

Abstract
It is now recognized that small noncoding RNA sequences have the ability to mediate transcriptional activation of specific target genes in human cells. Using bioinformatics analysis and functional screening, we screened short-activating RNA (saRNA) oligonucleotides designed to target the promoter regions of the pluripotency reprogramming factors, Kruppel-like factor 4 (KLF4) and c-MYC. We identified KLF4 and c-MYC promoter-targeted saRNA sequences that consistently induced increases in their respective levels of nascent mRNA and protein expression in a time- and dose-dependent manner, as compared with scrambled sequence control oligonucleotides. The functional consequences of saRNA-induced activation of each targeted reprogramming factor were then characterized by comprehensively profiling changes in gene expression by microarray analysis, which revealed significant increases in mRNA levels of their respective downstream pathway genes. Notably, the microarray profile after saRNA-mediated induction of endogenous KLF4 and c-MYC showed similar gene expression patterns for stem cell- and cell cycle-related genes as compared with lentiviral vector-mediated overexpression of exogenous KLF4 and c-MYC transgenes, while divergent gene expression patterns common to viral vector-mediated transgene delivery were also noted. The use of promoter-targeted saRNAs for the activation of pluripotency reprogramming factors could have broad implications for stem cell research.
Keywords: gene expression profiling, mesenchymal stem cells, pluripotency genes, RNA activation, saRNA
Introduction
Small RNA molecules are important in the regulation of various molecular and biological activities in the cell, and it is now well known that short RNA sequences play a critical role in regulating the expression levels of specific genes in a targeted manner. RNA interference (RNAi) has become widely recognized to be an important gene regulatory mechanism that causes sequence-specific downregulation of mRNAs.1 Acting through this mechanism, double-stranded short interfering RNAs (siRNAs) can knock down expression levels via the RNA-induced silencing complex, which mediates degradation or translational inhibition of the targeted mRNAs.2 Moreover, in addition to RNA-induced silencing complex-mediated regulation at the post-transcriptional level, RNAi can also modulate gene transcription itself. In fission yeast, homologs of the RNA-induced silencing complex can regulate chromatin through recruitment of histone-modifying proteins to loci transcribing small noncoding RNA,3 a mechanism also seen in plants, ciliates, nematodes, and flies. Small promoter-targeted RNAs have also been shown to repress transcription and induce epigenetic changes in eukaryotic cells through a mechanism called transcriptional gene silencing.4,5
In human cells, it has recently been reported that short RNAs targeted to the promoter regions of certain genes can activate expression at the transcriptional level.6,7 This phenomenon has been called RNA activation (RNAa), and has been shown to be conserved in other mammalian species, including mouse, rat, and nonhuman primates.8 Promoter-targeted small hairpin RNAs have also been shown to efficiently upregulate genes in vivo.9 While the mechanism is not completely understood, it appears that naturally occurring antisense transcripts arising from or near the same genetic locus are able to direct recruitment of Argonaute proteins and histone methyltransferases. Short-activating RNAs (saRNA) may regulate transcription by targeting these antisense transcripts for degradation, resulting in a reversal of this epigenetic silencing and upregulation of sense mRNA.10,11,12
Based on this approach, we have developed a method to design saRNAs for upregulation of specific cellular genes. In the present study, we focused on the feasibility and genetic consequences of upregulating important target genes involved in stem cell regulation and reprogramming. Combined expression of the transcription factors Kruppel-like factor 4 (KLF4), POU5F1 (also called OCT3/4), SOX2, and c-MYC has been shown to reprogram mouse and human fibroblasts into induced pluripotent stem (iPS) cells.13,14,15,16 In particular, KLF4 is important for maintenance of embryonic stem cells,17 and has been reported to be a master regulator in embryonic stem cells that controls the expression of other pluripotency factors including POU5F1, SOX2, c-MYC, and NANOG.18 It has previously been reported that direct reprogramming can be achieved in murine fibroblasts with only Klf4, Oct4, and Sox2.19 However, it has recently been shown that c-Myc is critical for efficient induction of pluripotency in the early phases of reprogramming, by altering the metabolic state of the cell.20
Accordingly, in the present study, we employed a genomic–bioinformatic approach to design saRNAs that specifically target two of these key reprogramming genes, KLF4 and MYC. These saRNAs were tested for their ability to upregulate the targeted reprogramming factors, as well as their respective downstream genes, in human mesenchymal stem cells (MSCs), adult bone marrow-derived tissue-specific stem cells that already have multilineage differentiation potential. The effects of saRNA transfection on endogenous gene expression profiles were compared with those resulting from lentiviral vector-mediated overexpression of the exogenous KLF4 and c-MYC transgenes. To date, there have been no studies comprehensively examining the cellular gene expression profiles after saRNA-mediated gene activation of endogenous genes, particularly pluripotency-related genes, and few studies comparing how different reprogramming methods might differentially affect various cellular pathways. These results indicate that the use of saRNA shows significant potential, both as a tool for studying stem cell biology, as well as a safe method to manipulate stem cell gene expression without altering the genome.
Results
Design of saRNA candidate sequences targeting KLF4 and c-MYC
To design saRNA candidates for activation of stem cell factors, we developed a novel bioinformatic approach. The KLF4 gene, located on chromosome 9 (9q31.2), and c-MYC gene, located on chromosome 8 (8q24.21), were our initial targets for activation (Figure 1a,b). To identify potential antisense transcripts from the KLF4 and c-MYC loci, we searched the genomic region surrounding each locus for spliced expressed sequence tags (ESTs) which mapped to the positive strand. Although it is usually difficult to determine the transcriptional orientation of ESTs, orientation can be determined by using splice site signatures of spliced ESTs.21 We found no spliced ESTs that overlapped KLF4, but the scan identified one antisense EST (DB461753) ~15 kb upstream of KLF4's annotated transcription start site (TSS). We were also unable to find a spliced ESTs that overlapped c-MYC, but identified one antisense EST (BC042052) ~2 kb upstream of c-MYC's TSS. These ESTs were then further investigated as potential candidates (Figure 1c).
Figure 1.

Design of KLF4 and c-MYC saRNA candidates. (a) KLF4 locus and potential antisense target candidates. The schematic shows the genomic location of KLF4, the structure of the KLF4 transcript, and the spliced ESTs reported from various cell types in the surrounding regions. Red boxes outline the KLF4 promoter region and the closest antisense EST upstream of KLF4 (DB461753). The antisense EST DB461753 initiates roughly 15 kb from KLF4's transcription start site (TSS) and terminates more than 25 kb away. Red arrows indicate the target sites for the short-activating RNA (saRNA) candidates. (b) MYC locus and potential antisense target candidates. The schematic shows the genomic location of MYC, the structure of the MYC transcript, and the spliced ESTs reported from various cell types in the surrounding regions. Red boxes outline the MYC promoter region and the closest antisense EST upstream of KLF4 (BC042052). The antisense EST BC042052 initiates roughly 2 kb from MYC's TSS and terminates 50 kb away. Red arrows indicate the target sites for the saRNA candidates. (c) saRNA candidates for KLF4 and MYC genes. The list shows the most promising saRNAs against the antisense EST DB461753, BC042052, and saRNAs targeting KLF4 or MYC sequences within a stretch of 500 bp either upstream or downstream of the TSS for each gene. EST, expressed sequence tag; n/a, not applicable.
