

Abstract
A series of tetrakis-azaaromatic quaternary ammonium salts was synthesized in order to identify compounds with higher affinity and selectivity as antagonists at neuronal nicotinic receptor subtypes that mediate nicotine-evoked DA release. A high hit rate was achieved in identifying potent analogues that inhibit these nAChRs. Three tetrakis analogues, 11j, 11f and 11g, were identified as potent (IC50 = 3, 28 and 56 nM, respectively) antagonists at these receptors. These compounds represent a novel structural class of nicotinic receptor antagonists.
Keywords: Nicotinic acetylcholine receptor, Quaternary ammonium, Dopamine release, Nicotine addiction
Nicotine evokes striatal dopamine release through stimulating neuronal nicotinic acetylcholine receptors (nAChRs) on dopaminergic neurons, which underlies reward produced by nicotine.1,2 nAChRs are believed to be homomeric or heteromeric pentamers composed of a heterogeneous family of subunits (α2–7, and β2–4) potentially forming a plethora of subtypes with a broad range of pharmacological and electrophysiological properties.3 Multiple subunit combinations also mediate nicotine-evoked dopamine (DA) release at striatal presynaptic DA terminals. The following subtypes have been implicated in mediating nicotine-evoked striatal dopamine release: α6β2β3*, α4α6β2β3*, α6β2*, α4α6β2*, α4β2* and α4α5β2*.4–6
Nicotine-evoked DA release is completely inhibited by the nonselective, noncompetitive nAChR antagonist mecamylamine.7 Mecamylamine has been investigated as a smoking cessation therapy, but peripheral side-effects precluded its clinical development.8–10 In our continued search for effective therapies to treat nicotine addiction, we have focused on the development of subtype-selective antagonists that target nAChRs that mediate nicotine-evoked DA release. Such a strategy may provide novel treatments which circumvent the untoward side-effects of mecamylamine.11–15
Modification of the nicotine molecule by quaternization of the pyridine-N atom with a lipophilic substituent to afford N-substituted analogues converts nicotine from a nAChR agonist to an antagonist.16a Moreover, emergence of subtype selectivity was obtained for some analogues depending upon the length of the N-n-alkyl group.16b Although N-n-dodecylnicotinium iodide (Fig. 1, NDDNI, 1a,) was the most potent (IC50 = 9 nM) inhibitor of nicotine-evoked DA overflow from striatal slices, this analogue had only 15-fold selectivity, since it also inhibited [3H]nicotine binding to the high affinity nAChR. An improvement in selectivity was obtained with N-n-octanylnicotinium iodide (NONI, 1b); however this analogue was 70-fold less potent than NDDNI.16b Second generation compounds based upon a bis-quaternary ammonium scaffold (see Fig. 1) identified N,N’-dodecane-1,12-diyl-bis-3-picolinium dibromide (Fig. 1, bPiDDB; 2) as a highly potent and selective antagonist for nAChR subtypes that mediate nicotine-evoked striatal DA release.17 bPiDDB had no affinity for either the α4β2* or α7* nAChR binding sites in rat brain membranes, but potently inhibited nicotine-evoked DA overflow (Ki = 2 nM), demonstrating high potency and selectivity for these receptors.17 bPiDDB has also been demonstrated subsequently to be brain bio-available and enters the CNS compartment by facilitated transport via the blood-brain barrier choline transporter.18 bPiDDB has similar affinity for this transporter as the natural substrate, choline.18 In behavioral studies, bPiDDB has been shown to decrease nicotine self-administration in rats.19
Figure 1.

Structures of three generations of azaaromatic quaternary ammoniun salts acting as nAChR antagonists at subtypes mediating nicotine-evoked DA release.
Further structural elaboration of the bPiDDB molecule resulted in the synthesis of a series of compounds in which a central phenyl core was utilized to append three linker units terminating in azaaromatic quaternary ammonium head groups. 20 This led to the identification of 1,3,5-tri-{5-[1-(3-picolinium)]-pent-1-ynyl}benzene tribromide (Fig. 1, tPy3PiB, 3), a compound which exhibited high potency and selectivity for nAChR subtypes mediating nicotine-evoked [3H]dopamine release with an IC50 of 0.2 nM.20
Continuing in the structural evolution of the above sub-libraries, i.e. the mono- to bis- to tris-azaaromatic quaternary ammonium analogues, an additional quaternary ammonium head group was introduced into the tris-scaffold to afford a series of tetrakis analogues. This modification was investigated to determine if analogues in the tetrakis series would have enhanced potency and selectivity due to the greater number of cationic interaction points with the receptor protein.
