

Abstract
Oxygen homeostasis represents an organizing principle for understanding metazoan evolution, development, physiology, and pathobiology. The hypoxia-inducible factors (HIFs) are transcriptional activators that function as master regulators of oxygen homeostasis in all metazoan species. Rapid progress is being made in elucidating homeostatic roles of HIFs in many physiological systems, determining pathological consequences of HIF dysregulation in chronic diseases, and investigating potential targeting of HIFs for therapeutic purposes.
Introduction
Oxygen is central to biology because of its utilization in the process of respiration. O2 serves as the final electron acceptor in oxidative phosphorylation, which carries with it the risk of generating reactive oxygen species (ROS) that react with cellular macromolecules and alter their biochemical or physical properties, resulting in cell dysfunction or death. As a consequence, metazoan organisms have evolved elaborate cellular metabolic and systemic physiological systems that are designed to maintain oxygen homeostasis. This review will focus on the role of hypoxia-inducible factors (HIFs) as master regulators of oxygen homeostasis and, in particular, on recent advances in understanding their roles in physiology and medicine. Due to space limitations and the remarkably pleiotropic effects of HIFs, the description of such roles will be illustrative rather than comprehensive.
O2 and Evolution, Part 1
Accumulation of O2 in Earth's atmosphere starting ~2.5 billion years ago led to evolution of the extraordinarily efficient system of oxidative phosphorylation that transfers chemical energy stored in carbon bonds of organic molecules to the high-energy phosphate bond in ATP, which is used to power physicochemical reactions in living cells. Energy produced by mitochondrial respiration is sufficient to power the development and maintenance of multicellular organisms, which could not be sustained by energy produced by glycolysis alone (Lane and Martin, 2010). The modest dimensions of primitive metazoan species were such that O2 could diffuse from the atmosphere to all of the organism's thousand cells, as is the case for the worm Caenorhabditis elegans. To escape the constraints placed on organismal growth by diffusion, systems designed to conduct air to cells deep within the body evolved and were sufficient for O2 delivery to organisms with hundreds of thousands of cells, such as the fly Drosophila melanogaster. The final leap in body scale occurred in vertebrates and was associated with the evolution of complex respiratory, circulatory, and nervous systems designed to efficiently capture and distribute O2 to hundreds of millions of millions of cells in the case of the adult Homo sapiens.
Hypoxia-Inducible Factors
Hypoxia-inducible factor 1 (HIF-1) is expressed by all extant metazoan species analyzed (Loenarz et al., 2011). HIF-1 consists of HIF-1α and HIF-1β subunits, which each contain basic helix-loop-helix-PAS (bHLH-PAS) domains (Wang et al., 1995) that mediate heterodimerization and DNA binding (Jiang et al., 1996a). HIF-1β heterodimerizes with other bHLH-PAS proteins and is present in excess, such that HIF-1α protein levels determine HIF-1 transcriptional activity (Semenza et al., 1996).
Under well-oxygenated conditions, HIF-1α is bound by the von Hippel-Lindau (VHL) protein, which recruits an ubiquitin ligase that targets HIF-1α for proteasomal degradation (Kaelin and Ratcliffe, 2008). VHL binding is dependent upon hydroxylation of a specific proline residue in HIF-1α by the prolyl hydroxylase PHD2, which uses O2 as a substrate such that its activity is inhibited under hypoxic conditions (Epstein et al., 2001). In the reaction, one oxygen atom is inserted into the prolyl residue and the other atom is inserted into the co-substrate α-ketoglutarate, splitting it into CO2 and succinate (Kaelin and Ratcliffe, 2008). Factor inhibiting HIF-1 (FIH-1) represses HIF-1α transactivation function (Mahon et al., 2001) by hydroxylating an asparaginyl residue, using O2 and α-ketoglutarate as substrates, thereby blocking the association of HIF-1α with the p300 coactivator protein (Lando et al., 2002). Dimethyloxalylglycine (DMOG), a competitive antagonist of α-ketoglutarate, inhibits the hydroxylases and induces HIF-1-dependent transcription (Epstein et al., 2001). HIF-1 activity is also induced by iron chelators (such as desferrioxamine) and cobalt chloride, which inhibit hydroxylases by displacing Fe(II) from the catalytic center (Epstein et al., 2001).
Studies in cultured cells (Jiang et al., 1996b) and isolated, perfused, and ventilated lung preparations (Yu et al., 1998) revealed an exponential increase in HIF-1α levels at O2 concentrations less than 6% (~40 mm Hg), which is not explained by known biochemical properties of the hydroxylases. In most adult tissues, O2 concentrations are in the range of 3-5% and any decrease occurs along the steep portion of the dose-response curve, allowing a graded response to hypoxia. Analyses of cultured human cells have revealed that expression of hundreds of genes was increased in response to hypoxia in a HIF-1-dependent manner (as determined by RNA interference) with direct binding of HIF-1 to the gene (as determined by chromatin immunoprecipitation [ChIP] assays); in addition, the expression of hundreds of genes was decreased in response to hypoxia in a HIF-1-dependent manner but binding of HIF-1 to these genes was not detected (Mole et al., 2009), indicating that HIF-dependent repression occurs via indirect mechanisms, which include HIF-1-dependent expression of transcriptional repressors (Yun et al., 2002) and microRNAs (Kulshreshtha et al., 2007). ChIP-seq studies have revealed that only 40% of HIF-1 binding sites are located within 2.5 kb of the transcription start site (Schödel et al., 2011).
In vertebrates, HIF-2α is a HIF-1α paralog that is also regulated by prolyl and asparaginyl hydroxylation and dimerizes with HIF-1β, but is expressed in a cell-restricted manner and plays important roles in erythropoiesis, vascularization, and pulmonary development, as described below. In D. melanogaster, the gene encoding the HIF-1α ortholog is designated similar and its paralog is designated trachealess because inactivating mutations result in defective development of the tracheal tubes (Wilk et al., 1996). In contrast, C. elegans has only a single HIF-1α homolog (Epstein et al., 2001). Thus, in both invertebrates and vertebrates, evolution of specialized systems for O2 delivery was associated with the appearance of a HIF-1α paralog.
O2 and Metabolism
The regulation of metabolism is a principal and primordial function of HIF-1. Under hypoxic conditions, HIF-1 mediates a transition from oxidative to glycolytic metabolism through its regulation of: PDK1, encoding pyruvate dehydrogenase (PDH) kinase 1, which phosphorylates and inactivates PDH, thereby inhibiting the conversion of pyruvate to acetyl coenzyme A for entry into the tricarboxylic acid cycle (Kim et al., 2006; Papandreou et al., 2006); LDHA, encoding lactate dehydrogenase A, which converts pyruvate to lactate (Semenza et al. 1996); and BNIP3 (Zhang et al. 2008) and BNIP3L (Bellot et al., 2009), which mediate selective mitochondrial autophagy (Figure 1). HIF-1 also mediates a subunit switch in cytochrome c oxidase that improves the efficiency of electron transfer under hypoxic conditions (Fukuda et al., 2007). An analogous subunit switch is also observed in Saccharomyces cerevisiae, although it is mediated by a completely different mechanism (yeast lack HIF-1), suggesting that it may represent a fundamental response of eukaryotic cells to hypoxia.
