

Abstract
BACKGROUND AND PURPOSE
Choline analogues, a new type of antimalarials, exert potent in vitro and in vivo antimalarial activity. This has given rise to albitiazolium, which is currently in phase II clinical trials to cure severe malaria. Here we dissected its mechanism of action step by step from choline entry into the infected erythrocyte to its effect on phosphatidylcholine (PC) biosynthesis.
EXPERIMENTAL APPROACH
We biochemically unravelled the transport and enzymatic steps that mediate de novo synthesis of PC and elucidated how albitiazolium enters the intracellular parasites and affects the PC biosynthesis.
KEY RESULTS
Choline entry into Plasmodium falciparum-infected erythrocytes is achieved both by the remnant erythrocyte choline carrier and by parasite-induced new permeability pathways (NPP), while parasite entry involves a poly-specific cation transporter. Albitiazolium specifically prevented choline incorporation into its end-product PC, and its antimalarial activity was strongly antagonized by choline. Albitiazolium entered the infected erythrocyte mainly via a furosemide-sensitive NPP and was transported into the parasite by a poly-specific cation carrier. Albitiazolium competitively inhibited choline entry via the parasite-derived cation transporter and also, at a much higher concentration, affected each of the three enzymes conducting de novo synthesis of PC.
CONCLUSIONS AND IMPLICATIONS
Inhibition of choline entry into the parasite appears to be the primary mechanism by which albitiazolium exerts its potent antimalarial effect. However, the pharmacological response to albitiazolium involves molecular interactions with different steps of the de novo PC biosynthesis pathway, which would help to delay the development of resistance to this drug.
Keywords: lipids, malaria, phospholipid synthesis, drug transport, Plasmodium, therapy
Introduction
Malaria is a major health problem, with more than 40% of the world's population at risk, and is responsible for 200–300 million clinical cases yearly and nearly a million deaths, mostly amongst children under 5 years of age in sub-Saharan Africa (Greenwood et al., 2008; WHO, 2009). The widespread resistance of Plasmodium falciparum, the most deadly malaria parasite, to most antimalarial drugs is a major obstacle to the eradication of this disease (RBM, 2008). The recently reported potential emergence of resistance to artemisinin (Carrara et al., 2009; Dondorp et al., 2010) is a major threat, thus highlighting the need for new chemotherapeutic approaches with novel mechanisms of action to treat P. falciparum infections.
When Plasmodium infects erythrocytes, the intracellular parasites need to synthesize considerable amounts of membranes for their growth and proliferation. P. falciparum membranes differ from other eukaryotic cells by the quasi-absence of cholesterol. Phospholipids (PL) are the major membranous lipid component, along with phosphatidylcholine (PC) and phosphatidylethanolamine (PE), which together constitute the bulk of malarial lipids (Vial and Ancelin, 1992; Vial et al., 2003; Vial and Ben Mamoun, 2005). PL biosynthesis is therefore essential for intraerythrocytic development of the parasite (Vial and Ben Mamoun, 2005), whereas these activities are absent from mature uninfected erythrocytes (Van Deenen and De Gier, 1975). P. falciparum possesses its own machinery to synthesize the large amount of PL it requires, the PL content increasing by sixfold during the parasite blood cycle (Vial et al., 2003).
An essential route of PC biosynthesis is the de novo CDP–choline pathway (also called the Kennedy pathway), which allows synthesis of PC from plasmatic choline. The three enzymes of the pathway are choline kinase (CK), which phosphorylates choline to form phosphocholine, CTP:phosphocholine cytidylyltransferase (CCT), which transfers a cytidine nucleotide to phosphocholine, and choline/ethanolamine phosphotransferase (CEPT), which produces PC by transferring phosphocholine onto diacylglycerol. Genes that encode these activities have been found in genome sequences from most malarial species and their biochemical activities have been characterized (Dechamps et al., 2010a, b).
In this regard, we designed and developed compounds that mimic the choline structure in order to target choline metabolism and thus PC biosynthesis. The compounds were optimized for in vitro antimalarial activity, and there was a very close correlation between the inhibition of parasite growth and specific inhibition of parasite de novo PC biosynthesis from choline (Calas et al., 1997; Ancelin et al., 1998; 2003b). The structure–activity relationships showed that bis-quaternary ammonium compounds are the most active, with an IC50 within sub-nanomolar concentrations (Calas et al., 2000; Hamze et al., 2005). Several generations of bis-quaternary ammonium compounds have been developed (Calas et al., 2000; Ancelin et al., 2003b), thus giving rise to bis-thiazolium compounds (Hamze et al., 2005) that, in addition to their potent antimalarial activity, may be delivered in prodrug form to improve their oral bioavailability (Vial et al., 2004).
Choline analogues inhibit P. falciparum asexual blood stages with concentrations in the low nanomolar range (Ancelin et al., 2003b; Hamze et al., 2005). They are also able to cure, without recrudescence, in vivo malaria infection at very low doses in rodent (<1 mg·kg−1 i.p.) and primate models (Wengelnik et al., 2002; Vial et al., 2004). The potency and specificity of these anti-PL effectors are probably due to their unique ability to accumulate in a non-reversible way inside the intraerythrocytic parasite (Wengelnik et al., 2002; Biagini et al., 2003; Vial et al., 2004). The bis-thiazolium salt T4 selectively inhibits de novo PC biosynthesis (Vial et al., 2004; Le Roch et al., 2008), while the interaction of T16 with haeme might contribute to antimalarial activity (Biagini et al., 2003).
The bis-thiazolium series provided the clinical candidate albitiazolium (albitiazolium bromide, formerly named T3 and SAR97276) (Figure 1) (Vial et al., 2004), that was successfully evaluated for toxicity and ADME (absorption, distribution, metabolism and excretion) properties in preclinical and human phase I clinical trials. Clinical phase II trials to treat severe malaria by the parenteral route are now being conducted by Sanofi (MMV, 2011).
Figure 1.
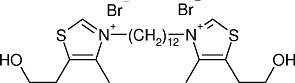
Structure of the choline analogue albitiazolium, formerly named T3 (3,3′-(dodecane-1,12-diyl)bis[5-(2-hydroxyethyl)-4-methyl-1,3-thiazol-3-ium] dibromide).
It is of the utmost interest to discover the precise mechanism of action of this new type of antimalarial. These studies aim at pinpointing the mechanism of action of this clinical candidate, which led us to characterize the entire de novo malaria PC synthesis pathway at the blood stage. We elucidated the mechanism mediating the entry of both choline and albitiazolium into P. falciparum-infected erythrocytes and into the intracellular parasite. We then showed that albitiazolium inhibits choline entry at each of these transport steps, while also affecting the enzymes conducting malaria PC synthesis.
Methods
Chemicals
[Methyl-3H]-choline, [methyl-14C]-choline, [1-3H]-ethan-1-ol-2-amine, phosphoryl-[methyl-14C]-choline, CDP-[methyl-14C]-choline and [8-3H]-hypoxanthine were purchased from GE Healthcare (Orsay, France). Choline (Cho), phosphoryl-Cho (P-Cho), CDP–choline (CDP-Cho), ethanolamine (Etn), PC and saponin were from Sigma (Saint-Quentin Fallavier, France). RPMI 1640 medium and modified RPMI 1640 without choline, serine, methionine, inositol and folic acid (called modified RPMI in the text) were obtained from Gibco (Saint Aubin, France). TLC silica gel 60 plates with a concentrating zone were from Merck (Darmstadt, Germany). Albitiazolium (albitiazolium bromide, 3,3′-dodecane-1,12-diylbis[5-(2-hydroxyethyl)-4-methyl-1,3-thiazol-3-ium] dibromide) and [thiazole-2,2′-14C]-albitiazolium were provided by Sanofi (Chilly-Mazarin, France).
Biological materials
P. falciparum 3D7 strain (MRA-102 from MR4) was cultured in human red blood cells (Etablissement Français du Sang, Montpellier, France) at 5% haematocrit in complete medium composed of RPMI 1640 medium supplemented with 25 mM HEPES buffer (pH 7.4) and 10% AB+ human serum (Trager and Jensen, 1976). Cultures were incubated at 37°C in a culture gas chamber under a gaseous mixture of 5% CO2, 5% O2 and 90% N2. Whenever required, parasites were synchronized with 5% sorbitol (Lambros and Vanderberg, 1979). For uptake experiments, mature P. falciparum-infected red blood cells (IRBC) were purified on a magnetic cell separator VarioMACS (CS column, Miltenyi Biotec, Paris, France) in HEPES-buffered RPMI 1640. Free parasites were prepared from mature infected erythrocytes by adding 10 volumes 0.02% (w v-1) saponin in cold PBS (116 mM NaCl, 8.3 mM Na2HPO4, 3.2 mM KH2PO4, pH 7.4) for 1 min, followed by two washes by centrifugation for 5 min at 4°C at 1260×g and resuspension in PBS. Free parasites were then resuspended in the appropriate medium. For all of these experiments, the exact number of cells in each sample was quantified on a Neubauer haemocytometer.