Thus a novel series of tetrakis-quaternary ammonium analogues have been synthesized and evaluated for their ability to inhibit nicotine-evoked striatal DA release. It should be noted that receptor kinetic studies within the bis-series of compounds indicated that such compounds interact in an orthosteric manner with the nAChR target,21 although this has not yet been established for the tris and tetrakis analogues
Two series of tetrakis-azaaromatic quaternary ammonium compounds, i.e., 1,2,4,5-tetrakis-(pent-1-ynyl-5-azaaromatic quaternary ammonium)-benzene salts (8 series, Scheme 1), and 1,2,4,5-tetrakis -(n-pentanyl-5-azaaromatic quaternary ammonium)-benzene salts (11 series), were prepared. The synthesis of these compounds is illustrated in Scheme 1. Sonagashira coupling of an appropriate bromobenzene precursor with an alkynol were employed extensively in the synthesis of the bis-quaternary ammonium and tris-quaternary ammonium analogues in our previous study (Fig. 1).20 However, the Sonagashira coupling of 1,2,4,5-tetrabromobenzene with 4-pentyn-1-ol (5) in triethylamine in the presence of tetrakis-(triphenylphospine)palladium(0) proved difficult, affording a mixture containing a plethora of components. This failure is likely due to the sluggish reactivity of tetrabromobenzene under Sonogashira coupling conditions. Thus, the alternative synthon, 1,2,4,5-tetraiodobenzene (4) was utilized.22 Coupling of 4 with 5 afforded the desired 1,2,4,5-benzene-tetrakis-1-pentyn-5-ol (6). The pentyn-5-ol side chains of this compound were then either directly transformed into the corresponding pentynyl bromide side chains to provide 7, or catalytically reduced to the corresponding pentan-1-ol side chains to produce 9. The resulting 9 was subsequently brominated to give 10. Reaction of 7 or 10 with an appropriate substituted pyridine or related azaaromatic precursor afforded the desired azaaromatic quaternary ammonium salt, 8 and 11, respectively. Characterization data for selected compounds are provided in the references and notes section.23
Scheme 1.

Synthesis of the tetrakis-azaaromatic quaternary ammonium compounds series 8 and 11.
The above tetrakis-azaaromatic quaternary ammonium compounds were initially evaluated for inhibition of nAChRs mediating nicotine-evoked [3H]DA release from superfused rat striatal slices using a probe concentration of 100 nM in a rapid screening assay. Also, interaction of these analogues with α4β2* and α7* nAChR subtypes was assessed in rapid-throughput [3H]nicotine and [3H]methyllycaconitine (MLA) binding assays, respectively, using the same probe concentration. The three most active antagonists at nAChRs mediating nicotine-evoked DA release (i.e. 11f, 11g and 11j) were then evaluated across a full concentration range to determine their IC50 values.
[3H]Nicotine and [3H]MLA binding assays were performed according to previously reported methods,16a using 3 nM and 2.5 nM concentrations of radioligand, respectively, and 10 µM cytisine and 10 µM nicotine, respectively, to assess nonspecific binding to whole brain membranes. Analogues were evaluated at a probe concentration of 100 nM. Amount of inhibition is presented as a percentage of radioligand binding in the absence of analogue (control).
[3H]DA release assays were performed according to a previously published method.16b Analogue-induced inhibition of nicotine-evoked [3H]DA release was determined using 10 µM nicotine and 100 nM analogue concentrations. Amount of inhibition is presented as a percentage of the response to nicotine under control conditions (in the absence of analog) and the values are provided in Table 1. Full concentration response (1 nM to 10 µM) was performed for the three most promising analogs and IC50 values were determined using an iterative nonlinear least squares curve-fitting program, PRISM version 4.0 (GraphPAD Software, Inc., San Diego, CA).
Table 1.