Recent deep sequencing experiments have revealed that antisense RNAs often are found in the region surrounding TSSs.22,23,24,25 Therefore, this region was chosen to design saRNA sequences that targeted potential antisense transcripts from the promoter region. We used the antisense sequence 500 nts upstream and downstream from the TSS (abbreviated KLF4_AS_TSS+/−500 and MYC_AS_TSS+/−500) as a second target candidate. Our goal was to design short sense RNAs that could potentially bind and degrade antisense RNAs generated from the two candidate sequences (ESTs and the regions surrounding TSSs) with the hypothesis that this binding and degradation of the antisense RNAs would activate gene expression. Candidate saRNAs targeting the 500 nt upstream of the TSS were designated PR1, whereas those targeting the 500 nt downstream of the TSS were designated PR2. To give effective antisense targeting and degradation and to minimize off-target effects, we used the GPboost siRNA design algorithm26 to identify potential short RNAs for downregulating the two candidate sequences.
From the lists of predicted siRNA candidates, we selected the two most promising non-overlapping siRNA target sites on the antisense EST DB461753 and BC042052, and the most promising siRNA target site on each side of the KLF4 and c-MYC TSS within the antisense promoter sequence (KLF4_AS_TSS+/−500 and MYC_AS_TSS+/−500). The candidate siRNAs were selected based on predicted efficacy score from GPboost.26 We found four potential saRNA candidates for activating each gene (Figure 1c).
Upregulation of target gene expression by saRNAs
Reasoning that activation of pluripotency factor gene expression might be more readily achieved in adult tissue-derived stem cells that retain restricted multilineage potential, we then tested whether these saRNA candidates could upregulate KLF4 or c-MYC expression, respectively, in primary human MSCs derived from adult bone marrow. Target gene expression levels were examined by quantitative PCR of reverse-transcribed mRNA from MSC cultures after transfection of each individual saRNA candidate oligonucleotide, as compared with transfection with an Alexa Fluor 555- or FAM-labeled negative control oligo. A transfection efficiency of >95% was determined by flow cytometry and by knockdown with control siRNAs using the same transfection conditions (See Supplementary Materials and Methods and Supplementary Figures S1 and S2). Initially, none of the saRNA oligos appeared to show any effect on target gene expression after 48 hours. However, upon continued transfection with KLF4-PR1 saRNA every other day, significant upregulation of KLF4 mRNA (Figure 2a) was observed, on the order of 2.5-fold over controls by day 4, and reaching approximately fourfold by day 6 of treatment (P < 0.01). Target gene mRNA levels after treatment with KLF4-PR1 were significantly higher when MSCs were exposed to saRNA at concentrations of 25 or 50 nmol/l, as compared with 5 nmol/l (Figure 2b). Increased Klf4 protein was confirmed in MSCs treated with KLF4-PR1 saRNA (Figure 2c, left panel) by western blot analysis, and densitometric quantitation of these blots showed over threefold upregulation of Klf4 protein relative to β-actin internal control (Figure 2c, right panel), correlating closely with the level of mRNA upregulation. This time-dependent upregulation of KLF4 gene expression by the KLF4 promoter-targeted PR1 saRNA was observed consistently over multiple experiments. None of the other three KLF4-targeted saRNA candidate sequences was able to upregulate target gene expression, although conversely, KLF4-DB1 appeared to reduce KLF4 mRNA levels in MSCs to about 60% that of scrambled oligo-treated controls by day 6.
Figure 2.

Screening of KLF4 and c-MYC short-activating RNA (saRNA) candidates in mesenchymal stem cells (MSCs). (a) RT-qPCR of KLF4 saRNAs treated in MSCs showing increase of KLF4 in KLF4-PR1. (b) RT-qPCR of KLF4 for KLF4-PR1–treated MSCs at the indicated saRNA concentrations for 6 days. (c) Western blot for Klf4 and β-actin protein and relative quantitation in control- and KLF4-PR1–treated MSCs. (d) RT-qPCR of MYC saRNAs treated in MSCs showing increase of c-MYC in MYC-PR1 and MYC-PR2. (e) RT-qPCR of MYC for MYC-PR1 and MYC-PR2–treated MSCs at the indicated saRNA concentrations for 6 days. (f) Western blot for c-Myc and β-actin protein and relative quantitation of c-Myc in control-, MYC-PR1-, and MYC-PR2–treated MSCs. Asterisks indicate statistical significance: *P < 0.05; **P < 0.01. con, control; RT-qPCR, reverse transcription-quantitative PCR.
Similarly, we found that among the four saRNA candidate sequences targeted to the c-MYC promoter and antisense ESTs, both MYC-PR1 and MYC-PR2 were able to induce consistent upregulation of MYC mRNA (Figure 2d) by up to 1.8-fold (P < 0.01) and 1.6-fold (P < 0.01), respectively, during the 6-day interval of treatment, as compared with scrambled sequence controls. While MYC-BC1 and MYC-BC2 also appeared to affect MYC mRNA levels to some extent, these effects were more modest, and were not consistent between days 4 and 6. Again, target gene mRNA levels after treatment with MYC-PR1 and MYC-PR2 were significantly higher when MSCs were exposed to saRNA at concentrations of 25 or 50 nmol/l, as compared with 5 nmol/l (Figure 2e). Image densitometry analysis of western blots from MSCs treated with MYC-PR1 or MYC-PR2 again confirmed upregulation of c-Myc protein by over 2.5-fold relative to β-actin internal control (Figure 2f). All western blots were performed in triplicate with replicates shown in Supplementary Figure S3.
To determine whether this upregulation of KLF4 and MYC is due to true transcriptional activation, expression levels of nascent RNA were assessed. MSCs were pulsed with ethynyl uridine (EU) during saRNA treatment and total RNA was isolated. Newly transcribed EU-RNA was separated from total RNA by biotinylation of EU in a copper-catalyzed “click” reaction, followed by purification on streptavidin magnetic beads. Quantitative PCR of reverse-transcribed nascent RNA showed that the level of nascent KLF4 mRNA was significantly upregulated with KLF4-PR1 treatment across a 6-day timecourse (Figure 3a). The level of nascent MYC mRNA was significantly upregulated with MYC-PR2 treatment, whereas no significant change in nascent MYC mRNA expression was seen with MYC-PR1 treatment (Figure 3b).
Figure 3.

Mechanism of activation by KLF4- and c-MYC–targeted short-activating RNAs (saRNAs). (a) RT-qPCR results from KLF4-PR1 saRNA-treated mesenchymal stem cells (MSCs), showing increases in newly transcribed KLF4 mRNA compared to total RNA. (b) RT-qPCR results from MYC-PR1- and MYC-PR2–treated MSCs showing increases in newly transcribed MYC mRNA compared to total RNA for MYC-PR2. Asterisks indicate statistical significance: *P < 0.05; **P < 0.01. (c) RT-qPCR results from KLF4- and MYC- saRNA-treated MSCs, showing no significant increase in mRNA levels of key interferon response genes. RT-qPCR, reverse transcription-quantitative PCR.