Figure 1. Regulation of Glucose Metabolism.
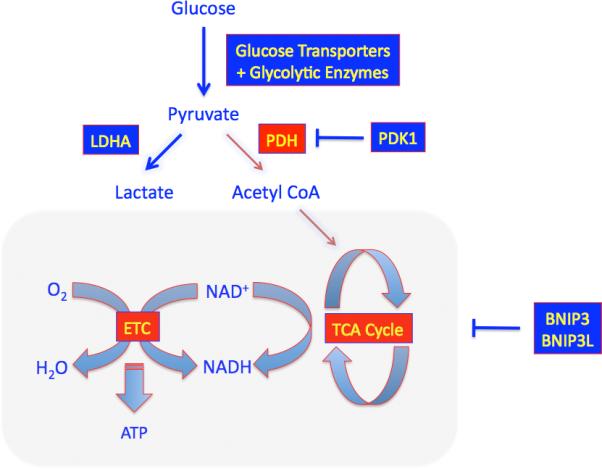
Under hypoxic conditions, HIF-1 activates the transcription of genes encoding: glucose transporters and glycolytic enzymes, which increase flux of glucose to pyruvate; PDK1 (encoding pyruvate dehydrogenase kinase), which inactivates PDH (pyruvate dehydrogenase), the mitochondrial enzyme that converts pyruvate to acetyl CoA for entry into the tricarboxylic acid (TCA/citric acid/Krebs) cycle; LDHA, which converts pyruvate to lactate; and BNIP3 and BNIP3L, which induce mitochondrial autophagy. The shunting of substrate away from the mitochondria reduces ATP production but prevents excess ROS production that occurs due to inefficient electron transport under hypoxic conditions.
It is conventional wisdom that cells switch to glycolysis when O2 becomes limiting for mitochondrial ATP production. Yet, HIF-1α-null mouse embryo fibroblasts, which do not down-regulate respiration under hypoxic conditions, have higher ATP levels at 1% O2 than wild-type cells at 20% O2, demonstrating that under these conditions O2 is not limiting for ATP production (Zhang et al., 2008). However, the HIF-1α-null cells die under prolonged hypoxic conditions due to ROS toxicity (Kim et al. 2006; Zhang et al., 2008). These studies have led to a paradigm shift with regard to our understanding of the regulation of cellular metabolism (Semenza, 2011): the purpose of this switch is to prevent excess mitochondrial generation of ROS that would otherwise occur due to the reduced efficiency of electron transfer under hypoxic conditions (Chandel et al., 1998). This may be particularly important in stem cells, in which avoidance of DNA damage is critical (Suda et al., 2011).
Role of HIFs in Development
Much of mammalian embryogenesis occurs at O2 concentrations of 1-5% and O2 functions as a morphogen (through HIFs) in many developmental systems (Dunwoodie, 2009). Mice that are homozygous for a null allele at the locus encoding HIF-1α die by embryonic day 10.5 with cardiac malformations, vascular defects, and impaired erythropoiesis, indicating that all three components of the circulatory system are dependent upon HIF-1 for normal development (Iyer et al., 1998; Yoon et al., 2011). Depending on the genetic background, mice lacking HIF-2α: die by embryonic day 12.5 with vascular defects (Peng et al., 2000) or bradycardia due to deficient catecholamine production (Tian et al., 1998); die as neonates due to impaired lung maturation (Compernolle et al., 2002); or die several months after birth due to ROS-mediated multi-organ failure (Scortegagna et al., 2003). Thus, while vertebrate evolution was associated with concomitant appearance of the circulatory system and HIF-2α, both HIF-1 and HIF-2 have important roles in circulatory system development. Conditional knockout of HIF-1α in specific cell types has demonstrated important roles in chondrogenesis (Schipani et al., 2001), adipogenesis (Yun et al., 2002), B-lymphocyte development (Kojima et al., 2002), osteogenesis (Wang et al., 2007), hematopoiesis (Takubo et al., 2010), T-lymphocyte differentiation (Dang et al., 2011), and innate immunity (Zinkernagel et al., 2007). While knockout mouse experiments point to the adverse effects of HIF-1 loss-of-function on development, it is also possible that increased HIF-1 activity, induced by hypoxia in embryonic tissues as a result of abnormalities in placental blood flow, may also dysregulate development and result in congenital malformations. For example, HIF-1α has been shown to interact with, and stimulate the transcriptional activity of, Notch, which plays a key role in many developmental pathways (Gustafsson et al., 2005).
Diseases in which HIF-1 Mediates Protective Responses
Coronary Artery Disease (CAD)
Heart disease is the major cause of mortality in the U.S. population. The formation of atherosclerotic plaques in the main coronary arteries leads to insufficient myocardial perfusion, especially when heart work and O2 consumption are increased. A physiological response to myocardial ischemia is the remodeling of collateral blood vessels to accept increased flow and thereby bypass stenotic regions. Two-thirds of patients with critical narrowing of a coronary artery (>70% reduction in diameter) have one or more collateral vessels. When rupture of atherosclerotic plaques results in complete coronary occlusion and myocardial infarction (MI), patients with collaterals are likely to have smaller infarcts and are more likely to survive. In patients with critical coronary stenosis, the frequency of a single nucleotide polymorphism (SNP) that changes proline to serine at residue 582 of HIF-1α was 5-fold greater among those without collaterals (Resar et al., 2005). This SNP and two others at the HIF1A locus were associated with stable exertional angina rather than MI as the initial clinical presentation in patients with CAD (Hlatky et al., 2007). Coronary stenosis and collaterals were not analyzed in this latter study, making interpretation of the data difficult. However, taken together these two clinical studies suggest that HIF1A is a major genetic modifier in CAD.
The remodeling of collateral vessels provides a means to increase O2 delivery and thereby eliminate hypoxia. However, in the short-term, cells must adapt to survive O2 deprivation, e.g. by shifting from oxidative to glycolytic metabolism as described above. The powerful nature of these adaptations is illustrated by the preconditioning phenomenon, in which exposure of the heart to short periods (e.g. 5 min [I5]) of ischemia followed by reperfusion (e.g. 5 min [R5]) protect the heart against subsequent episodes of prolonged ischemia (e.g. 30 min [I30]). Hearts from Hif1a+/- mice (heterozygous for a null allele at the Hif1a locus) show a complete loss of preconditioning (Cai et al., 2008). In addition to its effects on glucose metabolism, HIF-1 activates genes encoding enzymes that generate adenosine, a signaling molecule that mediates preconditioning (Eckle et al., 2008). The cardiac protection afforded by a preconditioning protocol lasting less than 1 hr (I5-R5-I5-R5-I30) is dependent on HIF-1, indicating that HIF-1-dependent transcription and subsequent translation occurs with remarkable rapidity.