Inhibition of [3H]NIC binding (probing α4β2* nAChRs) and [3H]MLA binding (probing α7* nAChRs) to rat brain membranes, nicotine-evoked [3H]DA release from superfused rat striatal slice.a
| Compounds | NIC binding (% inhibition at 100 nM) | MLA binding (% inhibition at 100 nM) | DA Release (% inhibition at 100 nM) | Compounds | NIC binding (% inhibition at 100 nM) | MLA binding (% inhibition at 100 nM) | DA Release (% inhibition at 100 nM) | ||
|---|---|---|---|---|---|---|---|---|---|
| 8a | 11 ± 11. % | 18 ± 7.8 % | 16 ± 13 % | 11a | 12 ± 4.4 % | 12 ± 6.9 % | 8 ± 4 % | ||
| 8b | 24 ± 5.5 % | 3.0 ± 3.0 % | 35 ± 14 % | 11b | 12 ± 8.3 % | 8.0 ± 5.3 % | 46 ± 5 % | ||
| 8c | 9.8 ± 1.2 % | 0.6 ± 0.6 % | 17 ± 6 % | 11c | 0.5 ± 0.5 % | 18 ± 10 % | 36 ± 11 % | ||
| 8d | 0 ± 0 % | 3.9 ± 3.9 % | 12 ± 5 % | 11d | 3.7 ± 3.7 % | 15 ± 8.0 % | 45 ± 14 % | ||
| 8e | 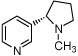 |
38 ± 7.4 % | 15 ± 2.0 % | 31 ± 13 % | 11e | 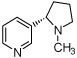 |
7.4 ± 4.7 % | 26 ± 14 % | 43 ± 21 % |
| 8f | 2.9 ± 2.9 % | 53 ± 3.5 % | 17 ± 17 % | 11f | 13 ± 3.3 % | 16 ± 8.0% | 54 ± 10 % IC5=28 ± 11 nMb |
||
| 8g | 0 ± 0 % | 4.9 ± 3.8 % | 39 ± 17 % | 11g | 6.5 ± 6.5 % | 24 ± 13 % | 63 ± 12 % IC50=56 ± 45 nMb |
||
| 8h | 9.7 ± 9.7 % | 17 ± 8.9 % | 24 ± 14.3 % | 11h | 0.9 ± 0.9 % | 27 ± 15 % | 27 ± 2% | ||
| 8i | 0.3 ± 0.3 % | 53 ± 0.8 % | 37 ± 15 % | 11i | 6.6 ± 4.5 % | 33 ± 17 % | 19 ± 15 % | ||
| 8j | 0 ± 0 % | 5.7 ± 5.7 % | 41 ± 15 % | 11j | 9.6 ± 5.2 % | 20 ± 13 % | 64 ± 15 % IC50=3.38 ± 2.62nMb |
||
| 2 | bPiDDB | Ki = 48.6 µMb | Ki > 100 µMb | IC50 = 2 nMb | 3 | tPy3PIB | 10 ± 1.9 % | 0 ± 0 % | 40 ± 12 % IC50=0.2 ± 0.07 nMb |
Percentage of inhibition at 100 nM are presented unless otherwise specified. Each value represents data from at least three independent experiments, each performed in duplicate.
IC50 from full concentration response assay; data from 4–5 independent experiments, each performed in duplicate.
The construction of the tetrakis-quaternary ammonium compounds are based on a skeleton incorporating four identical quaternary ammonium moieties attached to a central phenyl ring at the 1,2,4 and 5 positions, each through a 5 carbon linker unit. The underlying rational was that increasing the number of quaternary ammonium moieties in the molecule from two to three to four would increase progressively the ionic interaction of such molecules with the putative negatively charged binding sites on the target protein. Either a flexible or slightly more rigid linker that resulted in a more extensively planarized central aromatic core moiety, was utilized to probe the likely conformational orientation and the intramolecular distances between the head groups in the molecule. In addition, a variety of different quaternary ammonium head group structures was introduced into the tetrakis scaffold.