To verify that these saRNAs do not act through interferon response pathways, interferon response gene expression levels were assayed. No significant upregulation of interferon response genes was detected 24 hours after saRNA treatment (Figure 3c). In contrast, overexpression of KLF4 and c-MYC by lentiviral transduction showed dose-dependent induction of interferon response genes (Supplementary Figure S4).
To further elucidate the mechanism of activation by saRNA, the presence of promoter-associated antisense RNAs was investigated using 5′ Rapid Amplification of cDNA Ends (RACE). Since any antisense RNAs involved in regulating gene expression may not be polyadenylated,27 random hexamers were used to prime cDNA synthesis from total MSC RNA. Antisense strand-specific primers matching the KLF4-PR1, MYC-PR1, and MYC-PR2 saRNA sequences were used for 5′ RACE to amplify potential antisense RNAs that are targeted by each saRNA and may be involved in regulation of expression. RACE reactions were run on an agarose gel, followed by purification, cloning, and sequencing of any products (See Supplementary Materials and Methods). Despite the presence of bands from each saRNA primer, each was identified to be a known mRNA with homology to the 3′ end of the saRNA sequence, likely a result of mispriming at permissive annealing temperatures (Supplementary Figure S5). Repeats of this assay with more restrictive annealing temperatures yielded no products (data not shown).
To further rule out involvement of possible antisense RNAs arising downstream of each saRNA target sequence, cDNA was synthesized from total MSC RNA using antisense strand-specific primers for KLF4-PR1, MYC-PR1, and MYC-PR2. This cDNA was used as template for PCRs using primers downstream of the saRNA target sequence, which would be in the direction of the 5′ end of any putative antisense transcript. No antisense transcripts were amplified up to 525 bp from the KLF4-PR1 target site (Supplementary Figure S6a). Of the priming sites up to 667 bp from MYC-PR1, including and surpassing the MYC-PR2 target site, two products were amplified, cloned, and sequenced corresponding to the 63 and 136 bp downstream of MYC-PR1 (Supplementary Figure S6b). The expression level of this antisense transcript was significantly upregulated with MYC-PR2 treatment, whereas its upregulation with MYC-PR1 was not statistically significant (P = 0.0682) (Supplementary Figure S6c).
To assess the biological significance of saRNA activation of KLF4 and MYC in MSCs, cells were monitored for morphological changes during oligo treatment with KLF4-PR1 and MYC-PR2, which had been confirmed to be associated with increased nascent mRNA transcribed from their respective target genes. After 6 days of treatment, KLF4-PR1–treated MSCs showed marked differences in cell morphology compared with control, whereas MYC-PR2 had modest changes (Figure 4). In contrast to scrambled sequence oligo-treated controls, which appeared as progressively more densely packed fibroblastic cells over time, KLF4-PR1–treated MSCs were less confluent and predominantly arranged in clusters with epitheloid cell-like morphology. MYC-PR2–treated cells were less confluent than controls but appeared more heterogeneous with numerous epitheloid cells containing enlarged nuclei.
Figure 4.

Morphological changes with saRNA treatment. Phase contrast images of MSCs transfected with the indicated saRNA on the indicated day of a 6-day time course. MSC, mesenchymal stem cell; saRNA, short-activating RNA.
Thus, target gene mRNA levels after treatment with KLF-PR1 and MYC-PR2, were significantly higher when MSCs were exposed to these saRNA oligos at concentrations of 25 or 50 nmol/l over a 6-day treatment period. Hence, these promoter-targeted saRNA oligos consistently induced increases in both mRNA and protein expression of KLF4 and c-MYC, respectively, in a time- and dose-dependent manner. Neither of these saRNAs activated the interferon response, and both appear to act through specific activation of transcription. We therefore focused on these two positive saRNAs, using the 6-day treatment protocol, in further experiments examining how saRNA-mediated upregulation might affect downstream targets.
Gene expression profile analysis after saRNA-mediated upregulation of KLF4 and c-MYC
Microarray analysis was performed to determine the global gene expression profile and to investigate possible off-target effects with saRNA treatment. Differential gene expression profiles after upregulation of endogenous KLF4 and c-MYC by treatment with their respective saRNAs versus control oligo were examined, and compared with that after overexpression of exogenous KLF4 and c-MYC delivered by viral gene transfer using commercially available second-generation lentivirus vectors. MSCs were transduced with 0.1 pg p24 protein per cell as determined by p24 ELISA, a viral dose that does not result in significant induction of interferon response genes (Supplementary Figure S4). Normalized expression data for individual replicates are included in Supplementary Table S1. Only those cellular genes showing upward or downward changes in their expression levels at a significance level of at least P < 0.1, as compared with their levels in scrambled sequence oligo-treated controls, were included in these analyses.
Interestingly, analysis of the overall gene expression profile in human MSCs after treatment with KLF4 PR-1 (Figure 5a) and MYC-PR2 (Figure 5b) showed that the majority of cellular genes exhibited concordant changes in expression, but with some notable differences, between upregulation by saRNA and overexpression by lentiviral transduction. For MSCs treated with KLF4-PR1 saRNA, 68% of the cellular genes showing significant changes in their expression levels (971 out of 1,429 genes) showed the same pattern of regulation as Klf4 lentivirus-transduced MSCs. For MSCs treated with MYC-PR2, 64% (273 out of 429 genes) exhibited the same pattern of regulation.
Figure 5.

Differential gene expression of saRNA-transfected and virus-transduced samples compared to control. (a) Expression of all genes with corrected P value <0.05 and absolute fold change >1.5 for KLF4-PR1 and KLF4 virus samples. (b) Expression of all genes with corrected P value <0.1 and absolute fold change >1.5 for MYC-PR2 and c-MYC virus samples. (c) Expression of genes with stem cell-related gene ontology, corrected P value <0.1, and absolute fold change >1.5 for KLF4-PR1 and KLF4 virus samples. (d) Expression of genes with cell cycle-related gene ontology, corrected P value <0.1, and absolute fold change >1.5 for KLF4-PR1 and KLF4 virus samples. (e) Expression of genes with cell cycle-related gene ontology, corrected P value <0.1 and absolute fold change >1.5 for MYC-PR2 and c-MYC virus samples. saRNA, short-activating RNA.