Peripheral Arterial Disease (PAD)
Atherosclerotic stenosis of major conduit arteries in the legs results in ischemia that is manifested by intermittent claudication (leg pain during walking), which progresses to critical limb ischemia in which blood flow is not sufficient to maintain tissue viability, resulting in pain at rest and gangrene (tissue necrosis) that eventually necessitates limb amputation. The incidence of PAD in the general population is ~5%, whereas ~20% of individuals over 70 yr old have PAD and 1-2% of PAD patients develop critical limb ischemia. Limb ischemia induced in experimental animals by femoral artery ligation results in: increased HIF-1α protein levels; HIF-1-dependent expression of downstream target genes encoding multiple angiogenic growth factors, including vascular endothelial growth factor (VEGF), stromal-derived factor 1 (SDF-1), placental growth factor, angiopoietin 1, angiopoietin 2, platelet-derived growth factor B, and stem cell factor; recruitment of bone marrow-derived angiogenic cells (BMDACs); and recovery of tissue perfusion, all of which are impaired in older mice and in Hif1a+/- mice (Bosch-Marcé et al., 2007).
The failure of clinical trials targeting a single angiogenic factor (such as VEGF) and the role of HIF-1 as a master regulator responsible for the coordinated induction of multiple angiogenic factors suggested that HIF-1 may represent a better therapeutic target. A single intramuscular injection into the ischemic limb of AdCA5, a recombinant adenovirus encoding an engineered form of HIF-1α that is constitutively active, was sufficient to overcome the impaired recovery of blood flow following femoral artery ligation in 8-mon-old mice but was not effective in 13-mon-old mice (Bosch-Marcé et al., 2007; Rey et al., 2009). shRNA targeting PHD2 or PHD3 also increased ischemic vascularization in 3-mon-old mice (Loinard et al., 2009). 13-mon-old mice were rescued by a two-stage therapy consisting of intramuscular administration of AdCA5 followed 24 hr later by intravenous administration of BMDACs that were cultured for 4 days in the presence of angiogenic growth factors and DMOG. The rationale for this staged approach with local and systemic delivery was as follows. First, AdCA5 induces production of angiogenic factors and thereby provides a homing signal for BMDACs. Second, direct injection of a large number of cells into the ischemic tissue would increase cell death due to hypoxia, whereas systemic administration would select for the subpopulation of cells that were capable of homing to the ischemic tissue to participate in the vascular remodeling process. Combined therapy was ineffective if the BMDACs were not treated with DMOG. At the molecular level, activation of HIF-1 in BMDACs had two important consequences. First, HIF-1 induced expression of β2 integrins, which mediate increased adherence of circulating BMDACs to vascular endothelial cells, thereby increasing BMDAC retention within ischemic tissue (Rey et al., 2009). Second, HIF-1-mediated metabolic reprogramming increased BMDAC survival within ischemic tissue (Rey et al., 2011). Combined therapy resulted in limb salvage even when both BMDAC donor and ischemic recipient mice were 17 mon old, representing a model for autologous BMDAC administration to elderly patients with critical limb ischemia. A recombinant adenovirus, which encoded the HIF-1α bHLH-PAS domain fused to the herpes simplex virus VP16 transactivator protein, was administered to patients with critical limb ischemia by intramuscular injection without any adverse effects but the treatment had no significant therapeutic effect in a phase II trial (Creager et al., 2011). Although this trial involved administration of a potentially immunogenic non-native protein, which may have contributed to failure, the results suggest that combined gene and cell therapy may be required in humans as well as mice.
Wound Healing
The principal vascular response to heart or limb ischemia involves arteriogenesis, the remodeling of collateral blood vessels, whereas cutaneous wound healing requires angiogenesis, the budding of new capillaries from existing vessels, and vasculogenesis, in which non-resident cells are recruited to participate in de novo blood vessel formation. Both local and systemic responses to wounding contribute to the reparative vascularization process. Endothelial cells in preexisting vessels are activated to initiate angiogenesis in the injured tissue. In addition, the HIF-1-dependent release of cytokines from the wound elicits the mobilization and homing of BMDACs, which can participate in vasculogenesis or stimulate angiogenesis through paracrine mechanisms.
As in the case of CAD and PAD, impaired wound healing is associated with both aging and diabetes, with the combination having synergistic negative effects (Brem et al., 2007). Foot ulcers precede 85% of lower limb amputations in diabetic individuals. Compared to non-diabetic littermates, HIF-1α expression was markedly decreased in excisional skin wounds of young db/db mice (Mace et al., 2007). HIF-1 activity was inhibited in skin fibroblasts from db/db mice exposed to high glucose concentrations, which could be reversed by treatment with DMOG or desferrioxiamine, and topical application of desferrioxamine also improved wound vascularization and healing in db/db mice (Botusan et al., 2008; Thangarajah et al., 2009). Aging was associated with decreased expression of angiogenic factors and delayed wound healing in db/db mice (Liu et al., 2008). Expression of CA5 (Liu et al., 2008) or other constitutively active forms of HIF-1α (Mace et al., 2007; Botusan et al., 2008) resulted in increased angiogenic gene expression, BMDAC mobilization, wound vascularization, and rate of wound healing. Intramuscular administration of AdCA5 also improved recovery of perfusion and limb salvage after femoral artery ligation in db/db mice (Sarkar et al., 2009).
Burn injuries represent a major health problem affecting ~1 million Americans annually. In wild-type mice subjected to burn wounding, HIF-1α protein levels in the wound, SDF-1 levels in plasma, and BMDACs circulating in blood were increased on day 2, leading to increased wound vascularization and perfusion on day 7, whereas these angiogenic responses were impaired in Hif1a+/- mice (Zhang et al., 2010). Aging also impaired HIF-1α and SDF-1 expression in response to burn wounding. When BMDACs from young donor mice were injected intravenously, homing to burn wound tissue was impaired in old recipients, whereas the age of the BMDAC donor had no effect on homing (Zhang et al., 2011). In contrast, conditional knockout of HIF-1α in Tie2 lineage cells (which include BMDACs and endothelial cells) of the donor or recipient inhibits BMDAC homing to burn wounds (Sarkar et al., 2011).
Organ Transplant Rejection
Chronic rejection following lung transplantation, which is manifested as obliterative bronchiolitis (airway fibrosis), accounts for the 50% five-year survival, which is the worst of all organ allografts. The lung, which is perfused by the pulmonary arteries, pulmonary veins, and bronchial artery, is the only organ for which the arterial blood supply is not reestablished during transplant surgery (Wilkes, 2011). Autopsy data indicate that obliterative bronchiolitis is preceded by the loss of airway microvasculature (Luckraz et al., 2004). In an orthotopic tracheal transplantation model, analysis of knockout mice demonstrated that HIF-1-dependent recruitment of recipient Tie2+ angiogenic cells and repair of airway microvasculature was a critical determinant of graft survival and treatment of grafts with AdCA5 prior to transplantation increased graft perfusion, decreased fibrosis, and increased graft survival (Jiang et al., 2011). These results represent a paradigm shift in our understanding of chronic rejection from a purely immunological disease to one in which vascular responses to ischemia play a major role. Chronic rejection of renal transplants also involves destruction of the microvasculature and in an allogenic kidney transplant model, treatment of donor rats with a prolyl hydroxylase inhibitor prior to nephrectomy improved survival of recipients (Bernhardt et al., 2009). Two major classes of immunosuppressive drugs given post-transplant, calcineurin and mTOR inhibitors, have been shown to block HIF-1 activity (Laughner et al., 2001; Liu et al., 2007) and may thereby exert unintended counter-therapeutic effects. Finally, the role of aging-related impairment of HIF-1 activity in chronic rejection of solid organ transplants warrants investigation.