The results are summarized in Table 1. Overall, 7 out of the 20 tetrakis-analogues evaluated inhibited nicotine-evoked DA release in striatal slices by >40%, and an additional 5 inhibited nicotine-evoked DA release by 30–40%. This high hit rate of the tetrakis-analogues confirmed the feasibility of the SAR approach. There are note-worthy structure-activity relationships among these series of molecules. First, all the analogues in series 11 (i.e., analogues with fully saturated linkers), with the exception of 11a and 11i, exhibited greater inhibition of nicotine-evoked striatal DA release than their structural counterparts in the series 8 analogues (i.e., analogues with linkers containing an acetylene bond). Six out of seven of the most active inhibitors contained a saturated linker, i.e. 11b, 11d, 11e, 11f, 11g and 11j. It is interesting to note that the most active compound in the 8 series (i.e., 8j) and the most active compound in the 11 series (11j) both contained the same 3-(4-hydroxypropyl)pyridinium head group. Similarly, the second most active compound in both the 8 and 11 series (8g and 11g) both contained an isoquinolinium head group. Thus, this result indicates that the nature of the head group is important in the optimization of compounds inhibiting nicotine-evoked DA release. Interestingly, compounds 8f and 8i, both of which incorporate large lipophilic substituents into the pyridinium head groups, exhibited significant inhibition in the MLA assay, consistent with our previous findings with the structurally-related tris-azaaromatic quaternary ammonium compounds.20
Several compounds with head groups containing one or more lipophilic substituents on the pyridine ring exhibited good activity inhibiting nicotine-evoked striatal DA release. For example tetrakis-isoquinolinium, 4-picolinium and nicotinium heads with both saturated linkers (i.e., 8g, 8b and 8e) and unsaturated linkers (i.e., 11g, 11b and 11e) inhibited nicotine-evoked striatal DA release (35%–63%). This is consistent with our previous observations in the bis and tris-analogue series. Surprisingly, the 3-picolinium head group, which afforded quite robust antagonist activity in the previous bis- (bPiDDB) and tris- (tPy3PiB) quaternary ammonium scaffolds in their inhibition of nicotine-evoked striatal DA release, combined with either saturated or unsaturated linkers in the current study, turned out to be poor antagonists in the tetrakis series of compounds. However, it must be emphasized that the tetrakis-series of analogues do not contain within their structure the 1, 3, 5 phenyl substitution pattern present in the tris-scaffold.
Interestingly, introduction of a 1-hydroxypropanyl substituent into the pyridine ring resulted in the highly potent nAChR antagonists 8j and 11j. 11j is the most potent compound in the series 11 analogues, with an IC50 of 3 nM (Fig 2); whereas 8j is the most potent inhibitor in series 8 analogues. This observation uncovers an additional site of interaction with respect to the interaction of the quaternary ammonium head group substituent with the nAChR binding site(s). While ionic interaction of the head groups with the nAChR binding site may be one of the most important factors, it is possible that hydrogen bonding interactions involving the 1-hydroxypropanyl substituent may also play a significant role. This may open up another area of structural optimization worthy of investigation.
Fig 2.

Compound 11j inhibited S(−)-nicotine evoked [3H]DA overflow from rat striatal slices in concentration-dependent manner. Slices were superfused in the absence (control) or presence of analogue for 36 min prior to S(−)-nicotine addition to the buffer. Superfusion continued for 36 min with S(−)-nicotine added to the buffer. Control represents [3H]DA overflow in response to nicotine (10 µM). Data are expressed as mean ± SEM [3H]DA overflow. n = 4 rats/analogue.
It should be noted that evaluation of a molecule representing just the pyridinium headgroup moiety alone, i.e. N-methylpyridinium iodide (NMPi), afforded weak inhibitory properties in the dopamine release assay (Fig. 3). This indicates that the tetrakis scaffold can position four of these quaternary ammonium headgroups in a preferable orientation for favorable interaction with the receptor binding site, lowering the entropic barrier for receptor interaction.
Fig. 3.

S(−)-Nicotine-evoked fractional release of [3H]DA from rat striatal slices superfused with 100 nM N-methylpyridinium iodide (NMPi). Data are expressed as mean ± SEM fractional release as a percent of tissue tritium content. Control represents the amount of fractional release evoked by S(−)-nicotine in the absence of compound. The effect of mecamylamine (MEC) was determined as a positive control. Two-way ANOVA was performed to analyze the ability of MEC and NMPi (a between-subject factor) to inhibit S(−)-nicotine-evoked [3H]DA fractional release and time was a within-subject factor. Significant inhibition was obtained with MEC, but NMPi did not produce a significant inhibitory effect. n = 4–6 rats/analogue.
In conclusion, a novel series of tetrakis-quaternary ammonium compounds has been synthesized and evaluated for their ability to potently and selectivity inhibit striatal nicotine-evoked DA release. A high hit rate was achieved. The results suggest that the novel strategy to identify highly potent nAChR ligands through progressive introduction of cationic moieties around a common phenyl ring core structure can lead to potent inhibition of nAChR involved in nicotine-evoked DA release. Thus, compound 11j represents a tetrakis analogue with an unique 3-N-(3-hydoxypropanyl)-pyridinium head group, this compound and afforded the highest potency in this SAR study. This observation may offer the opportunity for further structural optimization in this interesting series of tetrakis analogues.
