

Abstract
We previously used changes in the near-UV circular dichroism and fluorescence spectra of DNA base analogue probes placed site specifically to show that the first three base pairs at the fork junction in model replication fork constructs are significantly opened by “breathing” fluctuations under physiological conditions. Here, we use these probes to provide mechanistic snapshots of the initial interactions of the DNA fork with a tight-binding replication helicase in solution. The primosome helicase of bacteriophage T4 was assembled from six (gp41) helicase subunits, one (gp61) primase subunit, and nonhydrolyzable GTPγS. When bound to a DNA replication fork construct this complex advances one base pair into the duplex portion of the fork and forms a stably bound helicase “initiation complex.” Replacement of GTPγS with GTP permits the completion of the helicase-driven unwinding process. Our spectroscopic probes show that the primosome in this stable helicase initiation complex binds the DNA of the fork primarily via backbone contacts and holds the first complementary base pair of the fork in an open conformation, whereas the second, third, and fourth base pairs of the duplex show essentially the breathing behavior that previously characterized the first three base pairs of the free fork. These spectral changes, together with dynamic fluorescence quenching results, are consistent with a primosome-binding model in which the lagging DNA strand passes through the central hole of the hexagonal helicase, the leading strand binds to the “outside” surfaces of subunits of the helicase hexamer, and the single primase subunit interacts with both strands.
Keywords: T4 DNA replication complex, helicase mechanisms, 2-aminopurine, DNA structure
Nucleic acid bases at single-strand/double-strand (ss–ds) DNA junctions of replication forks and polymerase elongation sites are prime binding targets for proteins and enzymes that manipulate and modify the DNA genome. Determining the conformational changes that occur at these junctions during DNA replication, repair, and transcription can illuminate the mechanisms of these central processes of gene expression. A unique feature of base pairs at and near these ss–dsDNA junctions is that they undergo substantial position-dependent spontaneous opening and closing (“breathing” or “fraying”) processes that are driven by thermal fluctuations (1). Proteins that bind more strongly to ss- than to dsDNA sequences can, in principle, use this differential binding free energy to bind these transiently open sequences as a first step in nucleic acid helicase activity, without the immediate expenditure of the chemical free energy of hydrolysis of nucleoside triphosphates (NTPs) (2). Thus, the interactions of genome regulatory proteins with thermally driven DNA breathing events are likely to be important in initiating reactions that involve the exposure and manipulation of single-stranded template sequences at replication forks or transcription bubbles within the normal dsDNA genome. Here, we examine the relationships between breathing fluctuations at the ss–dsDNA replication fork junction and the unwinding mechanism of the T4 primosome helicase.
The hexameric DNA replication helicases of T4 and higher organisms have much in common (3–5). Six subunits of the T4 helicase (gp41) form a hexagon in the presence of ATP or GTP and this helicase translocates 5′ → 3′ along the lagging DNA strand template in DNA replication (6), moving in synchrony with both the template-dependent DNA single-nucleotide addition cycle catalyzed by leading and lagging strand T4 DNA polymerases and the NTP binding and hydrolysis cycle that drives the helicase itself (7). The helicase activity of the T4 DNA replication complex resides in a helicase–primase (primosome) complex with a 6∶1 gp41–gp61 subunit ratio (8–10). The T4 primosome binds much more tightly to DNA replication forks than does the gp41 hexamer alone, functions processively, and is a significantly more active helicase. The primase (gp61) subunit of the primosome complex also catalyzes the template-directed synthesis of the short RNA primers that initiate new Okazaki fragments in the lagging strand synthesis at the replication fork (11).
Previous studies from our laboratory have shown that the site-specific incorporation of 2-aminopurine bases (2-AP, a fluorescent analogue of adenine) into DNA constructs does not significantly alter the structure or the stability of the DNA duplex and allows us to use near-UV spectroscopic methods to investigate mechanistically significant conformational changes at ss–dsDNA junctions in nucleic acid structures and nucleic acid–protein complexes (1, 12, 13). Here, we extend these studies to examine the reaction pathway used by the T4 primosome helicase to unwind the DNA duplex by monitoring the spectral (fluorescence and CD) changes that occur on initial binding of the functional primosome complex at 2-AP base probes substituted site specifically in the vicinity of the ds–ssDNA junction.
Results
DNA Constructs, Nomenclature, and Control Studies.