Colitis
The pathogenesis of chronic gastrointestinal inflammatory conditions such as Crohn's disease and ulcerative colitis includes microvascular abnormalities and tissue hypoxia. The severity of colitis induced by chemical (Karhausen et al., 2004) or bacterial (Hirota et al., 2010) toxins was increased in conditional knockout mice lacking HIF-1α expression in intestinal epithelial cells. Treatment of wild-type mice with DMOG reduced the severity of chemical (Cummins et al., 2008) or bacterial (Hirota et al., 2010) toxin-induced colitis.
Translational Prospects
Drug discovery programs have been initiated at many pharmaceutical and biotech companies to develop prolyl hydroxylase inhibitors (PHIs) that, as described above for DMOG, induce HIF activity for treatment of disorders in which HIF mediates protective physiological responses. Local and/or short term induction of HIF activity by PHIs, gene therapy, or other means are likely to be useful novel therapies for many of the diseases described above. In the case of ischemic cardiovascular disease, local therapy is needed to provide homing signals for the recruitment of BMDACs. Chronic systemic use of PHIs must be approached with great caution: individuals with genetic mutations that constitutively activate the HIF pathway (described below) have increased incidence of cardiovascular disease and mortality (Yoon et al., 2011). On the other hand, the profound inhibition of HIF activity and vascular responses to ischemia that are associated with aging suggest that systemic replacement therapy might be contemplated as a preventive measure for subjects in whom impaired HIF responses to hypoxia can be documented. In C. elegans, VHL loss-of-function increases lifespan in a HIF-1-dependent manner (Mehta et al., 2009), providing further evidence for a mutually antagonistic relationship between HIF-1 and aging.
Diabetes is an even more complicated condition: as described above, both impaired large-vessel responses to ischemia (CAD and PAD) and impaired small-vessel responses to cutaneous wounding due, at least in part, to loss of HIF activation are common. However, diabetes is also associated with excessive small-vessel proliferation (ocular neovascularization) and HIF-1 appears to play a key role in the pathogenesis of this complication (Yoshida et al., 2010).
Diseases in which HIF Activity Contributes to Pathogenesis
Hereditary Erythrocytosis
Individuals with excess red blood cell production due to germline mutations in the genes encoding VHL, PHD2, and HIF-2α have been identified, demonstrating the essential role of this pathway in regulating erythropoiesis (Yoon et al., 2011). These mutations impair hydroxylation and/or ubiquitination, thereby increasing the levels of HIF-1α and/or HIF-2α at any given PO2. Affected individuals manifest global physiologic changes that include altered ventilatory and pulmonary vascular responses to hypoxia (Smith et al., 2006) as well as altered metabolic responses to exercise (Formenti et al., 2011).
Cancer
Cancers contain hypoxic regions as a result of high rates of cell proliferation coupled with the formation of vasculature that is structurally and functionally abnormal. Increased HIF-1α and/or HIF-2α levels in diagnostic tumor biopsies are associated with increased risk of mortality in cancers of the bladder, brain, breast, colon, cervix, endometrium, head/neck, lung, ovary, pancreas, prostate, rectum, and stomach; these results are complemented by experimental studies, which demonstrate that genetic manipulations that increase HIF-1α expression result in increased tumor growth, whereas loss of HIF activity results in decreased tumor growth (Semenza, 2010). HIFs are also activated by genetic alterations, most notably, VHL loss of function in clear cell renal carcinoma (Majmunder et al., 2010). HIFs activate transcription of genes that play key roles in critical aspects of cancer biology, including stem cell maintenance (Wang et al., 2011), cell immortalization, epithelial-mesenchymal transition (Mak et al., 2010), genetic instability (Huang et al., 2007), vascularization (Liao and Johnson, 2007), glucose metabolism (Luo et al., 2011), pH regulation (Swietach et al., 2007), immune evasion (Lukashev et al., 2007), invasion and metastasis (Chan and Giaccia, 2007), and radiation resistance (Moeller et al., 2007). Given the extensive validation of HIF-1 as a potential therapeutic target, drugs that inhibit HIF-1 have been identified and shown to have anti-cancer effects in xenograft models (Table 1; Semenza, 2010).
Table 1.
Drugs that Inhibit HIF-1
| Process Inhibited | Drug Class | Prototype |
|---|---|---|
| HIF-1 α protein synthesis | Cardiac glycoside mTOR inhibitor Microtubule targeting agent Topoisomerase I inhibitor |
Digoxin Rapamycin 2-Methoxyestradiol Topotecan |
| HIF-1 α protein stability | HDAC inhibitor HSP90 inhibitor Calcineurin inhibitor Guanylate cyclase activator |
LAQ824 17-AAG Cyclosporine YC-1 |
| Heterodimerization | Antimicrobial agent | Acriflavine |
| DNA binding | Anthracycline Quinoxaline antibiotic |
Doxorubicin Echinomycin |
| Transactivation | Proteasome inhibitor Antifungal agent |
Bortezomib Amphotericin B |
| Signal transduction | BCR-ABL inhibitor Cyclooxygenase inhibitor EGFR inhibitor HER2 inhibitor |
Imatinib Ibuprofen Erlotinib, Gefitinib Trastuzumab |
Over 100 women die every day of breast cancer in the U.S. The mean PO2 is 10 mm Hg in breast cancer as compared to > 60 mm Hg in normal breast tissue and cancers with PO2 < 10 mm Hg are associated with increased risk of metastasis and patient mortality (Vaupel et al., 2004). Increased HIF-1α protein levels, as identified by immunohistochemical analysis of tumor biopsies, are associated with increased risk of metastasis and/or patient mortality in unselected breast cancer patients and in lymph node-positive, lymph node-negative, HER2+, or estrogen receptor+ subpopulations (Semenza, 2011). Metastasis is responsible for > 90% of breast cancer mortality. The requirement for HIF-1 in breast cancer metastasis has been demonstrated for both autochthonous tumors in transgenic mice (Liao et al., 2007) and orthotopic transplants in immunodeficient mice (Zhang et al., 2011; Wong et al., 2011). Primary tumors direct the recruitment of bone marrow-derived cells to the lungs and other sites of metastasis (Kaplan et al., 2005). In breast cancer, hypoxia induces the expression of lysyl oxidase (LOX), a secreted protein that remodels collagen at sites of metastatic niche formation (Erler et al., 2009). In addition to LOX, breast cancers also express LOX-like proteins 2 and 4. LOX, LOXL2, and LOXL4 are all HIF-1-regulated genes and HIF-1 inhibition blocks metastatic niche formation regardless of which LOX/LOXL protein is expressed, whereas available LOX inhibitors are not effective against all LOXL proteins (Wong et al., 2011), again illustrating the role of HIF-1 as a master regulator that controls the expression of multiple genes involved in a single (patho)physiological process.