Acknowledgement
This research was supported by NIH Grant U19DA017548.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Corrigall WA, Franklin KBJ, Coen KN, Clarke PBS. Psychopharmacology. 1992;107:285. doi: 10.1007/BF02245149. [DOI] [PubMed] [Google Scholar]
- 2.Dani JA, Ji D, Zhou FM. Neuron. 2001;31:349. doi: 10.1016/s0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- 3.Corringer PJ, Le Novere N, Changeux JP. Annu. Rev. Pharmacol. Toxicol. 2000;40:431. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- 4.Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. Mol. Pharmacol. 2004;65:1526. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- 5.Luetje WL. Mol Pharmacol. 2004;65:1333. doi: 10.1124/mol.65.6.1333. [DOI] [PubMed] [Google Scholar]
- 6.Gotti C, Moretti M, Clementi F, Riganti L, McIntosh JM, Collins AC, Marks MJ, Whiteaker P. Mol. Pharmacol. 2005;67:2007. doi: 10.1124/mol.105.011940. [DOI] [PubMed] [Google Scholar]
- 7.Grady SR, Marks MJ, Wonnacott S, Collins AC. J. Neurochem. 1992;59:848. doi: 10.1111/j.1471-4159.1992.tb08322.x. [DOI] [PubMed] [Google Scholar]
- 8.Rose JE, Behm FM, Westman EC, Levin ED, Stein RM, Ripka G, V Clin. Pharmacol. Ther. 1994;56:86. doi: 10.1038/clpt.1994.105. [DOI] [PubMed] [Google Scholar]
- 9.Rose JE. Annu. Rev. Med. 1996;47:493. doi: 10.1146/annurev.med.47.1.493. [DOI] [PubMed] [Google Scholar]
- 10.Rose JE, Westman EC, Behm FM, Johnson M, P, Goldberg JS. Pharmacol. Biochem. Behav. 1999;62:165. doi: 10.1016/s0091-3057(98)00153-1. [DOI] [PubMed] [Google Scholar]
- 11.Crooks PA, Ravard A, Wilkins LH, Teng LH, Buxton ST, Dwoskin LP. Drug. Dev. Res. 1995;36:91. [Google Scholar]
- 12.Dwoskin LP, Xu R, Ayers JT, Crooks PA. Exp. Opin. Ther. Pat. 2000;10:1561. [Google Scholar]
- 13.Dwoskin LP, Crooks PA. J. Pharmacol. Exp. Ther. 2001;298:395. [PubMed] [Google Scholar]
- 14.Crooks PA, Ayers JT, Xu R, Sumithran SP, Grinevich VP, Wilkins LH. Bioorg. Med. Chem. Lett. 2004;14:1869. doi: 10.1016/j.bmcl.2003.10.074. [DOI] [PubMed] [Google Scholar]
- 15.a Dwoskin LP, Wilkins LH, Pauly JR, Crooks PA. Annu. N Y Acad. Sci. 1999;868:617. doi: 10.1111/j.1749-6632.1999.tb11334.x. [DOI] [PubMed] [Google Scholar]; b Wilkins LH, Haubner A, Ayers JT, Crooks PA, Dwoskin LP. J. Pharmacol. Exp. Ther. 2002;301:1088. doi: 10.1124/jpet.301.3.1088. [DOI] [PubMed] [Google Scholar]
- 16.a Wilkins LH, Haubner A, Ayers JT, Crooks PA, Dwoskin LP. J. Pharmacol. Exp. Ther. 2002;301:1088. doi: 10.1124/jpet.301.3.1088. [DOI] [PubMed] [Google Scholar]; b Wilkins LH, Grinevich VP, Ayers JT, Crooks PA, Dwoskin LP. J. Pharmacol. Exp. Ther. 2003;304:400. doi: 10.1124/jpet.102.043349. [DOI] [PubMed] [Google Scholar]; c Dwoskin LP, Wilkins LH, Pauly JR, Crooks PA. Ann. N Y Acad. Sci. 1999;868:617. doi: 10.1111/j.1749-6632.1999.tb11334.x. [DOI] [PubMed] [Google Scholar]