However, this indicates that roughly a one-third of the cellular genes with significant changes in expression after saRNA transfection or lentiviral gene transfer showed discordant regulation between these two groups. To determine whether these differences were due to off-target effects or resulted from the different methods used to activate expression, we used MetaCore pathway analysis from GeneGo (Carlsbad, CA) to determine what pathways were significantly enriched in those genes that were differentially regulated between saRNA and virus samples. Notably, the pathways that were most significantly differentially regulated between KLF4-PR1 saRNA-treated MSCs and KLF4 virus-treated MSCs were those involved in cytoskeletal remodeling, macropinocytosis, and regulation of epithelial-to-mesenchymal transition (EMT), including transforming growth factor (TGF)-β induction of EMT (Table 1). The pathways that were most significantly differentially regulated between MYC-PR2 saRNA-treated MSCs and the c-MYC virus-treated MSCs included cell survival and proliferation pathways such as granzyme A signaling, TGF-β regulation, and telomere length, as well as macropinocytosis and EMT pathways (Table 2). That is, for KLF4-targeted saRNA versus KLF4-virus, as well as MYC-targeted saRNA versus MYC-virus, in both cases genes involved in the same pathways were found to be discordantly regulated in the same manner. The high degree of similarity in differentially regulated pathways observed with two different sets of saRNAs versus two different lentivirus vectors suggests that the discordant gene expression patterns primarily arise due to characteristic cellular changes in response to oligo transfection versus viral transduction, rather than off-target effects that are shared by each set of saRNAs.
Table 1. Top 10 most significantly enriched pathways among those genes that were differentially regulated between KLF4-PR1 and KLF4 virus samples.
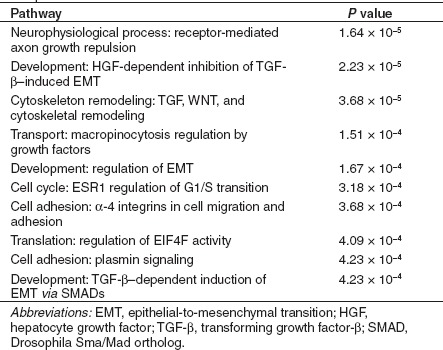
Table 2. Top 10 most significantly enriched pathways among those genes that were differentially regulated between MYC-PR2 and c-MYC virus samples.
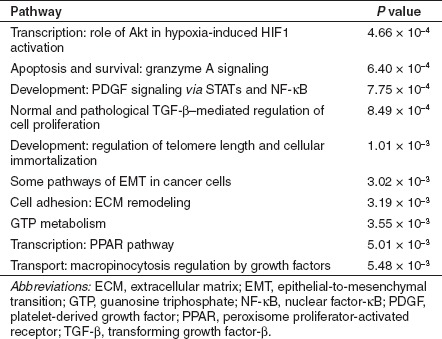
To determine whether narrowing the focus of the differential expression analysis to those types of genes we expect to be regulated by KLF4 and c-MYC would yield greater similarity, we generated lists of genes involved in stem cell maintenance, development, and proliferation, as well as cell cycle-related genes from the AmiGO gene ontology database.28 Lists of genes used in this analysis are included in Supplementary Tables S2 and S3. Heatmaps visualizing the differential expression profiles of KLF4 saRNA-treated and KLF4 virus-treated MSC compared with scrambled oligo-treated control MSC for stem cell-related genes (Figure 5c) and cell cycle-related genes (Figure 5d) showed much greater similarity, with 74% (26 out of 35) exhibiting concordant regulation for stem cell-related genes, and 80% (132 out of 165) exhibiting concordant regulation for cell cycle-related genes. Heatmaps focused on cell cycle-related genes in MYC-PR2 (Figure 5e) saRNA-treated MSC compared to MYC virus-treated MSC also showed a higher degree of similarity, with 67% of cell cycle-related genes (8 out of 12) for MYC-PR2 exhibiting concordant regulation.
Further DAVID gene ontology (GO) analysis29,30 of all genes revealed that the most significantly enriched GO terms in the KLF4-PR1 saRNA-treated samples were the same as those in the KLF4 virus-treated samples (Table 3). Similarly, the majority of the most significantly enriched GO terms in the MYC-PR2 saRNA-treated samples were also significantly enriched in the c-MYC virus-treated samples (Table 4).
Table 3. Top 10 most significantly enriched gene ontology terms among KLF4-PR1 and KLF4 virus samples from all genes with corrected P value <0.05 and absolute fold change >2.
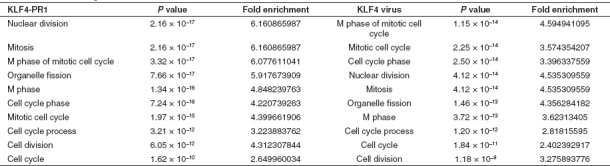
Table 4. Top 10 most significantly enriched gene ontology terms among MYC-PR2 samples from all genes with corrected P value <0.05 and absolute fold change >1.5.
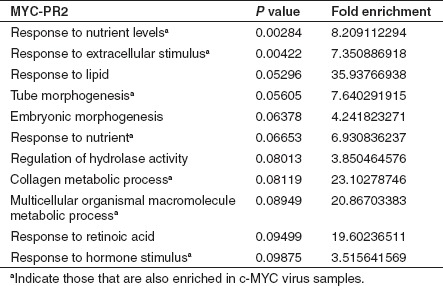
To further validate the gene expression profile, we chose several well-known transcriptional gene targets of Klf431 and c-Myc32 to verify the results seen in the microarray data by real-time PCR. We found that KLF4-PR1 saRNA transfection resulted in significantly increased expression of the Klf4 target genes cyclin D1, ornithine decarboxylase (ODC1), p21, and p53 (Figure 6a); KLF4 virus-transduced cells also showed significantly increased expression of cyclin D1 and p21 to a similar degree, although not of ODC1 or p53. Similarly, MYC-PR2 saRNA transfection resulted in significantly increased expression of c-Myc target genes ODC1, p21, and p53, as did transduction with c-MYC virus (Figure 6b).
Figure 6.

Validation of microarray gene expression of Klf4 and c-Myc target genes. (a) RT-qPCR of Klf4 target genes in KLF4-PR1 and Klf4 virus samples relative to control gene expression. (b) RT-qPCR of c-Myc target genes in MYC-PR2 and c-MYC virus samples relative to control gene expression. (c) RT-qPCR for KLF4-PR1 activation of stem cell and reprogramming factors relative to control gene expression. (d) RT-qPCR for KLF4-PR1 activation of OCT4 isoforms relative to control gene expression. (e) RT-qPCR for MYC-PR2 activation of KLF4. Asterisks indicate statistical significance: *P < 0.05; **P < 0.01. con, control; RT-qPCR, reverse transcription-quantitative PCR.
Finally, to assess the ability of the KLF4-PR1 saRNA to activate the expression of other reprogramming factors, we analyzed mRNA expression of OCT4, SOX2, and MYC, as well as the stem cell marker NANOG, by real-time PCR. We found that KLF4 activation by saRNA was also able to activate expression of OCT4, SOX2, MYC, and NANOG (Figure 6c). Further analysis of OCT4 mRNA isoforms showed that OCT4A was being significantly upregulated, whereas OCT4B showed no difference (Figure 6d).
Conversely, the ability of MYC-PR2 saRNA to activate endogenous KLF4 gene expression was also analyzed. In response to MYC-PR2 transfection, there was an approximately twofold increase in KLF4 mRNA levels (Figure 6e). Notably, this effect of MYC-PR2 saRNA was corroborated by the microarray analysis (Supplementary Table S1).