Traumatic Shock
Secondary lung injury is a major cause of death following severe abdominal trauma. Wild-type mice subjected to abdominal trauma/hemorrhagic shock develop intestinal villous necrosis and a severe pulmonary inflammatory response, whereas both gut and lung injury were attenuated in Hif1a+/- littermates (Feinman et al., 2010). Delineation of the mechanisms underlying maladaptive HIF-1-mediated responses to trauma, which are in contrast to the protective role of HIF-1 in the experimental colitis models described above, may identify novel therapeutic targets.
Pulmonary Arterial Hypertension (PAH)
Prolonged exposure to alveolar hypoxia, as occurs in individuals with chronic lung disease, results in a remodeling of the pulmonary vasculature leading to increased pulmonary arterial pressure and right ventricular hypertrophy. Multiple HIF-1 target genes that play key roles in the response of pulmonary artery smooth muscle cells to hypoxia have been identified (Table 2; Shimoda and Semenza, 2011). Hif1a+/- and Hif2a+/- mice are both protected from hypoxic pulmonary hypertension, indicating that HIF-1α and HIF-2α play key pathogenic roles (Yu et al., 1999; Brusselmans et al., 2003). HIFs are also implicated in chemically-induced and genetic forms of PAH in which hypoxia is not an inciting factor (Bonnet et al., 2006).
Table 2.
HIF Target Genes in Arterial Smooth Muscles in Pulmonary Hypertension
| Target Gene(s) | Protein Product(s) | Consequence |
|---|---|---|
| EDN1 | Endothelin | Contraction |
| KCNA5, KCNB1 | Voltage-gated K+ channels | Increased [K+]i |
| TRPC1, TRPC6 | Transient receptor potential Ca2+ channels | Increased [Ca2+]i |
| NHE1 | Sodium-hydrogen exchanger | Increased pHi |
| PDK1 | Pyruvate dehydrogenase kinase | Glycolysis |
Obstructive Sleep Apnea
In individuals with obstructive sleep apnea, pharyngeal soft tissue occludes the airway, resulting in hypoxemia, which is sensed by carotid body (CB) chemoreceptors, leading to sympathetic activation, arousal, clearing of the airway, and reoxygenation. The cycle of hypoxia and reoxygenation is repeated dozens of times per night and results in increased ROS levels in the CB and brain. Sympathetic activation and increased plasma catecholamine levels lead to systemic hypertension. Obstructive sleep apnea is associated with both hypoxia and hypercarbia, but exposure of rodents to chronic intermittent hypoxia (CIH) is sufficient to elicit hypertension (Fletcher et al., 1992). Treatment of mice with the superoxide scavenger MnTMPyP blocks CIH-induced increases in HIF-1α, catecholamines, and blood pressure, indicating that HIF-1 is downstream of ROS, but Hif1a+/- mice lack CIH-induced increases in ROS, catecholamines, and blood pressure, suggesting that HIF-1 is upstream of ROS (Peng et al., 2006). Recent data support the existence of a feed-forward loop in which ROS trigger HIF-1α induction, leading to transcription of the Nox2 gene encoding NADPH oxidase, which generates superoxide radicals (Yuan et al., 2011).
HIF-1α is expressed at low levels in the CB under normoxic conditions and is induced by CIH, whereas HIF-2α expression is high in the CB under normoxic conditions and decreased due to calpain-dependent degradation in response to CIH (Nanduri et al., 2009). Decreased HIF-2α levels were associated with decreased expression of the Sod2 gene encoding mitochondrial superoxide dismutase, which converts superoxide to hydrogen peroxide. Treatment of CIH-exposed rats with a calpain inhibitor blocks HIF-2α degradation, restores SOD2 activity, and prevents oxidative stress and hypertension. Thus, disruption of the balance between HIF-1α and HIF-2α levels in the CB is central to the pathogenesis of CIH-induced hypertension. It is striking that continuous hypoxia induces HIF-1α and HIF-2α, resulting in pulmonary hypertension (Figure 2A), whereas CIH induces HIF-1α but inhibits HIF-2α, resulting in systemic hypertension (Figure 2B).
Figure 2. Vascular Effects of Continuous and Intermittent Hypoxia.
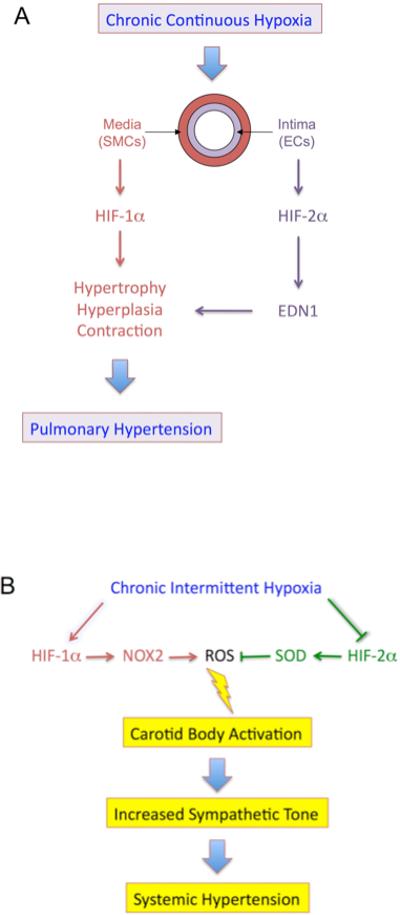
(A) Prolonged continuous alveolar hypoxia results in pulmonary arterial hypertension due to the activation of both HIF-1α and HIF-2α, leading to changes in pulmonary arterioles that reduce luminal diameter, thereby increasing pulmonary vascular resistance.
(B) Chronic intermittent hypoxia results in systemic arterial hypertension due to activation of HIF-1α and degradation of HIF-2α. Increased HIF-1α-dependent expression of NADPH oxidase 2 (NOX2), which generates superoxide anion, and decreased HIF-2α-dependent expression of superoxide dismutase 2 (SOD2), which consumes superoxide, result in increased ROS levels in the carotid body, leading to sympathetic nervous system activation and systemic hypertension.
When isolated CBs from wild-type mice are superfused with a hypoxic gas mixture, increased depolarization of the O2-sensing glomus cells and carotid sinus nerve activity was elicited, whereas these responses are absent in CBs from Hif1a+/- mice (Peng et al., 2006). In contrast, CBs from Hif2a+/- mice have augmented responses to acute hypoxia and the mice manifest increased catecholamines and blood pressure under normoxic conditions (Peng et al., 2011). MnTMPyP treatment of Hif2a+/- mice normalizes blood pressure and CB responses. Thus, even under normoxic conditions, the balance between HIF-1α and HIF-2α controls CB function and cardiovascular homeostasis. Functional antagonism has also been reported in the regulation of nitric oxide (Takeda et al., 2010) and VEGF (Eubank et al., 2011) signaling by macrophages. These findings provide novel insight into the logic underlying the acquisition of a HIF-1α paralog during vertebrate evolution.