Effect on PL and nucleic acid (NA) biosynthesis
The effect of albitiazolium on the incorporation of 20 µM [3H]-choline (specific activity, 0.5 µCi·nmol−1) or 2 µM [3H]-ethanolamine (specific activity, 2.5 µCi·nmol−1) into the PL fraction and of [3H]-hypoxanthine (1 µCi per well) into NA was assessed in microtitre plates. Infected erythrocyte suspensions (5–10% parasitaemia, 2% haematocrit) were incubated in 150 µL of modified RPMI 1640 without choline, supplemented with 25 mM HEPES in the presence or absence of albitiazolium at various concentrations. After 30 min at 37°C, radioactive precursors (50 µL) were added for 3 h at 37°C in a culture gas chamber. Incorporations were stopped by freezing the plates at −80°C. Parasite DNA and membrane PL were recovered on glass-fibre filter plates (Unifilter 96 GF/C, Perkin Elmer, Courtaboeuf, France) by harvesting the lysate using a FilterMate cell harvester (Packard Instruments, Meriden, CT, USA). Radioactivity incorporated into the macromolecules recovered on the filter was counted on a TopCount microplate scintillation counter (Packard Instruments). When choline was used as radioactive precursor, filters were pre-soaked in 0.05% polyethyleneimine to eliminate nonspecific binding to the filter. Control values were obtained by incubating an equal number of non-infected erythrocytes. The results are expressed as PL50 (PC50 or PE50) and NA50, corresponding to the drug concentrations that reduce the amount of synthesized PL (PC or PE) and NA by 50% respectively.
Effect of PL polar heads on albitiazolium antimalarial activity
The involvement of choline, ethanolamine and serine in the antimalarial activity of choline analogues was studied using the isobologram method (Berenbaum, 1978). The antimalarial activities of albitiazolium and of individual polar heads against the intra-erythrocytic growth of P. falciparum were first measured alone in microtitre plates according to a modified Desjardins test (1.5% final haematocrit, 0.6% parasitaemia) (Desjardins et al., 1979; Ancelin et al., 2003b). Then a P. falciparum-infected erythrocyte suspension containing or not PL polar heads at various concentrations were incubated for 48 h at 37°C in the presence of various albitiazolium concentrations. Then 0.6 µCi [3H]-hypoxanthine was added to each well for 18 h at 37°C. Reactions were stopped by freezing at −80°C, and [3H]-hypoxanthine incorporation was then quantified as described above. Antimalarial activity was expressed as IC50, which is the drug concentration leading to 50% parasite growth inhibition. Then the antimalarial activity of albitiazolium was determined in the presence of the various polar heads added at concentrations lower than their IC50. In these experiments, compound concentrations were expressed as a fraction of the IC50s of the compounds alone. These fractions were called fractional inhibitory concentrations (FIC) (Berenbaum, 1978; Gupta et al., 2002).
The type of interaction was determined by calculating the maximal sum of FIC50 of the two compounds combined for antimalarial characterization (ΣFIC50= FIC50 albitiazolium + FIC50 polar head). The additive effect of the compounds was deduced if ΣFIC was ≥1 and <2; while ΣFIC <1 denotes a synergy of the compounds for antimalarial activity, and ΣFIC ≥2 and <4 reflects a slight antagonism, which is marked when ΣFIC ≥4 (Gupta et al., 2002).
Transport and enzyme characterization
Each transport and enzymatic step conducting malarial PC synthesis was characterized on P. falciparum-infected erythrocytes at the mature stage. Preliminary time course experiments were performed to ensure that the uptake of radiolabelled compounds was measured under initial rate conditions throughout the studies, and that enzymatic reactions proceeded linearly with time and with the biological extracts.
Transport of choline into P. falciparum-infected erythrocytes
Purified mature IRBC were washed three times with modified RPMI and resuspended in modified RPMI at typically 5 × 108 IRBC·mL−1. Uptake of [14C]-choline was measured by incubating 0.5 × 108 IRBC in the presence of 0 to 250 µM [14C]-choline (specific activity, 6.7–50.4 nCi·nmol−1) at 37°C for 10 min. When furosemide was used to inhibit new permeability pathways (NPP) of the infected erythrocytes, IRBC were pre-incubated for 8 min in the presence of 150 µM furosemide before the addition of [14C]-choline for 10 min at 37°C. The reactions were stopped by the addition of 300 µL cold modified RPMI. To separate IRBC from the incubating medium, the suspensions were transferred onto 400 µL of oil (dibutyl phthalate; Sigma), and centrifuged (10 000×g, 30 s, 4°C), thus sedimenting the cells below the oil. Supernatants were removed, and the tube walls were washed. Then cell pellets were lysed by the addition of 100 µl distilled water, then solubilized and bleached by adding 100 µL of a cocktail containing 5 parts tissue solubilizer (solvable™; Perkin Elmer), 2 parts H2O2 (30%) and 2 parts glacial acetic acid. Radioactivity was determined using a Beckman LS6500 liquid-scintillation spectrometer after the addition of 3 mL of liquid scintillation cocktail (UltimaGold™; PerkinElmer).
Radioactive background was obtained by incubating the same amount of RBC or IRBC at 4°C and was deducted from the results obtained at 37°C so as to determine only the temperature-dependant uptake.
Entry of choline analogue into P. falciparum-infected erythrocytes
Entry of [14C]-albitiazolium into purified mature P. falciparum IRBC was measured by incubating 0.5 × 108 IRBC in 100 µL final HEPES-buffered RPMI 1640 in the presence of 0 to 250 µM [14C]-albitiazolium (specific activity, 23.8 nCi·nmol−1) for 8 min at 37°C. In some experiments, IRBC were pre-incubated in the presence of 150 µM of the NPP inhibitor furosemide for 8 min before adding [14C]-albitiazolium for 8 min at 37°C. The reactions were stopped by the addition of 300 µL cold RPMI, and the samples were treated as described above.
Transport of choline and choline analogue into P. falciparum-isolated parasites
All transport experiments were performed on trophozoite-stage parasites purified by lysis of the IRBC with 0.1% saponin for 1 min in HEPES-buffered RPMI 1640. Uptake of [14C]-choline and [14C]-albitiazolium into parasites was measured by incubating 108 parasites in 200 µL of modified RPMI in the presence of 0 to 250 µM [14C]-choline (specific activity, 0.4–50.4 nCi·nmol−1) or [14C]-albitiazolium (specific activity, 0.6–113 nCi·nmol−1) for 10 min at 37°C. For experiments designed to study the effects on the transport of effectors (choline and albitiazolium), suspensions of free parasites were pre-incubated with or without the effector for 8 min before the addition of [14C]-choline or [14C]-albitiazolium for 10 min at 37°C. The reactions were stopped by the addition of 300 µL cold modified RPMI, and the parasites were separated from the incubation medium by transferring the suspension onto 400 µL of oil (a 5:4 mixture of dibutyl phthalate/dioctyl phthalate). After centrifugation at 10 000×g for 30 s at room temperature, samples were treated as described above. Radioactive background was obtained by incubating the same amount of free parasites at 4°C and was deducted from the results obtained at 37°C so as to measure only the temperature-dependant uptake.
CK assay
CK activity was determined by a modification of the method of Ancelin and Vial (1986). Crude homogenates were prepared by lysing P. falciparum-infected erythrocytes with an equal volume of water followed by 30 s of sonication at 4°C (Branson Sonifier B30).
CK activity was determined in a final volume of 200 µL containing 125 mM Tris–HCl (pH 7.9), 10 mM MgCl2, 10 mM ATP, 5 mM EGTA and 5 to 600 µM [14C]-choline (1–56 nCi·nmol−1). The reaction was started by the addition of 20 µL of cellular extract corresponding to 0.1 × 107 to 1.7 × 107 infected cells. After 45 min at 37°C, the reaction was stopped by heating samples for 5 min at 95°C. For each sample, 20 µL of the reaction mix was spotted onto a TLC silica-gel plate, previously activated at 100°C for 1 h and developed using ethanol/2% ammonia (1:1, v v-1) as solvent. Radioactive spots were located by autoradiography, scraped off and counted by liquid scintillation. Spots were identified by migration and revelation of the choline and phosphorylcholine standards with iodine vapours.
CCT assay
P. falciparum CCT activity was measured on parasite extracts that were prepared by lysing pellets of mature P. falciparum-IRBC with the same volume of water with antiproteases (Roche, cedex, France).
CCT enzyme activity was assayed by monitoring the formation of CDP-Cho from the radiolabelled substrate [14C]-phosphocholine. Enzymatic reactions were carried out in 50 µL final volume containing 100 mM Tris (pH 8), 20 mM MgCl2, 20 mM CTP, 0.04–2 mM phosphoryl[methyl-14C]-choline (specific activity 1.6–61 nCi·nmol−1) and 100 µM oleic acid/egg PC (1:1, mol:mol) liposomes as activators. Liposomes were prepared as described in Yeo et al. (1997). The reactions were started by the addition of 10 µL of extract corresponding to 107 infected cells. After 30 min at 37°C in a shaking bath, the reactions were stopped by heating at 100°C for 5 min. For each sample, 20 µL of the reaction mix was spotted onto a TLC silica-gel plate, previously activated at 100°C for 1 h. Radiolabelled substrate and product were separated in 98% ethanol/0.5% NaCl/25% ammonia (50:50:1 by volume). Plates were exposed overnight to a storage phosphor screen (Molecular Biodynamics, Antony, France) and then analysed on a phosphorImager (Storm 840; Molecular Biodynamics). Radioactive spots identified by the migration of appropriate standards (P-Cho and CDP-Cho) were scraped directly into scintillation vials and counted by liquid scintillation.
CEPT assay
P. falciparum membranes were prepared and cholinephosphotransferase activity was assayed as described in Vial et al. (1984b) in a final volume of 210 µL containing 200 mM Tris–HCl (pH 8.1), 15 mM MnCl2, 0 to 300 µM CDP[methyl-14C]choline (specific activity, 1–2 nCi·nmol−1), 0.3 mM 1-palmitoyl-2-oleyl-sn-glycerol (Avanti Polar Lipids, Alabaster, AL, USA). Diacylglycerols were suspended in a solution of 500 mM Tris–HCl (pH 8.1) and 0.018% Triton X-100 and then strongly vortexed to obtain a slightly turbid solution (80 µL per assay). Reactions were initiated by the addition of membrane extracts equivalent to 4 × 107 infected cells.