Discussion
We have been able to identify and characterize two saRNAs, KLF4-PR1 and MYC-PR2, that specifically activate transcription of endogenous KLF4 and MYC genes, respectively, in primary human MSCs. Interestingly, in both cases, it was a promoter-targeted saRNA that gave the desired effect. Given that studies implicate antisense RNAs that overlap the gene of interest as the targets for degradation in RNAa,10,11 it is conceivable that these promoter-targeted saRNAs are targeting an antisense RNA that has not yet been discovered. However, we were unable to identify any antisense transcripts arising from the region of the KLF4-PR1 target site. Antisense transcripts have been reported to arise from the vicinity of the c-Myc promoter in human cells such as prostate cancer cell lines,33,34 and we were able to identify one such antisense RNA in primary human MSCs. However, this antisense RNA does not overlap the MYC-PR2 target site and is therefore unlikely to be the RNA target of MYC-PR2. Interestingly, this antisense RNA does overlap the MYC-PR1 target site, but cells transfected with MYC-PR1 saRNA did not show increased transcription of nascent MYC mRNA. Hence it is difficult to ascertain whether MYC-PR1 does target this antisense RNA, perhaps resulting in increased accumulation of MYC sense mRNA at a post-transcriptional level, or if the observed upregulation of MYC mRNA levels by MYC-PR1 is an off-target effect. Due to the relatively small activation of MYC, it would be difficult to completely rule out off-target effects even with the use of more sophisticated techniques such as chromatin immunoprecipitation analysis. Furthermore, specific saRNA-targeted effects on MYC expression by transcriptional or post-transcriptional mechanisms, and coexisting off-target effects, are not necessarily mutually exclusive. Although it may not be possible for every gene to be activated by RNAa, our results suggest that this method could be used to generate saRNA candidates for activation of other endogenous genes for which a promoter-associated antisense RNA has not yet been defined.
Focusing on stem cell- and cell cycle-related genes, which were expected to be altered by KLF4 and c-MYC expression, we found the similarities between saRNA-transfected and virus-transduced samples to be quite striking. This was confirmed by GO analysis, as the most significantly enriched terms were nearly identical in KLF4-PR1 oligo and KLF4 virus samples, and most of the top MYC-PR2 terms were also significantly enriched in the c-MYC virus samples. In addition, several well-known KLF4 and c-MYC target genes were validated by real-time PCR, and showed a similar pattern of expression in the saRNA-treated samples as in the virus-transduced samples. Furthermore, we have shown that KLF4 saRNA is able to activate expression of endogenous pluripotency factors including OCT4, SOX2, NANOG, and MYC. Activation of OCT4 was specific to OCT4A, which has been reported to be essential for stemness in human embryonic stem cells.35 The highly specific nature of OCT4A upregulation in response to transfection of saRNA targeting KLF4 makes this likely to be a true downstream event caused by specific activation of KLF4. Furthermore, the saRNA-treated MSCs showed marked differences in cell morphology, supporting our conclusion that this activation is biologically relevant.
Interestingly, for some downstream gene targets, our results were different from what was expected, as Klf4 is expected to downregulate expression of cyclin D1, ornithine decarboxylase, and p53,31 whereas c-Myc is expected to downregulate p21.32 However, these previously observed results are often cell- and tissue type-dependent, as evidenced by more recent reports showing activation of cyclin D1 and p53 by Klf4,36,37 and activation of p21 through p53 by c-Myc38 in different cell types. In addition, our data are consistent across both saRNA- and virus-treated samples in independent microarray and real-time PCR experiments.
As with RNAi, a primary concern for using RNAa to study biological processes is the minimization of potential off-target effects. To evaluate possible off-target effects, we compared the gene expression profile of saRNA activation of KLF4 and MYC to lentiviral-mediated expression. Some significant differences were observed in the total gene expression profile, but this is perhaps not surprising, as one method employed transfection of saRNA and the other lentiviral transduction. Indeed, the use of any transduction method involving introduction of long stretches of exogenous nucleic acids, including both viral vectors as well as plasmids, will likely activate a variety of innate signaling mechanisms.39,40 This may result in unwanted upregulation of interferons and related genes that may not only cause protein synthesis shutdown and effects on cell proliferation, but may also impair normal stem cell function, as has been described in hematopoietic stem cells.41 Notably, our interferon response gene expression analysis as well as our microarray analysis of gene functional annotation and differentially regulated pathways showed no evidence of interferon response upregulation after saRNA oligo transfection. In contrast, we observed significant induction of interferon responses upon viral transduction at a dose of 1 pg p24 per cell. This corresponds roughly to an multiplicity of infection of 10, which is within the range of multiplicity of infections typically used in reprogramming protocols. Hence, it may be highly advantageous that RNA duplexes <23 bp in length will not cause induction of interferon,42 while effecting transcriptional gene activation of endogenous pluripotency factors.
In fact, when we focused on the genes that were differentially expressed between the saRNA and virus samples, the most significantly enriched pathways implicated the use of lentiviral vector-mediated gene transfer as the cause of many of the expression profile changes that were discrepant. Notably, cytoskeletal remodeling, including TGF-mediated remodeling, were upregulated in both KLF4 virus- and c-MYC virus-treated samples. It is well known that retroviruses manipulate the host cytoskeleton to facilitate virus entry and integration.43 Furthermore, the presence of TGF signaling pathways is also not surprising, considering that carryover of HIV Tat can occur after packaging of second-generation lentivirus vectors, and that Tat protein has been associated with the induction of TGF-β,44 which likely serves multiple functions for the virus, including immunosuppression and facilitation of cytoskeletal remodeling.45,46 The differential regulation of macropinocytosis in both KLF4 virus- and c-MYC virus-treated samples is also likely due to viral entry, as both native HIV- and vesicular stomatitis virus-G (VSV-G)–pseudotyped HIV vectors have been reported to use macropinocytosis for entry.47,48 HIV Tat protein has also been shown to enter cells by macropinocytosis.49
Notably, several EMT regulation pathways were observed to be differentially regulated between saRNA- and virus-treated samples, irrespective of whether it was KLF4 or c-MYC being targeted or transduced. This was quite striking, as it has been reported that suppression of EMT signals is required for reprogramming mouse fibroblasts.8 Klf4 serves to activate the mesenchymal-to-epithelial transition (MET) that is required for reprogramming, whereas Oct4, Sox2, and c-Myc suppress TGF-β–induced EMT. The differential regulation of these pathways in our experiments fits well into this model, as in both cases Klf4 and c-Myc are acting in competition with the TGF-β induced by lentiviral infection. This suggests that using lentiviral vectors to activate reprogramming factors may actually hinder the reprogramming process, as these vectors have been shown to activate TGF-β signaling. This underscores the need for alternative methods of gene activation in reprogramming.