Translational Prospects
Small molecule inhibitors of HIF activity that have anti-cancer effects in mouse models have been identified (Table 1). Inhibition of HIF impairs both vascular and metabolic adaptations to hypoxia, which may decrease O2 delivery and increase O2 utilization. These drugs are likely to be useful (as components of multidrug regimens) in the treatment of a subset of cancer patients in whom high HIF activity is driving progression. As with all novel cancer therapeutics, successful translation will require the development of methods for identifying the appropriate patient cohort. Effects of combination drug therapy also need to be considered. VEGF receptor tyrosine kinase inhibitors, which induce tumor hypoxia by blocking vascularization, have been reported to increase metastasis in mouse models (Ebos et al., 2009), which may be mediated by HIF-1; if so, combined use of HIF-1 inhibitors with these drugs may prevent unintended counter-therapeutic effects.
HIF inhibitors may also be useful in the treatment of other diseases in which dysregulated HIF activity is pathogenic. Proof of principle has been established in mouse models of ocular neovascularization, a major cause of blindness in the developed world, in which systemic or intraocular injection of the HIF-1 inhibitor digoxin is therapeutic (Yoshida et al., 2010). Systemic administration of HIF inhibitors for cancer therapy would be contraindicated in patients who also have ischemic cardiovascular disease, in which HIF activity is protective. The analysis of SNPs at the HIF1A locus described above suggests that the population may include HIF hypo-responders, who are at increased risk of severe ischemic cardiovascular disease. It is also possible that HIF hyper-responders, such as individuals with hereditary erythrocytosis, are at increased risk of particularly aggressive cancer.
O2 and Evolution, Part 2
When lowlanders sojourn to high altitude, hypobaric hypoxia induces erythropoiesis, which is a relatively ineffective response because the problem is not insufficient red cells, but rather insufficient ambient O2. Chronic erythrocytosis increases the risk of heart attack, stroke, and fetal loss during pregnancy. Many high-altitude Tibetans maintain the same hemoglobin concentration as lowlanders and yet, despite severe hypoxemia, they also maintain aerobic metabolism. The basis for this remarkable evolutionary adaptation appears to have involved the selection of genetic variants at multiple loci encoding components of the oxygen sensing system, particularly HIF-2α (Beall et al., 2010; Simonson et al., 2010; Yi et al., 2010). Given that hereditary erythrocytosis is associated with modest HIF-2α gain-of-function, the Tibetan genotype associated with absence of an erythrocytotic response to hypoxia may encode reduced HIF-2α activity along with other alterations that increase metabolic efficiency. Delineating the molecular mechanisms underlying these metabolic adaptations may lead to novel therapies for ischemic disorders, illustrating the importance of oxygen homeostasis as a nexus where evolution, biology, and medicine converge.
ACKNOWLEDGMENTS
G.L.S. is the C. Michael Armstrong Professor at Johns Hopkins University School of Medicine and is supported by grants from the National Institutes of Health (HHSN268201000032C, P01-HL65608, U54-CA143868), American Cancer Society, and Japan Science and Technology Agency. The author is grateful to Larissa Shimoda, Rena Feinman, Peter Campochiaro, Robert Cole, Chi Dang, Joseph Garcia, Frank Gonzalez, Jun Liu, Mark Nicolls, Akhilesh Pandey, Jon Resar, John Harmon, Josef Prchal, and Nanduri Prabhakar for the opportunity to collaborate with them on studies that are described in this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, Li C, Li JC, Liang Y, McCormack M, Montgomery HE, Pan H, Robbins PA, Shianna KV, Tam SC, Tsering N, Veeramah KR, Wang W, Wangdui P, Weale ME, Xu Y, Xu Z, Yang L, Zaman MJ, Zeng C, Zhang L, Zhang X, Zhaxi P, Zheng YT. Natural selection on EPAS1 (HIF-2α) associated with low hemoglobin concentration in Tibetan highlanders. Proc. Natl. Acad. Sci. USA. 2010;107:11459–11464. doi: 10.1073/pnas.1002443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt WM, Gottmann U, Doyon F, Buchholz B, Campean V, Schödel J, Reisenbuechler A, Klaus S, Arend M, Flippin L, Willam C, Wiesener MS, Yard B, Warnecke C, Eckardt KU. Donor treatment with a PHD-inhibitor activating HIFs prevents graft injury and prolongs survival in an allogenic kidney transplant model. Proc. Natl. Acad. Sci. USA. 2009;106:21276–21281. doi: 10.1073/pnas.0903978106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- Bosch-Marcé M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, Zhang H, Strazza M, Rey S, Savino L, Zhou YF, McDonald KR, Na Y, Vandiver S, Rabi A, Shaked Y, Kerbel R, Lavallee T, Semenza GL. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ. Res. 101:1310–1318. doi: 10.1161/CIRCRESAHA.107.153346. [DOI] [PubMed] [Google Scholar]
- Botusan IR, Sunkari VG, Savu O, Catrina AI, Grünler J, Lindberg S, Pereira T, Ylä-Herttuala S, Poellinger L, Brismar K, Catrina SB. Stabilization of HIF-1α is critical to improve wound healing in diabetic mice. Proc. Natl. Acad. Sci. USA. 2008;105:19426–19431. doi: 10.1073/pnas.0805230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem H, Tomic-Canic M, Entero H, Hanflik AM, Wang VM, Fallon JT, Ehrlich HP. The synergism of age and db/db genotype impairs wound healing. Exp. Gerontol. 2007;42:523–531. doi: 10.1016/j.exger.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J. Clin. Invest. 2003;111:1519–1527. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Zhong H, Bosch-Marcé M, Fox-Talbot K, Wang L, Wei C, Trush MA, Semenza GL. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1α. Cardiovasc. Res. 2008;77:463–470. doi: 10.1093/cvr/cvm035. [DOI] [PubMed] [Google Scholar]
- Chan DA, Giaccia AJ. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 2007;26:333–339. doi: 10.1007/s10555-007-9063-1. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P. Loss of HIF-2α and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 2002;8:702–710. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- Creager MA, Olin JW, Belch JJF, Moneta GL, Henry TD, Rajagopalan S, Annex BH, Hiatt WR. Effect of hypoxia-inducible factor-1α gene therapy on walking performance in patients with intermittent claudication. Circulation. 2011;124:1765–1773. doi: 10.1161/CIRCULATIONAHA.110.009407. [DOI] [PubMed] [Google Scholar]
- Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–165. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwoodie SL. The role of hypoxia in development of the mammalian embryo. Dev. Cell. 2009;17:755–773. doi: 10.1016/j.devcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle T, Köhler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, Giaccia AJ. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubank TD, Roda JM, Liu H, O'Neil T, Marsh CB. Opposing roles for HIF-1α and HIF-2α in the regulation of angiogenesis by mononuclear phagocytes. Blood. 2011;117:323–332. doi: 10.1182/blood-2010-01-261792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinman R, Deitch EA, Watkins AC, Abungu B, Colorado I, Kannan KB, Sheth SU, Caputo FJ, Lu Q, Ramanathan M, Attan S, Badami CD, Doucet D, Barlos D, Bosch-Marcé M, Semenza GL, Xu DZ. HIF-1 mediates pathogenic inflammatory responses to intestinal ischemia-reperfusion injury. Am. J. Physiol. 2010;299:G833–G843. doi: 10.1152/ajpgi.00065.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, 3rd, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J. Appl. Physiol. 1992;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- Formenti F, Constantin-Teodosiu D, Emmanuel Y, Cheeseman J, Dorrington KL, Edwards LM, Humphreys SM, Lappin TR, McMullin MF, McNamara CJ, Mills W, Murphy JA, O'Connor DF, Percy MJ, Ratcliffe PJ, Smith TG, Treacy M, Frayn KN, Greenhaff PL, Karpe F, Clarke K, Robbins PA. Regulation of human metabolism by hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA. 2010;107:12722–12727. doi: 10.1073/pnas.1002339107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Hirota SA, Fines K, Ng J, Traboulsi D, Lee J, Ihara E, Li Y, Willmore WG, Chung D, Scully MM, Louie T, Medlicott S, Lejeune M, Chadee K, Armstrong G, Colgan SP, Muruve DA, MacDonald JA, Beck PL. Hypoxia-inducible factor signaling provides protection in Clostridium difficile-induced intestinal injury. Gastroenterology. 2010;139:259–269. doi: 10.1053/j.gastro.2010.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlatky MA, Quertermous T, Boothroyd DB, Priest JR, Glassford AJ, Myers RM, Fortmann SP, Iribarren C, Tabor HK, Assimes TL, Tibshirani RJ, Go AS. Polymorphisms in hypoxia inducible factor 1 and the initial clinical presentation of coronary disease. Am. Heart J. 2007;154:1035–1042. doi: 10.1016/j.ahj.2007.07.042. [DOI] [PubMed] [Google Scholar]
- Huang LE, Bindra RS, Glazer PM, Harris AL. Hypoxia-induced genetic instability -- a calculated mechanism underlying tumor progression. J. Mol. Med. 2007;85:139–148. doi: 10.1007/s00109-006-0133-6. [DOI] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996a;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 1996b;271:C1172–C1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- Jiang X, Khan MA, Tian W, Beilke J, Natarajan R, Kosek J, Yoder MC, Semenza GL, Nicolls MR. Adenovirus-mediated HIF-1á gene transfer promotes repair of mouse airway allograft microvasculature and attenuates chronic rejection. J. Clin. Invest. 2011;121:2336–2349. doi: 10.1172/JCI46192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive hematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Invest. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kojima H, Gu H, Nomura S, Caldwell CC, Kobata T, Carmeliet P, Semenza GL, Sitkovsky MV. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1α-deficient chimeric mice. Proc. Natl. Acad. Sci. USA. 2002;99:2170–2174. doi: 10.1073/pnas.052706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulshreshtha R, Ferracin M, Negrini M, Calin GA, Davuluri RV, Ivan M. A microRNA signature of hypoxia. Mol. Cell. Biol. 2007;27:1859–1867. doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N, Martin W. The energetics of genome complexity. Nature. 2010;467:929–934. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-inducible factor 1α is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007;67:563–572. doi: 10.1158/0008-5472.CAN-06-2701. [DOI] [PubMed] [Google Scholar]
- Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- Liu L, Marti GP, Wei X, Zhang X, Zhang H, Liu YV, Nastai M, Semenza GL, Harmon JW. Age-dependent impairment of HIF-1a expression in diabetic mice: correction with electroporation-facilitated gene therapy increases wound healing, angiogenesis, and circulating angiogenic cells. J. Cell. Physiol. 2008;217:319–327. doi: 10.1002/jcp.21503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YV, Hubbi ME, Pan F, McDonald KR, Mansharamani M, Cole RN, Liu JO, Semenza GL. Calcineurin promotes hypoxia-inducible factor 1α expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J. Biol. Chem. 2007;282:37064–37073. doi: 10.1074/jbc.M705015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loenarz C, Coleman ML, Boleininger A, Schierwater B, Holland PW, Ratcliffe PJ, Schofield CJ. The hypoxia-inducible transcription factor pathway regulates oxygen sensing in the simplest animal, Trichoplax adhaerens. EMBO Rep. 2011;12:63–70. doi: 10.1038/embor.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loinard C, Ginouvès A, Vilar J, Cochain C, Zouggari Y, Recalde A, Duriez M, Lévy BI, Pouysségur J, Berra E, Silvestre JS. Inhibition of prolyl hydroxylase domain proteins promotes therapeutic revascularization. Circulation. 2009;120:50–59. doi: 10.1161/CIRCULATIONAHA.108.813303. [DOI] [PubMed] [Google Scholar]
- Luckraz H, Goddard M, McNeil K, Atkinson C, Charman SC, Stewart S, Wallwork J. Microvascular changes in small airways predispose to obliterative bronchiolitis after lung transplantation. J. Heart Lung Transplant. 2004;23:527–531. doi: 10.1016/j.healun.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Lukashev D, Ohta A, Sitkovsky M. Hypoxia-dependent anti-inflammatory pathways in protection of cancerous tissues. Cancer Metastasis Rev. 2007;26:273–279. doi: 10.1007/s10555-007-9054-2. [DOI] [PubMed] [Google Scholar]
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace KA, Yu DH, Paydar KZ, Boudreau N, Young DM. Sustained expression of HIF-1α in the diabetic environment promotes angiogenesis and cutaneous wound repair. Wound Repair Regen. 2007;15:636–645. doi: 10.1111/j.1524-475X.2007.00278.x. [DOI] [PubMed] [Google Scholar]
- Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, Gouvin LM, Sharma VM, Mercurio AM. ERβ impedes prostate cancer EMT by destabilizing HIF-1a and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010;17:319–332. doi: 10.1016/j.ccr.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science. 2009;324:1196–1198. doi: 10.1126/science.1173507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller BJ, Richardson RA, Dewhirst MW. Hypoxia and radiotherapy: opportunities for improved outcomes in cancer treatment. Cancer Metastasis Rev. 2007;26:241–248. doi: 10.1007/s10555-007-9056-0. [DOI] [PubMed] [Google Scholar]
- Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, Ratcliffe PJ. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009;284:16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2α via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc. Natl. Acad. Sci. USA. 2009;106:1199–1204. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Peng J, Zhang L, Drysdale L, Fong GH. The transcription factor EPAS-1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc. Natl. Acad. Sci. USA. 2000;97:8386–8391. doi: 10.1073/pnas.140087397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Nanduri J, Khan SA, Yuan G, Wang N, Kinsman B, Vaddi DR, Kumar GK, Garcia JA, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 2α (HIF-2α) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc. Natl. Acad. Sci. USA. 2011;108:3065–3070. doi: 10.1073/pnas.1100064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marcé M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1α deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J. Physiol. 2006;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resar JR, Roguin A, Voner J, Nasir K, Hennebry TA, Miller JM, Ingersoll R, Kasch LM, Semenza GL. Hypoxia-inducible factor 1α polymorphism and coronary collaterals in patients with ischemic heart disease. Chest. 2005;128:787–791. doi: 10.1378/chest.128.2.787. [DOI] [PubMed] [Google Scholar]