Samples were incubated for 30 min in a shaking water-bath at 37°C. The reaction was stopped by the addition of 3 mL chloroform/methanol (2/1, v v-1), and the lipids were extracted according to the procedure of Folch (Folch et al., 1957; Vial et al., 1984b). The organic phase, containing radiolabelled PC, was brought to dryness in a scintillation vial then counted by liquid scintillation. Reaction products in the organic phase were characterized by TLC with authentic PL as standards, revealing that PC is the only reaction product in the organic phase.
Analysis of experimental results
In dose-response experiments, the concentration leading to 50% inhibition of parasite transport or enzyme activity (IC50) was measured when the substrate concentrations were close to their apparent Km and determined using a non-linear regression model (sigmoidal dose–response/variable slope; GraphPad Prism4®, GraphPad software, Inc., La Jolla, CA, USA). The experiments were performed at least twice, each at least in duplicate (with different batches of cells and at different time periods).
For transport and enzymatic studies, substrate- or inhibitor-dependent curves were analysed using GraphPad Prism 4® software and represented using the Michaelis–Menten equation for Km and Vmax determination. Ki values were calculated from Lineweaver–Burk linearization and plots of apparent Km as a function of the inhibitor concentration.
Results
Specific effect of albitiazolium on the de novo PC biosynthesis pathway
Bis-thiazolium salts such as albitiazolium have been designed as choline analogues to inhibit malarial PC biosynthesis. An effector is considered to be a specific inhibitor of de novo PC biosynthesis when it affects its biosynthesis without any simultaneous effect on PE or NA synthesis (Ancelin et al., 1998). As the extent of PL biosynthesis is largely dependent on the cell cycle, we determined the albitiazolium effect at the three stages of the parasite blood cycle. Effects were measured after a 3 h short incubation time to avoid toxic effects on parasite growth.
Figure 2 shows typical dose–response curves of albitiazolium. The PC50 of albitiazolium, the drug concentration that reduces the amount of PC synthesized from choline by 50%, was 2.8, 26 and 9.3 µM at ring, trophozoite and schizont stages, respectively. On the other hand, no significant effect was observed on ethanolamine incorporation up to 10 µM, and PE50 was higher than 1 mM at the three developmental stages. Effect on NA biosynthesis occurred at concentrations 13- to 80-fold higher than the PC50 values (NA50= 160–340 µM) (Figure 2 and Table 1). In the same conditions, pentamidine, another bis-cationic compound not considered to be a choline analogue but supposed to interact with DNA affected primarily NA synthesis and not PC synthesis (data not shown). These results indicate that albitiazolium specifically inhibits PC biosynthesis from choline at all blood stages.
Figure 2.
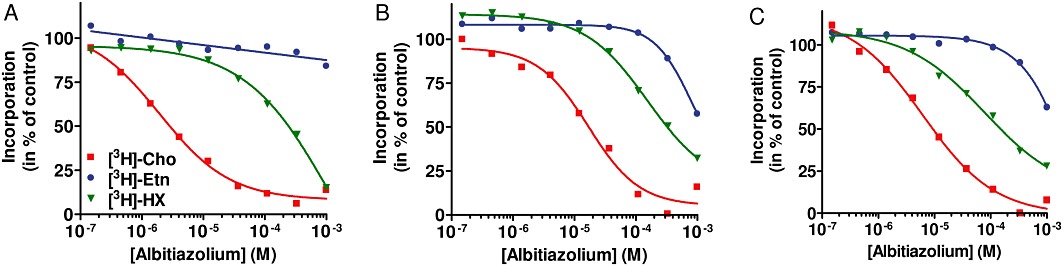
Effect of albitiazolium on PL and NA biosynthesis. P. falciparum-infected erythrocytes at the ring (A), trophozoite (B) and schizont stages (C) were pre-incubated with various concentrations of albitiazolium for 30 min. Then 20 µM [3H]-choline (Cho), 2 µM [3H]-ethanolamine (Etn) or [3H]-hypoxanthine (HX) were added for 3 h; and their incorporation into PL or NA was measured. Data are the mean of two independent experiments with different batches of cells, each performed in duplicate.
Table 1.
Comparative effect of albitiazolium on choline and ethanolamine incorporation into PL and on hypoxanthine incorporation into NA
| Parasite stage | PC50 (µM) | PE50 (µM) | NA50 (µM) |
|---|---|---|---|
| Ring | 2.8 | >1000 | 240 |
| Trophozoite | 26 | >1000 | 340 |
| Schizont | 9.3 | >1000 | 160 |
Concentrations inhibiting 50% of incorporation of precursors into PC (PC50), PE (PE50) and nucleic acids (NA50) were measured after 3 h incubation at high haematocrit (2%) and high parasitaemia (5–10%) to avoid the toxic effect of the choline analogue, as described in Figure 2. The results are means of two independent experiments (with different batches of cells) each conducted in duplicate.
Interaction between albitiazolium and PL precursors
The fact that albitiazolium specifically inhibits de novo PC biosynthesis does not necessarily imply that this effect is involved in its pharmacological antimalarial effect. To go further, we investigated whether choline could interfere with the pharmacological action of albitiazolium and antagonize its antimalarial activity.
We thus determined the antimalarial activity of albitiazolium alone or in combination with choline, ethanolamine and serine (i.e. the polar heads of the three structural PL) and the type of interaction exerted, using the isobologram method (Gupta et al., 2002). Polar heads had per se a very weak antimalarial activity, with IC50 of 12, 0.32 and 65 mM for choline, ethanolamine and serine, respectively (data not shown). The response parameters were determined, and the IC50 of the combined compounds were expressed as sums of the FICs (ΣFICs) of each added compound.
In two independent experiments, albitiazolium activity was highly antagonized by choline, with a sum of FIC50 of 12.7 and 28.3, respectively. Figure 3A,B indicate that the antimalarial activity of albitiazolium was decreased by 28-fold in the presence of 6 mM choline. On the other hand, when combined with the other major PL precursors, ethanolamine and serine, an additive effect was noted (i.e. no antagonism between the compounds) (Gupta et al., 2002). The FIC50 sums were lower than 1.7 for both ethanolamine and serine (Figure 3C,D).
Figure 3.
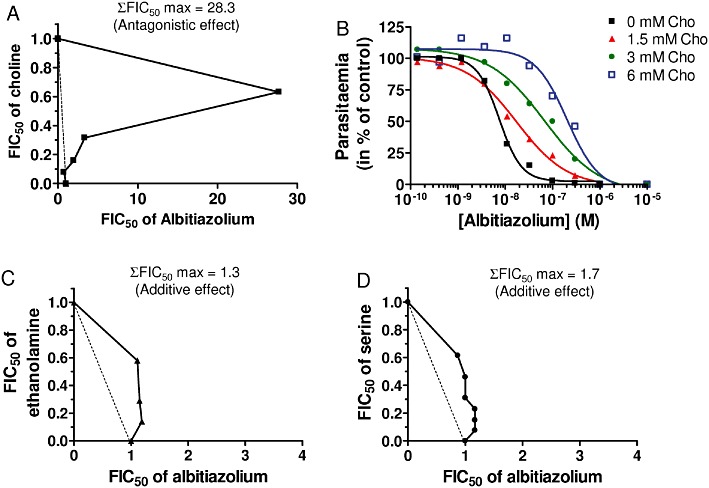
In vitro interaction between albitiazolium and the three PL precursors (choline, ethanolamine and serine) for its antimalarial activity. Effect of choline on antimalarial activity of albitiazolium is represented as an isobole plot (A) and as dose– response curves (B). Effect of ethanolamine (C) and serine (D) are represented as isobole plots. For isobole plots, the maximal sum of FICs of albitiazolium and the polar heads studied in combination for antimalarial activity (ΣFIC50) is indicated. Data presented are from one typical experiment, and each interaction experiment was conducted at least twice.
Thus, choline supplementation in the culture medium antagonized the antimalarial activity of albitiazolium, indicating that its pharmacological activity occurs through interference with choline metabolism whose main end product is the newly synthesized PC.
Characterization of the de novo PC biosynthesis pathway
Our next objective was to determine what step, involved in the de novo PC biosynthesis, was affected: choline entry into IRBC and then into the parasite, or one of the three enzymes involved in PC synthesis since all of them have a choline-containing substrate. In preliminary experiments, we had to develop biochemical tools to characterize each of these steps.
Choline entry into IRBC
First, we determined the mechanism by which choline enters IRBC and then the parasite to enable de novo PC synthesis. Choline penetrates the host erythrocyte membrane through the erythrocyte choline carrier (Ancelin et al., 1991; Staines and Kirk, 1998) or through parasite-induced NPP, which are channels induced by the parasite and present on the erythrocyte membrane (Ginsburg and Stein, 1987; 2004; Staines and Kirk, 1998; Kirk, 2001; Biagini et al., 2005). We dissected here the entry of choline into P. falciparum-IRBC and determined the proportion of choline entering the IRBC by each of the two pathways by using furosemide, a specific inhibitor of NPP (Kirk et al., 1994; Staines and Kirk, 1998; Staines et al., 2007).
Choline entry into normal erythrocytes or P. falciparum-IRBC was temperature-dependent and proceeded linearly with time for at least 20 min (data not shown). Choline entry into normal erythrocytes appeared to be mediated by a saturable process following Michaelis–Menten kinetics with a Km of 12.2 ± 2.2 µM (n= 4) and a maximal velocity (Vmax) of 0.43 ± 0.05 pmol per 107 RBC·min−1, which corresponds to transport via the remnant erythrocyte choline transporter (Martin, 1977) (Figure 4A).
Figure 4.