In recent years, invaluable information has been obtained from the use of RNAi to study stem cell biology by inhibiting expression of specific genes involved in the regulation of pluripotency within embryonic as well as somatic stem cells. Here, we have shown the potential of using saRNAs that, conversely, upregulate expression of endogenous genes in stem cells. As each gene can be selectively targeted for activation, the use of saRNAs may also provide a highly useful tool in studying the contribution of individual factors in iPS cell reprogramming. Several studies have used inducible systems to study the reprogramming process,20,50,51 but these have been limited by the inability to activate or repress the activity of each factor individually. Since RNAa is a transient process, it may be possible to develop an optimized protocol for iPS cell production wherein each factor can be activated when needed by the transfection of its specific saRNA, and similarly removed when it is no longer necessary.
In this context, current methods for upregulating KLF4 and c-MYC require transfection52 or viral transduction13,14,15,16 of KLF4 or c-MYC expression vectors into cells. As noted above, this study suggests that the TGF-β induced by lentiviral vectors may actually be detrimental to reprogramming. Further, oncogenic reactivation of stably integrated c-MYC transgenes poses a serious safety issue to the use of iPS cells.53 In addition, evidence that latent viral expression of reprogramming factors impairs normal differentiation of iPS cells,54 and intolerance to genomic damage caused by exogenous DNA or transposon integration39,40 further emphasizes the need for a method of iPS cell generation that uses endogenous cellular processes and requires no foreign DNA. In this regard, while reprogramming to full pluripotency has not, to date, been demonstrated with this method, other groups have recently shown saRNA-mediated upregulation of endogenous OCT4 in a breast cancer cell line,27 and endogenous KLF4 in prostate cancer cell lines.55 Notably, downstream gene expression and phenotypic changes induced by saRNA-mediated upregulation of KLF4 in prostate cancer cell lines were reported to be comparable to those obtained by retroviral vector transduction. This is consistent with our results obtained in primary human MSCs, and suggests that the use of synthetic saRNA oligos may prove highly advantageous as a safe and efficient alternative for upregulation of endogenous reprogramming genes.
Materials and Methods
Bioinformatics and saRNA design. The genes KLF4 and c-MYC were analyzed to design saRNA molecules. Four parameters were used: (i) download target gene annotations; (ii) identify antisense RNA target sequences; (iii) select promoter antisense sequences; and (iv) identify candidate saRNAs. First, the method downloads information about the target's genomic location, orientation, and transcriptional structure from available databases such as the RefSeq database at UCSC (University of California, Santa Cruz). Second, given a database of RNA transcripts with known read direction, such as the UCSC Spliced EST track, our method searches the database for transcripts that are antisense to and in the vicinity of the target gene. More specifically, the method identifies antisense transcripts that (i) overlap the target's promoter and the target mRNA's 5′ end; (ii) overlap the target mRNA; (iii) are at most 20–100 kb upstream of the target's TSS; or (iv) are at most 20–100 kb downstream of the target's polyadenylation site. The method uses these four criteria as hierarchical filters such that if it finds antisense transcripts that for example satisfy criterion (i), the method does not consider the three other criteria. Third, based on the target's TSS, the method downloads the antisense genomic sequence from a fixed size region upstream and downstream of the TSS. The typical region size used by the method is 500 nts upstream and downstream of TSS, but larger or smaller sizes can also be used. Fourth, the method designs siRNAs that give effective and specific downregulation of the antisense target sequence. The method (i) uses a siRNA design algorithm, such as GPboost,26 to identify candidate effective siRNAs; (ii) removes all candidate siRNAs with aaaa, cccc, gggg, or uuuu motifs and GC content <20% or >55%; (iii) removes all candidates that have Hamming distance <2 to all potential off-target transcripts; and (iv) returns a given number of remaining non-overlapping siRNAs sorted by their predicted siRNA knockdown efficacy. The method returns the two highest scoring saRNAs for a given antisense target sequence.
Cell culture. Bone marrow-derived adult human mesenchymal stem cells (Lonza, Basel, Switzerland) were cultured in the manufacturer's media as instructed. The KLF4, MYC, and control duplex RNA oligonucleotides were transfected into MSCs using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA) following the manufacturer's protocol with 30 pmol oligo to 1 µl reagent in a 24-well plate to a final oligo concentration of 50 nmol/l. Transfections were performed every other day for the duration of each experiment. The BLOCK-iT Alexa Fluor Red Fluorescent Control (Invitrogen) and Silencer FAM labeled Negative Control #1 siRNA (Applied Biosystems, Carlsbad, CA), which have no homology to any known gene, were used as negative controls and to assess transfection efficiency by fluorescence microscopy and flow cytometry. Images were taken at ×100 magnification on a Nikon TS100 microscope (Nikon Instruments, Melville, NY).
Plasmids and lentivirus vector production. The plasmids pSin-EF2-KLF4-Pur and pSin-EF2-c-MYC-Pur were generated by cloning human KLF4 and c-MYC transgenes from plasmids pMXs-hKLF4 or pMXs-hcMYC,56 respectively, into the pSin-EF2-Pur lentiviral vector backbone.57 VSV-G–pseudotyped second-generation lentivirus preparations were produced using standard protocols; briefly, packaging plasmids pMD2.G, psPAX2, and transfer vector were cotransfected into 293T cells with jetPRIME reagent (Polyplus-transfection, New York, NY), and 48 hours later virus-containing supernatant was collected, filtered, and concentrated by ultracentrifugation. Vector titers were determined by p24 ELISA, performed by the UCLA Virology Core.
Quantitative reverse transcription-PCR. Total RNA was isolated from MSCs using the RNeasy Micro Plus Kit to remove gDNA (QIAGEN, Valencia, CA). RNA was reverse-transcribed to cDNA using the High Capacity cDNA Kit (Applied Biosystems). For nascent RNA analysis, experiments were performed using the Click-iT Nascent RNA Capture Kit (Invitrogen) according to the manufacturer's protocol with a 1 hour EU pulse before sample collection on each day of the experiment. Quantitative real-time PCR was performed using Taqman Gene Expression Master Mix (Applied Biosystems) on a MyiQ2 thermal cycler (Bio-Rad, Hercules, CA) according to the manufacturer's standard protocols. The Taqman primer sets used were as follows: KLF4, Hs00358836_m1; POU5F1 (OCT4A and OCT4B isoform), Hs00999632_g1; POU5F1 (OCT4A isoform), Hs01895061_u1; POU5F1 (OCT4B isoform), Hs00742896_s1; SOX2, Hs00602736_s1; NANOG, Hs02387400_g1; MYC, Hs00153408_m1; CCND1, Hs00277039_m1; CDKN1A, Hs00355782_m1; ODC1, Hs00159739_m1; TP53, Hs99999147_m1; ACTB, Hs00357333_g1 (Applied Biosystems). For interferon response gene expression, primers from the Interferon Response Detection Kit (System Biosciences, Mountain View, CA) were used for SYBR Green real-time PCR with SsoFast EvaGreen Supermix (Bio-Rad). As suggested by the manufacturer's protocol, samples were collected for expression analysis 24 hours after saRNA transfection or viral transduction. Experiments were performed in triplicate wells with at least three replicate reactions per PCR. Expression of β-actin mRNA was used as an internal control and samples were normalized to the scrambled sequence control oligonucleotide or untreated samples. Statistical significance was determined by Student's t-test, with P values <0.05 considered significant.