- Rey S, Lee K, Wang CJ, Gupta K, Chen S, McMillan A, Bhise N, Levchenko A, Semenza GL. Synergistic effect of HIF-1α gene therapy and HIF-1-activated bone marrow-derived angiogenic cells in a mouse model of limb ischemia. Proc. Natl. Acad. Sci. USA. 2009;106:20399–20404. doi: 10.1073/pnas.0911921106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, Luo W, Shimoda LA, Semenza GL. Metabolic reprogramming by HIF-1 promotes the survival of bone marrow-derived angiogenic cells in ischemic tissue. Blood. 2011;117:4988–4998. doi: 10.1182/blood-2010-11-321190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar K, Fox-Talbot K, Steenbergen C, Bosch-Marce M, Semenza GL. Adenoviral transfer of HIF-1α enhances vascular responses to critical limb ischemia in diabetic mice. Proc. Natl. Acad. Sci. USA. 2009;106:18769–18774. doi: 10.1073/pnas.0910561106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar K, Rey S, Zhang X, Sebastian R, Marti GP, Fox-Talbot K, Cardona AV, Du J, Tan YS, Liu L, Lay F, Gonzalez FJ, Harmon JW, Semenza GL. Cardiovasc Res. 2011 Oct 25; doi: 10.1093/cvr/cvab020. 2011 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117:e207–e217. doi: 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, Marck BT, Matsumoto AM, Shelton JM, Richardson JA, Bennett MJ, Garcia JA. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat. Genet. 2003;35:331–340. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- Shimoda LA, Semenza GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011;183:152–156. doi: 10.1164/rccm.201009-1393PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson TS, Yang Y, Huff CD, Yun H, Qin G, Witherspoon DJ, Bai Z, Lorenzo FR, Xing J, Jorde LB, Prchal JT, Ge R. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329:72–75. doi: 10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, Liu C, Maxwell PH, McMullin MF, McNamara CJ, Percy MJ, Pugh CW, Ratcliffe PJ, Talbot NP, Treacy M, Robbins PA. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006;3:e290. doi: 10.1371/journal.pmed.0030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Swietach P, Vaughan-Jones RD, Harris AL. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007;26:299–310. doi: 10.1007/s10555-007-9064-0. [DOI] [PubMed] [Google Scholar]
- Takeda N, O'Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, Suda T. Regulation of the HIF-1α level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Thangarajah H, Yao D, Chang EI, Shi Y, Jazayeri L, Vial IN, Galiano RD, Du XL, Grogan R, Galvez MG, Januszyk M, Brownlee M, Gurtner GC. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc. Natl. Acad. Sci. USA. 2009;106:13505–13510. doi: 10.1073/pnas.0906670106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998;12:3320–3324. doi: 10.1101/gad.12.21.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P, Mayer A, Hockel M. Tumor hypoxia and malignant progression. Meth. Enzymol. 2004;381:335–354. doi: 10.1016/S0076-6879(04)81023-1. [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang B-H, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF-1〈 eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011;8:399–411. doi: 10.1016/j.stem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wan C, Deng L, Liu X, Cao X, Gilbert SR, Bouxsein ML, Faugere MC, Guldberg RE, Gerstenfeld LC, Haase VH, Johnson RS, Schipani E, Clemens TL. The hypoxia-inducible factor a pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Invest. 2007;117:1616–1626. doi: 10.1172/JCI31581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk R, Weizman I, Shilo BZ. trachealess encodes a bHLH-PAS protein that is an inducer of tracheal cell fates in Drosophila. Genes Dev. 1996;10:93–102. doi: 10.1101/gad.10.1.93. [DOI] [PubMed] [Google Scholar]
- Wilkes DS. Chronic lung allograft rejection and airway microvasculature: is HIF-1 the missing link? J. Clin. Invest. 2011;121:2155–2157. doi: 10.1172/JCI58329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CC, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, Fraley SI, Wong CM, Khoo US, Ng IO, Wirtz D, Semenza GL. Hypoxiainducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. USA. 2011;108:16369–16374. doi: 10.1073/pnas.1113483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, Zheng H, Liu T, He W, Li K, Luo R, Nie X, Wu H, Zhao M, Cao H, Zou J, Shan Y, Li S, Yang Q, Asan, Ni P, Tian G, Xu J, Liu X, Jiang T, Wu R, Zhou G, Tang M, Qin J, Wang T, Feng S, Li G, Huasang, Luosang J, Wang W, Chen F, Wang Y, Zheng X, Li Z, Bianba Z, Yang G, Wang X, Tang S, Gao G, Chen Y, Luo Z, Gusang L, Cao Z, Zhang Q, Ouyang W, Ren X, Liang H, Zheng H, Huang Y, Li J, Bolund L, Kristiansen K, Li Y, Zhang Y, Zhang X, Li R, Li S, Yang H, Nielsen R, Wang J, Wang J. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon D, Ponka P, Prchal JT. Hypoxia and hematopoiesis. Am. J. Physiol. 2011;300:C1215–C1222. doi: 10.1152/ajpcell.00044.2011. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Zhang H, Iwase T, Shen J, Semenza GL, Campochiaro PA. Digoxin inhibits retinal ischemia-induced HIF-1α expression and ocular neovascularization. FASEB J. 2010;24:1759–1767. doi: 10.1096/fj.09-145664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AY, Frid MG, Shimoda LA, Wiener CM, Stenmark K, Semenza GL. Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am. J. Physiol. 1998;275:L818–L826. doi: 10.1152/ajplung.1998.275.4.L818. [DOI] [PubMed] [Google Scholar]
- Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J. Clin. Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell. Physiol. 2011;226:2925–2933. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun Z, Maecker HL, Johnson RS, Giaccia AJ. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev. Cell. 2002;2:331–341. doi: 10.1016/s1534-5807(02)00131-4. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marcé M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang H, Wong CC, Wei H, Gilkes DM, Korangath P, Chaturvedi P, Schito L, Chen J, Krishnamachary B, Winnard PT, Jr., Raman V, Zhen L, Mitzner WA, Sukumar S, Semenza GL. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2011 Aug 22; doi: 10.1038/onc.2011.365. 2011 10.1038/onc.2011.365. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang X, Liu L, Wei X, Tan YS, Tong L, Chang R, Ghanamah MS, Reinblatt M, Marti GP, Harmon JW, Semenza GL. Impaired angiogenesis and mobilization of circulating angiogenic cells in HIF-1α heterozygous-null mice after burn wounding. Wound Repair Regen. 2010;18:193–201. doi: 10.1111/j.1524-475X.2010.00570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Sarkar K, Rey S, Sebastian R, Andrikopoulou E, Marti GP, Fox-Talbot K, Semenza GL, Harmon JW. Aging impairs the mobilization and homing of bone marrow-derived angiogenic cells to burn wounds. J. Mol. Med. 2011;89:985–995. doi: 10.1007/s00109-011-0754-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel AS, Johnson RS, Nizet V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J. Mol. Med. 2007;85:1339–1346. doi: 10.1007/s00109-007-0282-2. [DOI] [PubMed] [Google Scholar]