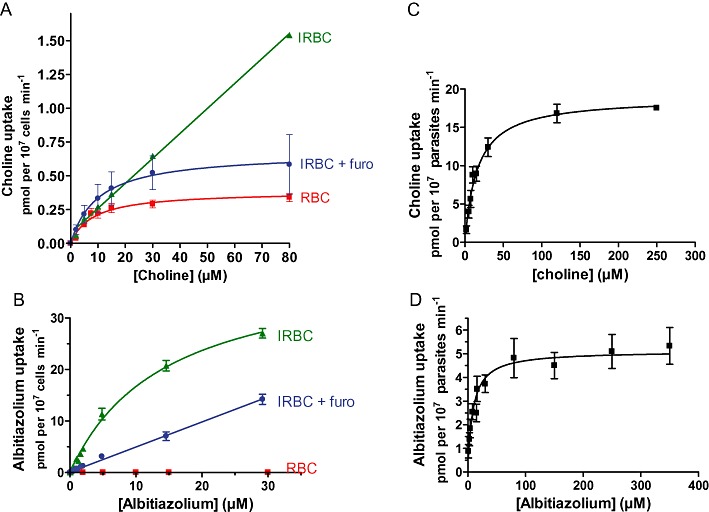
Concentration-dependent uptake of choline and albitiazolium by P. falciparum IRBC and isolated parasites. The entry of choline (A) or albitiazolium (B) in purified mature-stage IRBC was measured after 10 min (choline) or 8 min (albitiazolium) incubation in the presence or absence of 150 µM furosemide. When furosemide was used, IRBC were pre-incubated for 8 min in the presence of furosemide before the addition of choline or albitiazolium. Uptake into normal RBC was measured only in the absence of furosemide. The entry of choline (C) and albitiazolium (D) into saponin-free parasites was measured after 10 min (choline) or 8 min (albitiazolium) incubation at 37°C. The results are expressed as means ± SEM (n≥ 3).
In IRBC, choline uptake seemed to occur through a non-saturable process (Figure 4A), reaching a rate of 1.5 pmol per 107 IRBC·min−1 at an external concentration of 80 µM choline (i.e. about fourfold the physiological concentration of choline in plasma) (Das et al., 1986). When furosemide was added to inhibit NPP, choline uptake was markedly decreased, thus highlighting a saturable component and Michaelis–Menten type kinetics, with a Km of 11.0 ± 2.4 µM and a Vmax of 0.69 ± 0.23 pmol per107 IRBC·min−1 (Figure 4A). These parameters appear to be close to those corresponding to choline entry into erythrocytes via the choline carrier (similar Km). Moreover, we can state that, at physiological choline concentration (15 µM) (Das et al., 1986), choline entered P. falciparum-IRBC at 75% by a saturable process, probably via the erythrocytic choline carrier, while the remaining part (25%) entered via furosemide-sensitive NPP.
Choline entry into P. falciparum
Once in the erythrocyte cytosol, choline has to enter the intracellular parasite. This was investigated by measuring choline entry into saponin-freed P. falciparum parasites. A preliminary time course showed that choline entry was linear for at least 20 min (data not shown); thus, our incubation time was set at 10 min to ensure that choline uptake was measured under initial rate conditions throughout. Choline entry into the parasite occurred as a saturable process exhibiting Michaelis–Menten kinetics with a Km of 18.5 ± 3.3 µM and a Vmax of 23.3 ± 4.5 pmol per 107 parasites·min−1 (Figure 4C and Table 2), indicating that choline enters the intracellular parasite via a dedicated transporter.
Table 2.
Kinetic parameters of choline and albitiazolium entry into P. falciparum free parasites and their cross-inhibition
| Choline entry | Albitiazolium entry | |
|---|---|---|
| Km (µM) | 18.5 ± 3.3 | 14.2 ± 2.6 |
| Vmax (pmol per 107 cells·min−1) | 23.3 ± 4.5 | 6.1 ± 1.1 |
| Inhibitors | Albitiazolium | Choline |
|---|---|---|
| IC50 (µM) | 2.3 ± 0.2 | 808 |
| Ki (µM) | 0.53 | 321 |
| Type of inhibition | Competitive | Competitive |
Values are means of at least three independent experiments performed in duplicate (SEM is indicated). Otherwise, values are means of two independent experiments. IC50s were determined using 15 µM choline and 16 µM albitiazolium; concentrations that correspond to their Km.
Enzymes of the de novo PC biosynthesis pathway
After penetration into the intracellular parasite, choline is incorporated into the end-product PC, leading us to characterize the three related enzymatic activities, CK, the CCT and the CEPT in P. falciparum parasite extracts. Activities were measured by the formation of the enzyme product from a radiolabelled substrate. At the initial linear rate of the enzyme reactions, the activities of the three enzymes were determined as a function of increasing concentrations of the choline-containing substrates and in the presence of a saturating concentration of the second substrate (ATP, CTP or DAG, respectively). These concentrations were previously determined (data not shown and experimental procedure). All three, CK, CCT and CEPT, exhibit saturable curves reaching a plateau that is characteristic of Michaelis–Menten kinetics. These curves allowed us to determine Km values for choline-containing substrates of 31.2 ± 4.1, 511.0 ± 24.8 and 13.2 ± 0.7 µM for CK, CCT and CEPT, respectively (Table 3).
Table 3.
Kinetic parameters of the steps mediating the de novo biosynthesis of PC from exogenous choline (erythrocyte and parasite choline carrier and Kennedy pathway enzymes) and inhibitory effect of albitiazolium
| Transporter/enzyme | Erythrocyte choline entry | Parasite choline entry | CK | CCT | CEPT |
|---|---|---|---|---|---|
| Km (µM) | 11.0 ± 2.4 | 18.5 ± 3.3 | 31.2 ± 4.1 | 511.0 ± 24.8 | 13.2 ± 0.7 |
| Vmax (pmol per 107 IRBC·min−1) | 0.7 ± 0.2 | 23.3 ± 4.5 | 63.3 ± 11.6 | 5.2 ± 1.4 | 17.0 ± 1.5 |
| IC50 of albitiazolium (µM) | 2.9 ± 0.8* | 2.3 ± 0.2 | 1500 | 2500 | 4200 |
Choline entry into infected erythrocytes and parasites followed Michaelis–Menten kinetics. Inhibitory effect of albitiazolium on each step is expressed as IC50 and measured when choline (transporter) or the choline-containing metabolites (enzymes) were close to their apparent respective Km (i.e. one to twofold their respective Km).
For the endogenous choline carrier, inhibition was achieved in the presence of 150 µM furosemide to inhibit choline entry by furosemide-sensitive NPP.
Values are means of at least three independent experiments performed in duplicate (SEM is indicated). Otherwise, values are means of two independent experiments.
Albitiazolium entry
Whatever its mechanism of action, albitiazolium must first enter the infected erythrocyte then the intracellular parasite to exert its pharmacological activity. We thus characterized these two essential steps.
Entry into P. falciparum IRBC
Albitiazolium transport was determined both in uninfected and infected erythrocytes at pharmacologically active concentrations (nM range) (Vial et al., 2004) and at a concentration of 30 µM, which is beyond the plasma concentration observed in humans during phase I and II clinical trials (unpublished observations). In both types of cells, entry at 0°C was not significant compared with radioactive background. At 37°C, albitiazolium entry proceeded linearly as a function of time for at least 10 min (data not shown).
After 8 min of incubation at 37°C, albitiazolium entry into normal erythrocytes was lower than 0.01 pmol per 107 RBC·min−1, even in the presence of 30 µM albitiazolium (Figure 4B), which corresponds to less than twofold the radioactive background observed after incubation of RBC at 0°C (data not shown). This result indicates that a non-significant amount of albitiazolium enters the RBC, probably by diffusion, and that albitiazolium is probably not a substrate of the choline carrier of the erythrocyte. On the other hand, at 37°C, high albitiazolium entry into IRBC was detected and this proceeded linearly as a function of time for at least 10 min (data not shown). Albitiazolium entry appeared to be linear from 9 nM to 5 µM and then tended to plateau, indicating a saturable process with an apparent Km of 20.6 ± 4.3 µM and a Vmax of 47.9 ± 4.9 pmol per 107 IRBC·min−1 (Figure 4B). The addition of furosemide at 150 µM (Staines and Kirk, 1998) reduced albitiazolium uptake by about 70% (Figure 4C). Measuring the entry of 50 µM albitiazolium into IRBC in the presence of much higher furosemide concentrations (Figure S1) revealed that furosemide was unable to completely abolish drug entry, with 15% of albitiazolium uptake remaining even in the presence of 5 mM furosemide. Albitiazolium probably enters IRBC mainly through a furosemide-sensitive route, which corresponds to NPPs while the residual non-saturable mechanism remains to be clarified.
Entry into P. falciparum parasites
Albitiazolium entry into isolated P. falciparum parasites was measured after 10 min of incubation in order to be within the linear phase of transport. Albitiazolium entry appeared to occur through a saturable Michaelis–Menten process (Figure 4D), with an apparent Km of 14.15 ± 2.6 µM and a Vmax of 6.1 ± 1.1 pmol per 107 parasites·min−1. The Km of albitiazolium uptake into isolated parasites was found to be similar to that exhibited by choline uptake in the same conditions (Table 2).
Effect of albitiazolium on the de novo PC biosynthesis pathway
Effect on choline entry into P. falciparum IRBC
We demonstrated that choline entered IRBC via both the remnant erythrocyte choline carrier and furosemide-sensitive NPP. We thus detailed the effect of albitiazolium on the total choline entry into IRBC (i.e. choline carrier + NPP) and on the carrier-mediated choline entry using furosemide to inhibit choline entry via NPP.