Western blot. Cells were lysed and protein concentration was determined using Coomassie Plus Assay Reagent (Thermo Scientific, Waltham, MA). Each sample was loaded onto a NuPAGE Bis-Tris Gel (Invitrogen) at 30 µg/well and electrophoresed and transferred according to the manufacturer's specifications. Primary antibodies used were GKLF (sc-20691; Santa Cruz Biotechnology, Santa Cruz, CA) and c-Myc (sc-764; Santa Cruz Biotechnology). β-Actin primary antibody (ab8227; Abcam, Cambridge, MA) was used as an internal control and for quantitation. Protein was detected using anti-rabbit HRP conjugated secondary antibody (HAF008; R&D Systems, Minneapolis, MN), developed using Immun-Star WesternC Reagent (Bio-Rad), and visualized on a ChemiDoc XRS+ (Bio-Rad). Blots shown are representative from three replicates. Protein quantitation was performed using Image Lab software (Bio-Rad). Statistical significance was determined by Student's t-test, with P values <0.05 considered significant.
Microarray and data analysis. Total RNA was isolated from treated MSCs as described. RNA was processed and hybridized to a GeneChip Human Gene 1.0 ST array (Affymetrix, Santa Clara, CA) in triplicate by the City of Hope Microarray Core Facility (Duarte, CA). Data analysis was performed by the UCLA DNA Microarray Core Facility. Samples were normalized using the ExonRMA16 summarization algorithm and filtered on expression percentile in the raw data (20–100%). Differential expression analysis compared to control samples was performed using an unpaired t-test with asymptotic P value computation and Benjamini-Hochberg multiple testing correction. Heatmaps were generated using hierarchical clustering using centroid linkage and Euclidean similarity measure. For pathway analysis, lists of genes differentially regulated between saRNA and virus samples were used to generate significantly enriched pathways in MetaCore (version 6.3 build 25177 by GeneGo). For GO analysis, lists of differentially expressed genes with the indicated adjusted P values and absolute fold changes were generated for the saRNA- and virus-treated samples versus control. These lists were used to generate functional annotation charts using DAVID bioinformatic analysis with the GOTERM_BP_FAT category on a HuGene-1_0-st-v1 background.29,30
SUPPLEMENTARY MATERIAL Figure S1. Transfection efficiency of red fluorescent control oligonucleotide by flow cytometry in MSCs. Figure S2. Transfection efficiency of positive control siRNAs as evaluated by RT-qPCR for expression levels of targeted genes. Figure S3. Replicates of western blots from Figure 2c,f. Figure S4. RT-qPCR of Klf4 and c-Myc virus transduction in MSCs for interferon response genes at the indicated p24 amount per cell. Figure S5. 5′-RACE for identification of promoter-associated antisense RNAs. Figure S6. PCR to identify promoter-associated antisense RNAs. Table S1. Normalized microarray expression data for each individual replicate used in this study. Table S2. List of genes with stem cell-related gene ontology, used to generate Figure 3c. Table S3. List of genes with cell cycle-related gene ontology, used to generate Figure 3d–f. Materials and Methods.
Acknowledgments
The authors thank the City of Hope Bioinformatics Core Facility as well as Ascia Eskin and the UCLA DNA Microarray Core Facility for expert technical assistance with microarray experiments and data analyses, the UCLA Vector Core and Shared Resource Facility for assistance with lentiviral vector construction and production, and the UCLA Virology Core Facility for assistance with p24 ELISA assays. We also would like to thank Kathrin Plath and James Byrne at the UCLA Broad Stem Cell Research Center for their expert advice and very helpful discussion. The authors declared no conflict of interest.
Supplementary Material
Transfection efficiency of red fluorescent control oligonucleotide by flow cytometry in MSCs.
Transfection efficiency of positive control siRNAs as evaluated by RT-qPCR for expression levels of targeted genes.
Replicates of western blots from Figure 2c,f.
RT-qPCR of Klf4 and c-Myc virus transduction in MSCs for interferon response genes at the indicated p24 amount per cell.
5′-RACE for identification of promoter-associated antisense RNAs.
PCR to identify promoter-associated antisense RNAs.
Normalized microarray expression data for each individual replicate used in this study.
List of genes with stem cell-related gene ontology, used to generate Figure 3c.
List of genes with cell cycle-related gene ontology, used to generate Figure 3d–f.
REFERENCES
- Hannon GJ., and, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- Castanotto D., and, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D. Small RNAs in transcriptional gene silencing and genome defence. Nature. 2009;457:413–420. doi: 10.1038/nature07756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke M, Aufsatz W, Kanno T, Daxinger L, Papp I, Mette MF.et al. (2004Genetic analysis of RNA-mediated transcriptional gene silencing Biochim Biophys Acta 1677129–141. [DOI] [PubMed] [Google Scholar]
- Hawkins PG., and, Morris KV. RNA and transcriptional modulation of gene expression. Cell Cycle. 2008;7:602–607. doi: 10.4161/cc.7.5.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LC, Okino ST, Zhao H, Pookot D, Place RF, Urakami S.et al. (2006Small dsRNAs induce transcriptional activation in human cells Proc Natl Acad Sci USA 10317337–17342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE., and, Corey DR. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol. 2007;3:166–173. doi: 10.1038/nchembio860. [DOI] [PubMed] [Google Scholar]
- Huang V, Qin Y, Wang J, Wang X, Place RF, Lin G.et al. (2010RNAa is conserved in mammalian cells PLoS ONE 5e8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen MP, Lehtola T, Heinonen SE, Assefa GS, Korpisalo P, Girnary R.et al. (2009Efficient regulation of VEGF expression by promoter-targeted lentiviral shRNAs based on epigenetic mechanism: a novel example of epigenetherapy Circ Res 105604–609. [DOI] [PubMed] [Google Scholar]
- Morris KV, Santoso S, Turner AM, Pastori C., and, Hawkins PG. Bidirectional transcription directs both transcriptional gene activation and suppression in human cells. PLoS Genet. 2008;4:e1000258. doi: 10.1371/journal.pgen.1000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Younger ST, Nguyen NB, Hardy DB, Monia BP, Corey DR.et al. (2008Antisense transcripts are targets for activating small RNAs Nat Struct Mol Biol 15842–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP.et al. (2012Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation Nat Biotechnol 30453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., and, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K.et al. (2007Induction of pluripotent stem cells from adult human fibroblasts by defined factors Cell 131861–872. [DOI] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA.et al. (2008Reprogramming of human somatic cells to pluripotency with defined factors Nature 451141–146. [DOI] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K.et al. (2007In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state Nature 448318–324. [DOI] [PubMed] [Google Scholar]