- 17.Dwoskin LP, Sumithran SP, Zhu J, Deaciuc AG, Ayers JT, Crooks PA. Bioorg. Med. Chem. Lett. 2004;14:1863. doi: 10.1016/j.bmcl.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 18.Lockman PR, Manda VK, Geldenhuys WJ, Mittapalli RK, Thomas F, Albayati ZF, Crooks PA, Dwoskin LP, Allen DD. J. Pharmacol. Exp. Ther. 2008;324:284. doi: 10.1124/jpet.107.130906. [DOI] [PubMed] [Google Scholar]
- 19.Neugebauer NM, Zhang Z, Crooks PA, Dwoskin LP, Bardo MT. Psychopharmacol. 2006;184:426. doi: 10.1007/s00213-005-0163-8. [DOI] [PubMed] [Google Scholar]
- 20.Zheng G, Sumithran SP, Deaciuc AG, Dwoskin LP, Crooks PA. Bioorg. Med. Chem. Lett. 2007;17:6701. doi: 10.1016/j.bmcl.2007.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dwoskin LP, Wooters TE, Sumethran SP, Siripurapu KB, Joyce BM, Lockman PR, Manda VK, Ayers JT, Zhang Z, McIntosh JM, Crooks PA, Bardo MT. J. Pharmacol. Exp. Ther. 2008;326:563. doi: 10.1124/jpet.108.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Kort M, Correa V, Valentijn ARPM, Van der Marel GA, Potter BVL, Taylor CW, Van Boom JH. J. Med. Chem. 2000;43:3295. doi: 10.1021/jm000957c. [DOI] [PubMed] [Google Scholar]
- 23.Compound 8j: A mixture of 5,5’,5’’,5’’’-(1,2,4,5-benzentetrayl)tetrakis-[1-bromo-4-pentyne] (7) (300 mg, 0.46 mmol) and 3-(1-hydroxypropyl)pyridine (340 mg, 2.0 mmol) was heated at 60–70 °C for 18 hrs. The resulting mixture was treated with diethyl ether and then dissolved in water (15 mL), the aqueous solution was extracted extensively with chloroform (30 mL × 5). Water was removed by lyophilization to afford 350 mg of 8j. Yield: 63 %. 1H NMR (300 MHz, CD3OD) δ 9.14 (s, 4H), 9.04 (d, J = 6.0 Hz, 4H), 8.45 (d, J = 8.1 Hz, 4H), 8.05 (dd, J1 = 6.0 Hz, J2 = 8.1 Hz, 4H), 7.39 (s, 2H), 4.90 (t, J = 6.9 Hz, 8H), 3.61 (t, J = 6.0 Hz, 8H), 2.96 (t, J = 7.8 Hz, 8H), 2.75 (t, J = 6.6 Hz, 8H), 2.36−2.45 (m, 8H), 1.88–1.97 (m, 8H) ppm. ESI-MS: m/z. 1125.1, 1127.1, 1128.1. [M-Br]1+ (calcd for [C58H70Br3N4O4+]: 1125.29, 1126.30, 1127.29, 1128.29.) m/z 522.1, 523.1, 523.6. (calcd for [C58H70Br2N4O42+]: 522.19, 523.18, 523.69.) m/z 321.8, 322.1, 322.4. (calcd for [C58H70BrN4O43+]: 321.82, 322.15, 322.48.) m/z 221.7, 221.9, 222.4. (calcd for [C58H70N4O44+]: 221.63, 221.89, 222.39.) Compound 11f: A mixture of 5,5’,5’’,5’’’-(1,2,4,5- benzentetrayl)tetrakis-[1-bromopentane] (10) (330 mg, 0.49 mmol) and 3-benzyl-pyridine (390 mg, 2.3 mmol) was heated at 60–70 °C for 18 hrs. The resulting mixture was treated with diethyl ether and then dissolved in water (15 mL), the aqueous solution was extracted extensively with chloroform (30 mL × 5). Water was removed through lyophilization to afford 575 mg of the title compound. Yield: 87 %. 