When infected erythrocytes were incubated in the presence of 15 µM choline (i.e. a concentration corresponding to a choline carrier Km value), albitiazolium significantly inhibited choline entry from 4 µM and by 50% (IC50) at 19 µM (Figure 5A); complete inhibition was not reached at 36 µM. In the presence of furosemide, albitiazolium inhibition occurred with an IC50 of 2.9 ± 0.8 µM and, interestingly, was complete at 12 µM (Table 3 and Figure 5A). Next we measured choline entry in the presence of furosemide and found increasing albitiazolium concentrations increased the slope of the primary plots [1/V=f(1/[choline]), where V is the velocity] and all lines intersected on the ordinate axis (Figure 5B), indicating competitive inhibition. The inhibition constant (Ki), calculated from Lineweaver–Burk linearization and from the plot of apparent Km as a function of albitiazolium concentration, was 3.1 µM.
Figure 5.
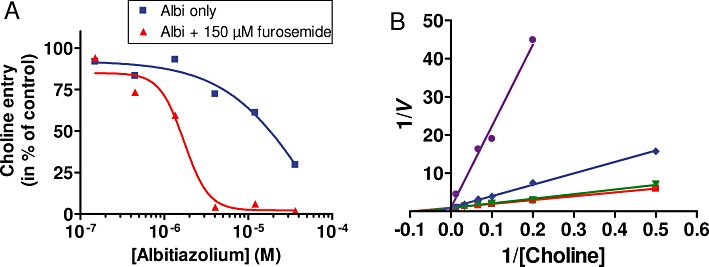
Effect of albitiazolium on choline transport in P. falciparum-infected erythrocytes. (A) Inhibition of 15 µM choline entry into P. falciparum-infected erythrocytes by albitiazolium (albi) was determined after 10 min of incubation in presence or not of 150 µM furosemide. (B) The initial velocities of choline uptake into P. falciparum IRBC in the presence of increasing concentrations of albitiazolium are shown as Lineweaver–Burk double-reciprocal plots. Inhibitor concentrations were 0, 4, 6 and 10 µM albitiazolium. Experiments were repeated twice.
Inhibition of choline entry into P. falciparum parasites
We also measured the effect of albitiazolium on the parasite choline transporter characterized above using choline at 15 µM, which corresponds to its Km value. In these conditions, albitiazolium inhibited choline entry into the parasite with an IC50 of 2.3 ± 0.2 µM (Table 2), and complete inhibition was achieved at 10 µM (Figure 6A). Figure 6B presents the Lineweaver–Burk plot of choline entry in the presence of four albitiazolium concentrations and shows that the intersection of all lines at the 1/V axis coincide, thus indicating that the inhibition of choline entry by albitiazolium was competitive, with a Ki of 0.53 µM (Figure 6B and Table 2).
Figure 6.
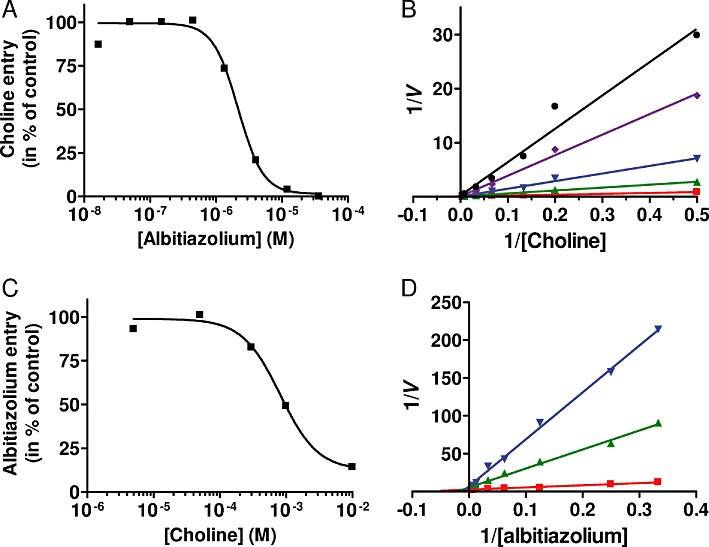
Transport of choline and albitiazolium into P. falciparum saponin-free parasites and substrate competition. (A) Inhibition of 15 µM choline entry into P. falciparum saponin-free parasites by albitiazolium was determined after 10 min of incubation. (B) The initial velocities of choline uptake into P. falciparum saponin-free parasites in the presence of increasing concentrations of albitiazolium are shown as Lineweaver–Burk double-reciprocal plots. (C) Inhibition of 16 µM albitiazolium entry into P. falciparum saponin-free parasites by choline was determined after 10 min of incubation. (D) Lineweaver–Burk representation of albitiazolium entry into saponin-free parasites in the presence of increasing choline concentrations. Inhibitor concentrations were 0, 3, 6, 9 and 12 µM albitiazolium for (B) and 0, 0.8 and 2 mM choline for (D). Experiments were repeated twice.
Cross-inhibition of choline and albitiazolium transport
The above experiments showed that albitiazolium entered the parasite via a saturable Michaelian kinetic process, and that albitiazolium competitively inhibits choline entry into the intracellular parasite. To determine whether albitiazolium and choline followed similar route(s) to enter the parasite, we evaluated the capacity of choline to inhibit albitiazolium entry into the parasite. When albitiazolium was used at a concentration corresponding to its Km value (16 µM), choline inhibited entry of the choline analogue with an IC50 of 808 µM (Table 2 and Figure 6C) and complete inhibition was achieved at higher concentrations. The type of choline inhibition of albitiazolium transport was also determined. Figure 6D presents a Lineweaver–Burk plot of albitiazolium entry at two choline concentrations and indicates a competitive inhibition with a calculated Ki value of 321 µM (Table 2). Thus, in addition to the fact that albitiazolium inhibited choline entry into the parasite, these results indicate that, reciprocally, choline competitively inhibits albitiazolium entry into the parasite. This cross-inhibition indicates that choline and albitiazolium probably enter the parasite via similar processes.
Effects of choline analogues on Kennedy pathway enzymes
Finally, we also assessed whether albitiazolium could affect each of the three enzymatic steps involved in the de novo PC biosynthesis pathway (CK, CCT and CEPT; see Figure 7). This was determined under optimal conditions for enzymatic catalysis as determined above, except that choline-containing substrates were used at concentrations close to their Km (between 1 and 2 Km). In these conditions, albitiazolium completely inhibited each of the three enzymes, with complete inhibition being achieved at 100 mM. The concentrations at which 50% of the activity of the enzyme (IC50) was inhibited were in the millimolar range; 1.5, 2.5 and 4.2 mM for CK, CCT and CEPT, respectively (Table 3).
Figure 7.
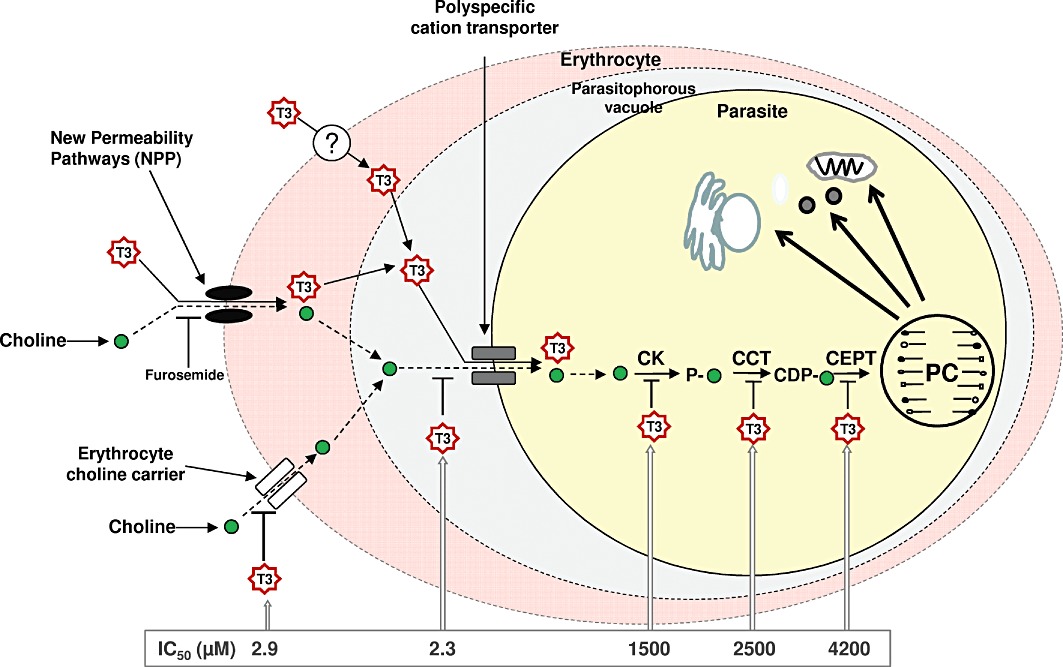
The fate of albitiazolium and choline in the P. falciparum-infected erythrocyte and albitiazolium-induced inhibition of PC biosynthesis. Choline enters the infected erythrocyte essentially through the erythrocyte choline carrier and a small amount via furosemide-sensitive NPPs. Choline is then transported into the parasite by the parasite choline carrier and used to synthesize PC via the Kennedy pathway. The choline analogue albitiazolium enters the infected erythrocyte via furosemide-sensitive NPPs and a furosemide-insensitive pathway. Albitiazolium inhibits choline transport by the erythrocyte choline carrier but is not transported by it. The choline analogue is taken up by the parasite via the parasite choline carrier, thus inhibiting choline transport. Albitiazolium is accumulated in the parasite and exerts its activity principally on the different steps of the de novo PC biosynthesis pathway. It should be noted that albitiazolium is abbreviated to ‘T3’ for convenience in this diagram.