- Li Y, McClintick J, Zhong L, Edenberg HJ, Yoder MC., and, Chan RJ. Murine embryonic stem cell differentiation is promoted by SOCS-3 and inhibited by the zinc finger transcription factor Klf4. Blood. 2005;105:635–637. doi: 10.1182/blood-2004-07-2681. [DOI] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, Wang J., and, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Cassady JP., and, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell. 2008;2:10–12. doi: 10.1016/j.stem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Sridharan R, Tchieu J, Mason MJ, Yachechko R, Kuoy E, Horvath S.et al. (2009Role of the murine reprogramming factors in the induction of pluripotency Cell 136364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A.et al. (2003Widespread occurrence of antisense transcription in the human genome Nat Biotechnol 21379–386. [DOI] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ., and, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Vogelstein B, Velculescu VE, Papadopoulos N., and, Kinzler KW. The antisense transcriptomes of human cells. Science. 2008;322:1855–1857. doi: 10.1126/science.1163853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK.et al. (2008RNA exosome depletion reveals transcription upstream of active human promoters Science 3221851–1854. [DOI] [PubMed] [Google Scholar]
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA.et al. (2008Divergent transcription from active promoters Science 3221849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetrom P., and, Snøve O., Jr A comparison of siRNA efficacy predictors. Biochem Biophys Res Commun. 2004;321:247–253. doi: 10.1016/j.bbrc.2004.06.116. [DOI] [PubMed] [Google Scholar]
- Hawkins PG., and, Morris KV. Transcriptional regulation of Oct4 by a long non-coding RNA antisense to Oct4-pseudogene 5. Transcription. 2010;1:165–175. doi: 10.4161/trns.1.3.13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM.et al. (2000Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat Genet 2525–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC.et al. (2003DAVID: Database for Annotation, Visualization, and Integrated Discovery Genome Biol 4P3. [PubMed] [Google Scholar]
- Huang da W, Sherman BT., and, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Evans PM., and, Liu C. Roles of Krüpel-like factor 4 in normal homeostasis, cancer and stem cells. Acta Biochim Biophys Sin (Shanghai) 2008;40:554–564. doi: 10.1111/j.1745-7270.2008.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA., and, Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli S, Pastori C, Magistri M, Carbone GM., and, Catapano CV. Promoter-specific transcriptional interference and c-myc gene silencing by siRNAs in human cells. EMBO J. 2009;28:1708–1719. doi: 10.1038/emboj.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celano P, Berchtold CM, Kizer DL, Weeraratna A, Nelkin BD, Baylin SB.et al. (1992Characterization of an endogenous RNA transcript with homology to the antisense strand of the human c-myc gene J Biol Chem 26715092–15096. [PubMed] [Google Scholar]
- Cauffman G, Liebaers I, Van Steirteghem A., and, Van de Velde H. POU5F1 isoforms show different expression patterns in human embryonic stem cells and preimplantation embryos. Stem Cells. 2006;24:2685–2691. doi: 10.1634/stemcells.2005-0611. [DOI] [PubMed] [Google Scholar]
- Zhu S, Tai C, MacVicar BA, Jia W., and, Cynader MS. Glutamatergic stimulation triggers rapid Krüpple-like factor 4 expression in neurons and the overexpression of KLF4 sensitizes neurons to NMDA-induced caspase-3 activity. Brain Res. 2009;1250:49–62. doi: 10.1016/j.brainres.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Wassmann S, Wassmann K, Jung A, Velten M, Knuefermann P, Petoumenos V.et al. (2007Induction of p53 by GKLF is essential for inhibition of proliferation of vascular smooth muscle cells J Mol Cell Cardiol 43301–307. [DOI] [PubMed] [Google Scholar]
- Felsher DW, Zetterberg A, Zhu J, Tlsty T., and, Bishop JM. Overexpression of MYC causes p53-dependent G2 arrest of normal fibroblasts. Proc Natl Acad Sci USA. 2000;97:10544–10548. doi: 10.1073/pnas.190327097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S.et al. (2009A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity Nature 4601149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Lin C, Lu D, Ning Z, Cox T, Melvin D.et al. (2008Chromosomal transposition of PiggyBac in mouse embryonic stem cells Proc Natl Acad Sci USA 1059290–9295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Onai N, Yoshihara H, Arai F, Suda T., and, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med. 2009;15:696–700. doi: 10.1038/nm.1973. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Anderson EM, Vermeulen A, Fedorov Y, Robinson K, Leake D.et al. (2006Induction of the interferon response by siRNA is cell type- and duplex length-dependent RNA 12988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fackler OT., and, Kräusslich HG. Interactions of human retroviruses with the host cell cytoskeleton. Curr Opin Microbiol. 2006;9:409–415. doi: 10.1016/j.mib.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Poggi A., and, Zocchi MR. HIV-1 Tat triggers TGF-β production and NK cell apoptosis that is prevented by pertussis toxin B. Clin Dev Immunol. 2006;13:369–372. doi: 10.1080/17402520600645712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold D, Wrenger S, Kähne T., and, Ansorge S. HIV-1 Tat: immunosuppression via TGF-beta1 induction. Immunol Today. 1999;20:384–385. doi: 10.1016/s0167-5699(99)01497-8. [DOI] [PubMed] [Google Scholar]
- Mandal S, Johnson KR., and, Wheelock MJ. TGF-beta induces formation of F-actin cores and matrix degradation in human breast cancer cells via distinct signaling pathways. Exp Cell Res. 2008;314:3478–3493. doi: 10.1016/j.yexcr.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Mercer J., and, Helenius A. Virus entry by macropinocytosis. Nat Cell Biol. 2009;11:510–520. doi: 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- Maréchal V, Prevost MC, Petit C, Perret E, Heard JM., and, Schwartz O. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J Virol. 2001;75:11166–11177. doi: 10.1128/JVI.75.22.11166-11177.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia JS, Stan RV., and, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Breault DT., and, Hochedlinger K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H.et al. (2008Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells Cell Stem Cell 2151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Nakagawa M, Hyenjong H, Ichisaka T., and, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- Okita K, Ichisaka T., and, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Papapetrou EP, Tomishima MJ, Chambers SM, Mica Y, Reed E, Menon J.et al. (2009Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation Proc Natl Acad Sci USA 10612759–12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Place RF, Huang V, Wang X, Noonan EJ, Magyar CE.et al. (2010Prognostic value and function of KLF4 in prostate cancer: RNAa and vector-mediated overexpression identify KLF4 as an inhibitor of tumor cell growth and migration Cancer Res 7010182–10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R.et al. (2008Generation of human induced pluripotent stem cells from dermal fibroblasts Proc Natl Acad Sci USA 1052883–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S.et al. (2007Induced pluripotent stem cell lines derived from human somatic cells Science 3181917–1920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transfection efficiency of red fluorescent control oligonucleotide by flow cytometry in MSCs.
Transfection efficiency of positive control siRNAs as evaluated by RT-qPCR for expression levels of targeted genes.
Replicates of western blots from Figure 2c,f.
RT-qPCR of Klf4 and c-Myc virus transduction in MSCs for interferon response genes at the indicated p24 amount per cell.
5′-RACE for identification of promoter-associated antisense RNAs.
PCR to identify promoter-associated antisense RNAs.
Normalized microarray expression data for each individual replicate used in this study.
List of genes with stem cell-related gene ontology, used to generate Figure 3c.
List of genes with cell cycle-related gene ontology, used to generate Figure 3d–f.