peptide FSLLRY and found that fibrosis was prevented. However, this treatment did not prevent LV hypertrophy or hypertension itself. This result is identical to what we have shown previously with the mast cell stabilizer, nedocromil;1 strengthening the concept that mast cell tryptase is a major driver of fibrosis in the hypertensive heart. This increase in collagen is a result of increased synthesis of both collagen types I and III by myofibroblasts and an imbalance of collagens metabolism by matrix metalloproteinases in the SHR.25 Myofibroblasts’ ability to modulate collagen turnover contributes to tissue remodeling in the hypertensive heart, which ultimately leads to fibrosis.25 The phenotypic change of fibroblast to myofibroblast can be detected by the presence α-SMA and ED-A fibronectin in the fibroblast.26, 27 We found a trend towards increased α-SMA and a significant increase in ED-A fibronectin in isolated cardiac fibroblasts treated with tryptase. These markers are indicative of fibroblasts transitioning to a myofibroblastic phenotype, and suggest that tryptase and MAPK pathways can cause phenoconversion of the cardiac fibroblast to myofibroblast, which was prevented by blockade of PAR-2 and MAPK signaling pathway.
In summary, the results of this study reveal a previously unidentified pathway of cardiac fibroblast activation. This pathway is initiated by tryptase stimulation of PAR-2 and leads to induction of MAPK activation in the form of phosphorylation of ERK1/2. The phosphorylation of ERK leads to fibroblast conversion to a myofibroblastic phenotype, and ultimately collagen production and accumulation within the heart. It is concluded that mast cell tryptase plays a critical role in the fibrosis of the hypertensive heart.
Perspectives
Since tryptase is derived almost exclusively from mast cells, the results of this study reinforce the importance of mast cells in the development of fibrosis in the hypertensive heart which further extends our knowledge of how tryptase induces fibrosis in the heart. Our data now establishes tryptase activation of PAR-2 in cardiac fibroblasts and the subsequent selective activation of ERK1/2 as being involved in the transition of fibroblast to myofibroblastic phenotype and increasing collagen synthesis. Thus, this study represents another indicator of the importance of mast cells to myocardial remodeling by describing a previously unknown role for tryptase/PAR-2. Further, this study also highlights the importance of developing novel therapeutics that can target this receptor.
Supplementary Material
Acknowledgments
Sources of Funding This work was supported by a UNCF-Merck Graduate Science Research Dissertation Fellowship (to J.L.M), National Heart, Lung, and Blood Institute grants R01-HL-62228 and R01-HL-073990 (to J.S.J.) and 1K99HL093215-01 (to S.P.L).
Footnotes
Disclosures. None.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension. 2009;53:1041–1047. doi: 10.1161/HYPERTENSIONAHA.108.123158. [DOI] [PubMed] [Google Scholar]
- 2.Akers IA, Parson M, Hill MR, Hollenburg MD, Sanjar S, Laurent GJ, McAnulty RJ. Mast cell tryptase stimulates human lung fibroblast proliferation via protease activate receptor-2. Am J Physiol - Lung Cell Mol Physiol. 2000;278:L193–L201. doi: 10.1152/ajplung.2000.278.1.L193. [DOI] [PubMed] [Google Scholar]
- 3.Sirvent AE, Gonzalez C, Enriquez R, Fernandez J, Millan I, Barber X, Amoros F. Serum tryptase level and markers of renal dysfunction in a population with chronic kidney disease. Journal of Nephrology. 2010;23:282–290. [PubMed] [Google Scholar]
- 4.Frungieri MB, Weidinger S, Meineke V, Kohn FM, Mayerhofer A. Proliferative action of mast-cell tryptase is mediated by PAR2, COX2, prostaglandins, and PPARgamma : Possible relevance to human fibrotic disorders. PNAS. 2002;99:15072–15077. doi: 10.1073/pnas.232422999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nystedt S, Ramakrishnan V, Sundelin J. The proteinase-activated receptor 2 is induced by inflammatory mediators in human endothetial cells: comparison with the thrombin receptor. J Biol Chem. 1996;271:14910–14915. doi: 10.1074/jbc.271.25.14910. [DOI] [PubMed] [Google Scholar]
- 6.Frungieri MB, Albrecht M, Raemsch R, Mayerhofer A. The action of the mast cell product tryptase on cyclooxygenase-2 (COX2) and subsequent fibroblast proliferation involves activation of the extracellular signal-regulated kinase isoforms 1 and 2 (erk1/2) Cellular Signalling. 2005;17:525–533. doi: 10.1016/j.cellsig.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Burgess ML, Terracio L, Hirozane T, Borg TK. Differential integrin expression by cardiac fibroblasts from hypertensive and exercise-trained rat hearts. Cardiovascular Pathology. 2003;11:78–87. doi: 10.1016/s1054-8807(01)00104-1. [DOI] [PubMed] [Google Scholar]