Discussion
In the fight against malaria, new antimalarial drugs with original mechanisms of action are urgently needed (RBM, 2008). Our laboratory is developing new classes of antimalarial drugs designed to target malarial membrane biogenesis (Vial et al., 2011). The most successful approach is based on choline analogues to prevent de novo CDP–Cho-mediated PC synthesis of the parasite at its blood stage, which occurs at the expense of plasmatic choline (Dechamps et al., 2010b). Optimization of this class of molecules (Wengelnik et al., 2002; Ancelin et al., 2003a, b; Vial et al., 2004; Hamze et al., 2005) led to the selection of albitiazolium as clinical candidate, which is currently in phase II clinical trials to treat severe malaria by the parenteral route (MMV, 2011). Given the importance of the mechanism of action sustaining the toxic effect for the malaria parasite, this work aims to answer two questions: (1) Is de novo biosynthesis of PC involved in the antimalarial activity of albitiazolium? (2) How does the drug affect the biosynthetic pathway?
We have shown here that albitiazolium specifically inhibits cellular de novo PC synthesis from choline at all the parasite blood stages while not affecting synthesis of PE, the other major structural malarial PL (Figure 2). Of utmost interest, choline is able to strongly antagonize the antimalarial activity of albitiazolium (Figure 3A,B). This highlights the involvement of choline metabolism in the antimalarial activity of albitiazolium.
To further elucidate the biochemical mechanism through which interactions between this drug and its target(s) result in the pharmacological response, we have, for the first time, unravelled and biochemically characterized the entire de novo PC synthesis pathway of P. falciparum. Here we analysed the two transport steps mediating choline entry into the IRBC and then into the intracellular parasite as well as the three enzymatic reactions (Figure 7), with the aim of determining the effect of albitiazolium on each step of this pathway. Partial data on de novo PC synthesis have been reported for other Plasmodium species such as the simian parasite Plasmodium knowlesi (Vial et al., 1984b; Ancelin and Vial, 1989; Ancelin et al., 1991) and the rodent parasite Plasmodium vinckei (Staines and Kirk, 1998). However, these parasites have different hosts, different times of blood cycle (24 or 48 h) and certain differences in their lipid metabolism (Dechamps et al., 2010a).
The previously reported enhancement of choline entry into Plasmodium-infected erythrocytes appears to occur through a saturable process in P. knowlesi (Ancelin et al., 1991; Ancelin and Vial, 1992), through a non-saturable process in P. falciparum (Kirk et al., 1991), and through the presence of two components in P. vinckei-infected erythrocytes (Staines and Kirk, 1998). Here, we showed that P. falciparum-infected erythrocytes exhibit a markedly increased rate of choline entry compared with control erythrocytes. Choline entry into IRBC occurred via two processes – a saturable process with a Km similar to that of the choline carrier of normal erythrocytes and a Vmax twofold higher than that observed in uninfected cells, and a non-saturable process, that can be attributed to furosemide-sensitive NPPs (Kirk et al., 1994) (Figure 4A). At 15 µM, its physiological concentration, 75% of the choline entering the IRBC appears to be by the saturable process and 25% by the non-saturable process.
Choline entry into the saponin-freed P. falciparum parasite exhibits temperature-dependent Michaelis–Menten kinetics, with an apparent Km of 18.5 ± 3.3 µM and a Vmax of 23.3 ± 4.5 pmol per 107 parasites·min−1 (Figure 4C and Table 3), in agreement with the findings of Biagini et al. (2004). In intact infected erythrocytes, total choline uptake is 1.5 pmol per 107 IRBC·min−1 in the presence of 80 µM choline (Figure 4A), while choline entry via the remnant choline transporter has a Vmax of 0.7 ± 0.2 pmol per 107 IRBC·min−1. The transport capacity of choline inside the parasite is thus much higher than the choline flux through both NPPs and the erythrocyte choline carrier. Choline entry into the infected erythrocyte is therefore probably the rate-limiting step of choline influx from the plasma to the intracellular parasite, and the parasite choline transporter has been likened to a choline ‘vacuum cleaner’ for the host cell (Biagini et al., 2004).
So far, the enzymatic activities of the de novo PC synthesis pathway have only been characterized in P. knowlesi (Vial et al., 1984a; Ancelin and Vial, 1989), except for the P. falciparum CK, which has been characterized as a native recombinant enzyme (Ancelin and Vial, 1986; Alberge et al., 2009). The kinetic parameters obtained here for the three P. falciparum enzymes (Table 3) revealed that the affinity of CCT for the substrate phosphocholine (Km= 511 µM) was by far the lowest compared to those of CK and CEPT for their respective choline substrates, which were 31 and 13 µM, respectively. When measured in optimal conditions, the maximal specific activity decreased in the order CK, CEPT and CCT. Figure 7 summarizes the steps involved in P. falciparum de novo PC synthesis, and the corresponding kinetic parameters are indicated in Table 3.
We also characterized the entry of albitiazolium. It was striking that albitiazolium did not enter normal erythrocytes to a significant extent (<0.01 pmol per 107 RBC·min−1, i.e. close to the radioactive background, at 30 µM albitiazolium) (Figure 4B), indicating that albitiazolium cannot be transported by the erythrocyte choline carrier. On the other hand, albitiazolium entered infected erythrocytes to a high extent (47.9 ± 4.9 pmol per 107 IRBC·min−1 at maximal capacity), and this occurred via a saturable process involving at least two components. The major component represented up to 85% and occurred via a furosemide-dependent and saturable process (Figure S1), suggesting that this entry was mediated via furosemide-sensitive NPPs (Figure 4B) even if transport via NPP is usually described as a non-saturable process at physiological concentrations of the transported molecule (Kirk, 2001). Albitiazolium entry appeared to proceed linearly for concentrations up to 5 µM before reaching saturation at higher concentrations. This concentration has to be compared to its pharmacological concentration, taking into account its binding to human plasma-proteins which was found to be 33% at 20 ng·mL−1 and no saturation was observed until 5000 ng·mL−1 (unpublished observations). Thus, the concentration was more than 100-fold higher than those that are effective at treating malaria in vitro (∼10 nM) (Vial et al., 2004). and 10-fold higher than the ∼500 nM concentration in plasma (during at least 4 h) needed to get a complete cure in rodents and humans (unpublished observations). In addition, NPP are usually described as non-saturable channels for low molecular weight solutes (Kirk, 2001; Martin et al., 2005), but they can accommodate compounds with a high molecular mass (300–600 Da) (Saliba and Kirk, 1998), even though it has not been shown whether this latter transport involves saturable or non-saturable processes. A second, less important, component (∼15%) of albitiazolium entry appeared to be furosemide-insensitive and non-saturable (Figures 4B and S1). This could be mediated by a different type of NPP that is not sensitive to furosemide or another uncharacterized process (Ginsburg and Stein, 2005).
Albitiazolium (Wein et al.. in preparation), like other bis-thiazolium salts (Biagini et al., 2003; Vial et al., 2004) and bis-cationic choline analogues (Wengelnik et al., 2002) accumulate to high extent within infected erythrocytes with a cellular accumulation ratio (CAR) of ∼400 at mature stages. This accumulation appears to be restricted to hematozoan-infected erythrocytes, including Plasmodium and Babesia species (Biagini et al., 2003; Vial et al., 2004; Richier et al., 2006). The fact that albitiazolium is not transported by the choline transporter and enters infected erythrocytes by NPPs, which are absent in normal cells as shown here, probably explains why the drug specifically accumulates within infected erythrocytes.
Once inside the IRBC, albitiazolium as choline enters the intracellular parasite via a saturable process following Michaelis–Menten kinetics (Figure 4D and Table 3). The cross-inhibition between choline and albitiazolium (Figure 6) suggests that the PC polar head and its analogue use similar organic cation transporter(s) to enter the parasite, as corroborated by their equal sensitivity to ionic dependence and various transport effectors (Biagini et al., 2004). Figure 7 illustrates the entry of albitiazolium to the intracellular parasite, and Table 2 shows that, under optimal conditions (i.e. within the linear phase of transport), the maximal velocity of albitiazolium entry was fourfold lower than for choline entry.
Since high concentrations of choline can overcome its antiplasmodial properties, the therapeutic effect of albitiazolium probably relies on the metabolic fate of choline. Biochemically characterizing all steps mediating choline incorporation in its only known end-product PC allowed us to decipher the molecular mechanism of action of albitiazolium.
Albitiazolium barely inhibited the NPP-mediated choline entry but it inhibited choline entry by the remnant erythrocyte choline carrier (Ki= 3.1 µM) (Figure 5B), even though the bulky choline analogue, composed of two cationic heads linked by an alkyl chain of 12 methylene groups (Figure 1), is not itself transported. Indeed, Deves and Krupka (1979) showed that lengthening the alkyl chain of choline analogues increased their affinity for the erythrocyte choline carrier due to hydrophobic bonding in a region near the substrate site but prevented the molecules from being transported.
Albitiazolium also inhibits choline entry into the intracellular parasite (Figure 6A,B and Table 2). Cross-inhibition between choline and albitiazolium appears to be competitive, even though the capacity of albitiazolium to inhibit choline entry is far greater (Ki= 0.53 µM) than that of choline to prevent albitiazolium entry (Ki of 321 µM) (Table 2). Finally, albitiazolium also appeared to inhibit the three enzymes involved in the incorporation of choline into PC. It was striking that the choline analogue could completely inhibit the three enzymatic activities, even though the IC50 values were in the millimolar range (Table 3).