1H NMR (300 MHz, CD3OD) δ 9.20 (s, 4H), 8.97 (d, J = 6.0 Hz, 4H), 8.41 (d, J = 8.1 Hz, 4H), 8.00 (dd, 4H), 7.19–7.34 (m, 20H), 6.91 (s, 2H), 4.70 (t, J = 7.5 Hz, 8H), 4.27 (s, 8H), 2.51–2.57 (m, 8H), 2.01–2.11 (m, 8H), 1.56–1.62 (m, 8H), 1.43–1.50 (m, 8H) ppm. 13C NMR, 145.61, 144.26, 143.36, 142.56, 138.18, 137.35, 130.18, 129.13, 129.06, 128.01, 127.17, 61.81, 38.03, 32.11, 31.56, 31.07, 26.23 ppm. ESI-MS: m/z. 1169.3, 1270.3, 1271.2, 1272.1. (Calcd for [C74H86Br3N4+], 1269.44, 1270.44, 1271.44, 1272.44.) m/z 594.2, 595.2, 595.7. calcd for C74H86Br2N42+, 594.26, 595.26, 595.76.) m/z 369.9, 370.2, 370.5,. (calcd for C74H86BrN43+, 369.87, 370.20, 370.53.) Compound 11g:A mixture of 5,5’,5’’,5’’’-(1,2,4,5-benzentetrayl)-tetrakis-[1-bromopentane] (10) (330 mg, 0.49 mmol) and 3-(3-hydroxypropanyl)-pyridine (300 mg, 2.3 mmol) was heated at 60–70 °C for 18 hrs. The resulted mixture was treated with diethyl ether and then dissolved in water (15 mL), the aqueous solution was extracted extensively with chloroform (30 mL × 5). Water was removed through lyophilization to afford 460 mg of the title compound. Yield: 79 %. 1H NMR (300 MHz, CD3OD) δ 10.15 (s, 4H), 8.78–8.81 (m, 4H), 8.49–8.54 (m, 8H), 8.27–8.32 (m, 4H), 8.17–8.28 (m, 4H), 8.00–8.05 (m, 4H), 6.80 (s, 2H), 4.87 (t, J = 7.5 Hz, 8H), 2.44–2.51 (m, 8H), 2.15–2.22 (m, 8H), 1.46–1.58 (m, 16H) ppm. 13C NMR 149.62, 137.62, 137.18, 137.12, 134.77, 131.40, 130.42, 130.07, 127.84, 127.39, 126.38, 61.72, 31.97, 31.38, 30.94, 26.23 ppm. ESI-MS: m/z. 1109.1, 1110.1, 1111.1, 1112.1. (Calcd for [C62H70Br3N4+]: 1109.31, 1110.32, 1111.31, 1112.31.) m/z 316.4, 316.8, 317.1. (calcd for [C62H70BrN43+]: 316.49, 316.83, 317.16.) Compound 11j: A mixture of 5,5’,5’’,5’’’-(1,2,4,5-benzentetrayl)tetrakis-[1-bromopentane] (330 mg, 0.49 mmol) and 3-(3-hydroxypropanyl)-pyridine (325 mg, 2.3 mmol) was heated at 60–70 °C for 18 hrs. The resulting mixture was treated with diethyl ether and then dissolved in water (15 mL), the aqueous solution was extracted extensively with chloroform (30 mL × 5). Water was removed through lyophilization to afford 371 mg of the title compound. Yield: 62 %. 1H NMR (300 MHz, CD3OD) δ 9.07 (s, 4H), 8.94 (d, J=6.0 Hz, 4H), 8.50 (d, J = 8.4 Hz, 4H), 8.05 (dd, 4H), 6.90 (s, 2H), 4.69 (t, J = 7.5 Hz, 8H), 3.62 (t, J = 6.0 Hz, 8H), 2.99 (t, J = 7.8 Hz, 8H), 2.57 (t, J = 7.5 Hz, 8H), 2.05–2.12 (m, 8H), 1.93–1.98 (m, 8H), 1.57–1.62 (m, 8H), 1.46–1.50 (m, 8H) ppm. 13C NMR, 145.53, 144.24, 143.98, 142.24, 137.30, 130.15, 127.80, 61.77, 60.48, 33.08, 32.05, 31.55, 31.06, 29.12, 26.20 ppm. ESI-MS: m/z. 1141.3, 1142.3, 1143.2, 1144.1. (Calcd for [C58H86Br3N4O4+]: 1141.42, 1142.42, 1143.42, 1144.42.) m/z 531.2, 530.2, 531.7. (Calcd for [C58H86Br2N4O42+]: 531.25, 530.25, 531.75.) m/z 327.2, 327.5, 327.8. (Calcd for [C58H86BrN4O43+]: 327.19, 327.53, 327.86).