- 8.Edwards CA, O’Brien WDJ. Modified assay for determination of hydroxyproline in a tissue hydrolyate. Clinic Chimica Acta. 1980;104:161–167. doi: 10.1016/0009-8981(80)90192-8. [DOI] [PubMed] [Google Scholar]
- 9.Levick SP, Loch D, Rolfe B, Fairlie DP, Taylor SM, Brown L. Antifibrotic activity of an inhibitor of group iia secretory phospholipase a2 in young spontaneous hypertensive rat. Journal of Immunology. 2006;176:7000–7007. doi: 10.4049/jimmunol.176.11.7000. [DOI] [PubMed] [Google Scholar]
- 10.Al-Ani B, Saifeddine M, Kawabata A, Renaux B, Mokashi S, Hollenburg MD. Proteinase activated receptor 2 (PAR2): development of a ligand-binding assay correlating with activation of PAR-2 by PAR-1 and PAR-2-derived peptide ligands. Journal Pharmacology. Experimental Therapeutic. 1999;290:753–760. [PubMed] [Google Scholar]
- 11.Otsubo S, Nitta K, Uchida K, Yumura W, Nihei H. Mast cells and tubulointerstitial fibrosis in patients with ANCA-associated glomerulonephritis. Clinical and Experimental Nephrology. 2003;7:0041–0047. doi: 10.1007/s101570300005. [DOI] [PubMed] [Google Scholar]
- 12.Adbelkarim S, Short J, Guo J, Steinberg SF. Protease-Activated Receptor-1 mediated DNA synthesis in cardiac fibroblast is via epidermal growth factor receptor transactivation: Distinct PAR-1 signaling pathways in cardiac fibroblast and cardiomyocytes. Circulation Research. 2002;91:532–539. doi: 10.1161/01.res.0000035242.96310.45. [DOI] [PubMed] [Google Scholar]
- 13.Jenkins AL, Chinni C, De Niese MR, Blackhart B, Mackie EJ. Expression of protease activated receptor 2 (PAR2) during embryonic development. Developmental Dynamics. 2000;218:465–471. doi: 10.1002/1097-0177(200007)218:3<465::AID-DVDY1013>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, Kahn M, Nelken NA, Coughlin SR, Payan DG, Bunnett NW. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J. 1996;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrington T, Johnson G. Organization and regulation of mitogen-activated protein kinase signaling pathways. Current Opinion in Cell Biology. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 16.Blenis J. Signal transduction via the MAP kinases:proceed to your own RSK. PNAS. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. Journal Biological Chemistry. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 18.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. Journal Biological Chemistry. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 19.Brennner DA, Rippe RA, Veloz L. Analysis of collagen alpha 1 (I) promoter. Nucleic Acids Research. 1989;7:6055–6064. doi: 10.1093/nar/17.15.6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katai H, Stephenson JD, Simkevich CP, Thompson JP, Raghow R. An AP-1 like motif in the first intron of human Pro alpha 1 (I) collagen gene is a critical determinant of its transcriptional activity. Molecular and Cellular Biochemistry. 1992;118:119–129. doi: 10.1007/BF00299391. [DOI] [PubMed] [Google Scholar]
- 21.Rossert J, Eberspaecher H, de Crombrugghe B. Separate cis-acting DNA elements of the mouse pro-alpha 1(I) collagen promoter direct expression of reporter genes to different type I collagen-producing cells in transgenic mice. The Journal of Cell Biology. 1995;129:1421–1432. doi: 10.1083/jcb.129.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diez J, Panizo A, Gil MJ, Monreal I, Hernandez M, Mindan JP. Serum markers of collagen type I metabolism in spontaneously hypertensive rats : relation to myocardial fibrosis. Circulation. 1996;93:1026–1032. doi: 10.1161/01.cir.93.5.1026. [DOI] [PubMed] [Google Scholar]
- 23.Querejeta R, Varo N, Lopez B, Larman M, Artinano E, Etayo JC, Ubago JLM, Gutierrez-Stampa M, Emparanza JI, Gil MJ, Monreal I, Mindan JP, Diez J. Serum carboxy-terminal propeptide of procollagen type I is a marker of myocardial fibrosis in hypertensive heart disease. Circulation. 2000;101:1729–1735. doi: 10.1161/01.cir.101.14.1729. [DOI] [PubMed] [Google Scholar]
- 24.Querejeta R, Lopez B, Gonzalez A, Sanchez E, Larman M, Ubago JL Martinez, Diez J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation. 2004;110:1263–1268. doi: 10.1161/01.CIR.0000140973.60992.9A. [DOI] [PubMed] [Google Scholar]
- 25.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. Journal of Clinical Investigation. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sappino AP, Schurch W, Gabbiani G. Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulations. Laboratory Investigation. 1990;63:144–161. [PubMed] [Google Scholar]
- 27.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-B1. Journal of Cell Biology. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.