Biochemical data thus indicate that two transport steps and the three enzymatic activities involved in the de novo PC synthesis are potentially inhibited by this choline analogue. The next questions that must be addressed are which step(s) are really affected in cellulo and which ones are most likely to be involved in its antimalarial properties. For this, the in situ interaction between the drug and its targets for its catalytic activity, as determined here, is dependent on the concentration of endogenous substrates and of the pharmacological compound in the vicinity of the target transporter(s) or enzyme(s). At the physiological choline concentration of 15 µM (Das et al., 1986) and at the pharmacological albitiazolium concentration of 100 nM, the drug cannot exert significant inhibition of choline entry mediated by the erythrocyte choline carrier because external concentrations required to inhibit these steps are in the micromolar range (Table 3 and Figure 5A).
Because of its high accumulation inside IRBC, the level of albitiazolium in the vicinity of intracellular target(s) is much higher than its external concentration. For example, at an external concentration of 100 nM, a 400-fold accumulation of albitiazolium is observed in P. falciparum-IRBC (Wein et al., unpublished observations); the albitiazolium concentration inside the IRBC is 40 µM when an equal distribution of the drug occurs inside the IRBC.
Accumulation of the closely related T16 antimalarial compound localized to 23% in the erythrocyte cytosol and to 77% inside the parasite, where a substantial part was recovered in the small digestive vacuole compartment (Biagini et al., 2003) as well as an unidentified compartment (Richier et al., 2006). Accordingly, and based on respective volumes of 75 fl (Saliba et al., 1998) and 50 fl (Mauritz et al., 2009) for IRBC and mature parasites, the albitiazolium concentration would reach 28 µM in the erythrocyte cytosol surrounding the parasite for an external concentration of 100 nM. Since a concentration of 2.3 µM albitiazolium inhibits 50% of choline entry into isolated parasites (Table 3), it is therefore likely that at 28 µM, albitiazolium strongly inhibits choline entry into the parasite. Under the same conditions, we can also estimate the concentration of the drug in the intracellular parasite compartment(s) where the drug localized (assuming its volume is ∼3% that of the infected erythrocyte) at equilibrium (CAR ∼400), which would reach approximately 1 mM. Thus, due to the combined effects of accumulation, which allowed the drug to reach millimolar concentrations in a low volume compartment of accumulation, and the inhibition of cellular choline entry, which results in low levels of the downstream metabolites P-Cho and CDP-Cho, the three enzymes are potentially, at least partially, inhibited in cellulo.
Altogether, there are many biochemical features through which molecular interactions of albitiazolium with the different steps of the de novo PC biosynthesis pathway can contribute to its potent pharmacological response, but its effect on parasite choline entry appears to be the primary target. The ability of albitiazolium to affect more than one target in the same metabolic pathway allows for a synergistic effect leading to reinforced inhibition of PC synthesis and thus to potent antimalarial activity.
In addition, previous studies have shown that bis-thiazolium compounds accumulated in IRBC were partly recovered in the Plasmodium food vacuole, where they were found to associate with metabolites of haemoglobin degradation. This interaction with haemozoin was shown to contribute to the antimalarial activity of the bis-thiazolium (Biagini et al., 2003). This multiple mode of action, distinct from available antimalarial agents, is a major strength of this inhibitor class, and it could also help to delay the development of resistance.
Finally, multiple impacts on the same metabolic pathway as well as a dual mechanism of action could be very important with respect to delaying the emergence of resistance to this antimalarial drug. This hypothesis is supported by experimental works using a 1 year drug pressure through a validated protocol (Rathod et al., 1997) with the choline analogue G25 (Wengelnik et al., 2002) in which the emergence of resistant parasites was not observed (data not shown).
Acknowledgments
The research leading to these results benefitted from funding from the European Community's Framework Programme, Antimal Integrated Project (LSHP-CT-2005-018834) and the EviMalar Network of Excellence (FP7/2007-2013, N° 242095), Sanofi and the Innomad/Eurobiomed programme of Pole of competitiveness (Région Languedoc-Roussillon/FEDER/OSEO). Sweta Maheshwari was funded by InterMalTraining FP7-Marie Curie ITN (PITN-GA-2008-215281) The P. falciparum 3D7 strain was obtained through the Malaria Research and Reference Reagent Resource Center (MR4; http://mr4.org/). Laurent Fraisse is full-time employee of Sanofi.
Glossary
- CCT
CTP:phosphocholine cytidylyltransferase
- CDP-Cho
CDP–choline
- CEPT
choline/ethanolamine phosphotransferase
- Cho
choline
- CK
choline kinase
- Etn
ethanolamine
- FIC
fractional inhibitory concentration
- IRBC
infected red blood cell
- Ki
inhibition constant
- Km
Michaelis constant
- NA
nucleic acids
- NPP
new permeability pathways
- PC
phosphatidylcholine
- P-Cho
phosphoryl-choline
- PE
phosphatidylethanolamine
- PL
phospholipids
- RBC
red blood cell
- Vmax
maximal velocity
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Inhibition of albitiazolium entry into IRBC by furosemide. Entry of 50 μM albitiazolium into P. falciparum-infected erythrocytes was measured after 8 min of incubation in presence of increasing concentrations of furosemide.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alberge B, Gannoun-Zaki L, Bascunana C, Tran van Ba C, Vial H, Cerdan R. Comparison of the cellular and biochemical properties of Plasmodium falciparum choline and ethanolamine kinases. Biochem J. 2009;425:149–158. doi: 10.1042/BJ20091119. [DOI] [PubMed] [Google Scholar]
- Ancelin ML, Vial HJ. Choline kinase activity in Plasmodium-infected erythrocytes: characterization and utilization as a parasite-specific marker in malarial fractionation studies. Biochim Biophys Acta. 1986;875:52–58. doi: 10.1016/0005-2760(86)90010-x. [DOI] [PubMed] [Google Scholar]
- Ancelin ML, Vial HJ. Regulation of phosphatidylcholine biosynthesis in Plasmodium-infected erythrocytes. Biochim Biophys Acta. 1989;1001:82–89. doi: 10.1016/0005-2760(89)90310-x. [DOI] [PubMed] [Google Scholar]
- Ancelin ML, Vial HJ. Saturable and non-saturable components of choline transport in Plasmodium-infected mammalian erythrocytes: possible role of experimental conditions. Biochem J. 1992;283((Pt 2)):619–621. doi: 10.1042/bj2830619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancelin ML, Parant M, Thuet MJ, Philippot JR, Vial HJ. Increased permeability to choline in simian erythrocytes after Plasmodium knowlesi infection. Biochem J. 1991;273((Pt 3)):701–709. doi: 10.1042/bj2730701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancelin ML, Calas M, Bompart J, Cordina G, Martin D, Ben Bari M, et al. Antimalarial activity of 77 phospholipid polar head analogs: close correlation between inhibition of phospholipid metabolism and in vitro Plasmodium falciparum growth. Blood. 1998;91:1426–1437. [PubMed] [Google Scholar]
- Ancelin ML, Calas M, Bonhoure A, Herbute S, Vial HJ. In vivo antimalarial activities of mono- and bis quaternary ammonium salts interfering with Plasmodium phospholipid metabolism. Antimicrob Agents Chemother. 2003a;47:2598–2605. doi: 10.1128/AAC.47.8.2598-2605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancelin ML, Calas M, Vidal-Sailhan V, Herbute S, Ringwald P, Vial HJ. Potent inhibitors of Plasmodium phospholipid metabolism with a broad spectrum of in vitro antimalarial activities. Antimicrob Agents Chemother. 2003b;47:2590–2597. doi: 10.1128/AAC.47.8.2590-2597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenbaum MC. A method for testing for synergy with any number of agents. J Infect Dis. 1978;137:122–130. doi: 10.1093/infdis/137.2.122. [DOI] [PubMed] [Google Scholar]
- Biagini GA, Richier E, Bray PG, Calas M, Vial H, Ward SA. Heme binding contributes to antimalarial activity of bis-quaternary ammoniums. Antimicrob Agents Chemother. 2003;47:2584–2589. doi: 10.1128/AAC.47.8.2584-2589.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagini GA, Pasini EM, Hughes R, De Koning HP, Vial HJ, O'Neill PM, et al. Characterization of the choline carrier of Plasmodium falciparum: a route for the selective delivery of novel antimalarial drugs. Blood. 2004;104:3372–3377. doi: 10.1182/blood-2004-03-1084. [DOI] [PubMed] [Google Scholar]
- Biagini GA, Ward SA, Bray PG. Malaria parasite transporters as a drug-delivery strategy. Trends Parasitol. 2005;21:299–301. doi: 10.1016/j.pt.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Calas M, Cordina G, Bompart J, Ben Bari M, Jei T, Ancelin ML, et al. Antimalarial activity of molecules interfering with Plasmodium falciparum phospholipid metabolism. Structure-activity relationship analysis. J Med Chem. 1997;40:3557–3566. doi: 10.1021/jm9701886. [DOI] [PubMed] [Google Scholar]
- Calas M, Ancelin ML, Cordina G, Portefaix P, Piquet G, Vidal-Sailhan V, et al. Antimalarial activity of compounds interfering with Plasmodium falciparum phospholipid metabolism: comparison between mono- and bisquaternary ammonium salts. J Med Chem. 2000;43:505–516. doi: 10.1021/jm9911027. [DOI] [PubMed] [Google Scholar]
- Carrara VI, Zwang J, Ashley EA, Price RN, Stepniewska K, Barends M, et al. Changes in the treatment responses to artesunate-mefloquine on the northwestern border of Thailand during 13 years of continuous deployment. PLoS ONE. 2009;4:e4551. doi: 10.1371/journal.pone.0004551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, de Belleroche J, Moore CJ, Rose FC. Determination of free choline in plasma and erythrocyte samples and choline derived from membrane phosphatidylcholine by a chemiluminescence method. Anal Biochem. 1986;152:178–182. doi: 10.1016/0003-2697(86)90138-7. [DOI] [PubMed] [Google Scholar]
- Dechamps S, Maynadier M, Wein S, Gannoun-Zaki L, Marechal E, Vial HJ. Rodent and non-rodent malaria parasites differ in their phospholipid metabolic pathways. J Lipid Res. 2010a;51:81–96. doi: 10.1194/jlr.M900166-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechamps S, Shastri S, Wengelnik K, Vial HJ. Glycerophospholipid acquisition in Plasmodium– a puzzling assembly of biosynthetic pathways. Int J Parasitol. 2010b;40:1347–1365. doi: 10.1016/j.ijpara.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother. 1979;16:710–718. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deves R, Krupka RM. The binding and translocation steps in transport as related to substrate structure. A study of the choline carrier of erythrocytes. Biochim Biophys Acta. 1979;557:469–485. doi: 10.1016/0005-2736(79)90344-4. [DOI] [PubMed] [Google Scholar]
- Dondorp AM, Yeung S, White L, Nguon C, Day NPJ, Socheat D, et al. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol. 2010;8:272–280. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Ginsburg H, Stein WD. Biophysical analysis of novel transport pathways induced in red blood cell membranes. J Membr Biol. 1987;96:1–10. doi: 10.1007/BF01869329. [DOI] [PubMed] [Google Scholar]
- Ginsburg H, Stein WD. The new permeability pathways induced by the malaria parasite in the membrane of the infected erythrocyte: comparison of results using different experimental techniques. J Membr Biol. 2004;197:113–134. doi: 10.1007/s00232-003-0646-7. [DOI] [PubMed] [Google Scholar]
- Ginsburg H, Stein WD. How many functional transport pathways does Plasmodium falciparum induce in the membrane of its host erythrocyte? Trends Parasitol. 2005;21:118–121. doi: 10.1016/j.pt.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Greenwood BM, Fidock DA, Kyle DE, Kappe SH, Alonso PL, Collins FH, et al. Malaria: progress, perils, and prospects for eradication. J Clin Invest. 2008;118:1266–1276. doi: 10.1172/JCI33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Thapar MM, Wernsdorfer WH, Bjorkman A. In vitro interactions of artemisinin with atovaquone, quinine, and mefloquine against Plasmodium falciparum. Antimicrob Agents Chemother. 2002;46:1510–1515. doi: 10.1128/AAC.46.5.1510-1515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamze A, Rubi E, Arnal P, Boisbrun M, Carcel C, Salom-Roig X, et al. Mono- and bis-thiazolium salts have potent antimalarial activity. J Med Chem. 2005;48:3639–3643. doi: 10.1021/jm0492608. [DOI] [PubMed] [Google Scholar]
- Kirk K. Membrane transport in the malaria-infected erythrocyte. Physiol Rev. 2001;81:495–537. doi: 10.1152/physrev.2001.81.2.495. [DOI] [PubMed] [Google Scholar]
- Kirk K, Wong HY, Elford BC, Newbold CI, Ellory JC. Enhanced choline and Rb+ transport in human erythrocytes infected with the malaria parasite Plasmodium falciparum. Biochem J. 1991;278((Pt 2)):521–525. doi: 10.1042/bj2780521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk K, Horner HA, Elford BC, Ellory JC, Newbold CI. Transport of diverse substrates into malaria-infected erythrocytes via a pathway showing functional characteristics of a chloride channel. J Biol Chem. 1994;269:3339–3347. [PubMed] [Google Scholar]
- Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65:418–420. [PubMed] [Google Scholar]
- Le Roch KG, Johnson JR, Ahiboh H, Chung DW, Prudhomme J, Plouffe D, et al. A systematic approach to understand the mechanism of action of the bisthiazolium compound T4 on the human malaria parasite, Plasmodium falciparum. BMC Genomics. 2008;9:513. doi: 10.1186/1471-2164-9-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K. Membrane Transport in Red Cells. London: Academic Press; 1977. [Google Scholar]
- Martin RE, Henry RI, Abbey JL, Clements JD, Kirk K. The ‘permeome’ of the malaria parasite: an overview of the membrane transport proteins of Plasmodium falciparum. Genome Biol. 2005;6:R26. doi: 10.1186/gb-2005-6-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauritz JM, Esposito A, Ginsburg H, Kaminski CF, Tiffert T, Lew VL. The homeostasis of Plasmodium falciparum-infected red blood cells. PLoS Comput Biol. 2009;5:e1000339. doi: 10.1371/journal.pcbi.1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MMV. Global Malaria Portfolio, 3Q 2011. Geneva, Switzerland: Medicine for Malaria Venture; 2011. http://www.mmv.org/research-development/science-portfolio. [Google Scholar]
- Rathod PK, McErlean T, Lee PC. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc Natl Acad Sci U S A. 1997;94:9389–9393. doi: 10.1073/pnas.94.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RBM. Roll Back Malaria, The Global Malaria Action Plan for a Malaria-Free World. Geneva, Switzerland: WHO; 2008. http://www.rollbackmalaria.org/gmap/index.html. [Google Scholar]
- Richier E, Biagini GA, Wein S, Boudou F, Bray PG, Ward SA, et al. Potent antihematozoan activity of novel bisthiazolium drug T16: evidence for inhibition of phosphatidylcholine metabolism in erythrocytes infected with Babesia and Plasmodium spp. Antimicrob Agents Chemother. 2006;50:3381–3388. doi: 10.1128/AAC.00443-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba KJ, Kirk K. Uptake of an antiplasmodial protease inhibitor into Plasmodium falciparum-infected human erythrocytes via a parasite-induced pathway. Mol Biochem Parasitol. 1998;94:297–301. doi: 10.1016/s0166-6851(98)00077-2. [DOI] [PubMed] [Google Scholar]
- Saliba KJ, Horner HA, Kirk K. Transport and metabolism of the essential vitamin pantothenic acid in human erythrocytes infected with the malaria parasite Plasmodium falciparum. J Biol Chem. 1998;273:10190–10195. doi: 10.1074/jbc.273.17.10190. [DOI] [PubMed] [Google Scholar]
- Staines HM, Kirk K. Increased choline transport in erythrocytes from mice infected with the malaria parasite Plasmodium vinckei vinckei. Biochem J. 1998;334((Pt 3)):525–530. doi: 10.1042/bj3340525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staines HM, Alkhalil A, Allen RJ, De Jonge HR, Derbyshire E, Egée S, et al. Electrophysiological studies of malaria parasite-infected erythrocytes: current status. Int J Parasitol. 2007;37:475–482. doi: 10.1016/j.ijpara.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Van Deenen LL, De Gier J. Lipids of the red cell membrane. In: Surgenor G, editor. The Red Blood Cell. New York: Academic Press; 1975. pp. 147–211. [Google Scholar]
- Vial HJ, Ancelin ML. Malarial lipids. An overview. Subcell Biochem. 1992;18:259–306. [PubMed] [Google Scholar]
- Vial HJ, Ben Mamoun C. Plasmodium lipids: metabolism and function. In: Sherman IW, editor. Molecular Approaches to Malaria. Washington, D.C.: ASM Press; 2005. pp. 327–352. [Google Scholar]
- Vial HJ, Thuet MJ, Ancelin ML, Philippot JR, Chavis C. Phospholipid metabolism as a new target for malaria chemotherapy. Mechanism of action of D-2-amino-1-butanol. Biochem Pharmacol. 1984a;33:2761–2770. doi: 10.1016/0006-2952(84)90693-2. [DOI] [PubMed] [Google Scholar]
- Vial HJ, Thuet MJ, Philippot JR. Cholinephosphotransferase and ethanolaminephosphotransferase activities in Plasmodium knowlesi-infected erythrocytes. Their use as parasite-specific markers. Biochim Biophys Acta. 1984b;795:372–383. doi: 10.1016/0005-2760(84)90088-2. [DOI] [PubMed] [Google Scholar]
- Vial HJ, Eldin P, Tielens AG, van Hellemond JJ. Phospholipids in parasitic protozoa. Mol Biochem Parasitol. 2003;126:143–154. doi: 10.1016/s0166-6851(02)00281-5. [DOI] [PubMed] [Google Scholar]
- Vial HJ, Wein S, Farenc C, Kocken C, Nicolas O, Ancelin ML, et al. Prodrugs of bisthiazolium salts are orally potent antimalarials. Proc Natl Acad Sci U S A. 2004;101:15458–15463. doi: 10.1073/pnas.0404037101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial HJ, Penarete D, Wein S, Caldarelli S, Fraisse L, Peyrottes S. Lipids as drug targets for malaria therapy. In: Becker K, editor. Drug Discovery in Infectious Diseases, Volume 2. Apicomplexan Parasites: Molecular Approaches toward Targeted Drug Development. Weinheim, Germany: Wiley-Blackwell; 2011. pp. 137–162. [Google Scholar]
- Wengelnik K, Vidal V, Ancelin ML, Cathiard AM, Morgat JL, Kocken CH, et al. A class of potent antimalarials and their specific accumulation in infected erythrocytes. Science. 2002;295:1311–1314. doi: 10.1126/science.1067236. [DOI] [PubMed] [Google Scholar]
- WHO. World Malaria Report 2009. Geneva, Switzerland: World Health Organization; 2009. http://www.who.int/malaria/publications/atoz/9789241563901/en/index.html. [Google Scholar]
- Yeo HJ, Larvor MP, Ancelin ML, Vial HJ. Plasmodium falciparum CTP:phosphocholine cytidylyltransferase expressed in Escherichia coli: purification, characterization and lipid regulation. Biochem J. 1997;324((Pt 3)):903–910. doi: 10.1042/bj3240903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.