The overall structure of the DNA replication fork constructs used in this study is shown below and the individual structures of ![]() the constructs, labeled with 2-AP base analogue probes in either the lagging or the leading strand, are shown in Table S1. Each construct consists of a pair of noncomplementary strands (on the left) and a base-paired duplex (on the right). Negative numbers designate base positions in ssDNA segments and positive numbers designate base-pair positions in the dsDNA. Table S1 shows both the forked constructs that contain base analogue probe(s) (indicated by X) in the lagging (5′) strand and the constructs with probes in the leading (3′) strand. The sequence contexts of the DNA strands were maintained throughout the study to avoid sequence-dependent changes in DNA breathing or local conformation at the probe positions, and constructs were labeled in both strands to detect helicase interaction asymmetries. Unwinding assays were performed to show that the T4 helicase activity is not inhibited by the substitution of 2-AP for adenine (SI Text and Fig. S1).
the constructs, labeled with 2-AP base analogue probes in either the lagging or the leading strand, are shown in Table S1. Each construct consists of a pair of noncomplementary strands (on the left) and a base-paired duplex (on the right). Negative numbers designate base positions in ssDNA segments and positive numbers designate base-pair positions in the dsDNA. Table S1 shows both the forked constructs that contain base analogue probe(s) (indicated by X) in the lagging (5′) strand and the constructs with probes in the leading (3′) strand. The sequence contexts of the DNA strands were maintained throughout the study to avoid sequence-dependent changes in DNA breathing or local conformation at the probe positions, and constructs were labeled in both strands to detect helicase interaction asymmetries. Unwinding assays were performed to show that the T4 helicase activity is not inhibited by the substitution of 2-AP for adenine (SI Text and Fig. S1).
We had previously shown that the introduction of 2-AP probes into DNA fork constructs at specific lagging strand positions at or near the ss–dsDNA junction can be used to monitor DNA breathing fluctuations at these positions (1). To provide the “free construct” (no helicase) background for the present study we summarize in Fig. 1 the normalized fluorescence intensities and CD spectra obtained for monomer and dimer 2-AP probes located at various positions in the fork constructs used. Fig. 1 A–D shows for both strands that the normalized fluorescent intensities of 2-AP monomer probes located at the -1 and 1 positions in free DNA constructs, as well as for 2-AP dimer probes located at the -2,-1, and -1,1 positions (green and black bars), are all greater than those of their ssDNA counterparts (red bars).
Fig. 1.

Spectroscopic properties of DNA constructs as a function of 2-AP probe position relative to the ss–dsDNA junction. Fluorescence intensities at 370 nm for constructs with single 2-AP probes in the (A) lagging and (B) leading DNA strands. All constructs are shown in Table S1 and identified by construct name below each fluorescence intensity bar in Fig. 1 A–D and by color in the spectra of Fig. 1 E and F. Fluorescence intensities at 370 nm are shown for constructs with 2-AP dimer probes in the (C) lagging and (D) leading DNA strands. Fluorescence intensity changes have been normalized to the intensity of the probe signal in the control ssDNA, and full fluorescence spectra are shown in Fig. S2. CD spectra of forked DNA constructs with 2-AP dimer probes at defined positions on the (E) lagging and (F) leading strands.
We had previously observed such increases for constructs with 2-AP probes in the lagging (5′ → 3′) strand near ss–dsDNA junctions in forked and primer/template DNA constructs, and attributed the increased fluorescence of the {-1} and {-2,-1} probes to the uniquely exposed (unstacked) conformations of unpaired bases located immediately upstream of the ss–dsDNA junction, and the increased fluorescence intensity of the {1} and {-1,1} probes to the extensive breathing of the first base pair at the junction (1). We note here, in particular, that the fluorescence intensity of the {1} probe in the lagging (but not the leading) strand is significantly greater than that of the {-1} probe. This differs from what we had seen before (1), with lagging strand constructs with much shorter ssDNA “dangling end” sequences, and suggests that the increased “unstacked” character of the {1} position here could reflect an increase in the “flexing” of the construct at this position, perhaps reflecting the length of the attached ssDNA sequence.
Fig. 1 A–D also shows that the fluorescent intensities of DNA constructs labeled with isolated 2-AP monomer probes in the dsDNA segment do not change significantly beyond position 3 and are essentially constant beyond positions 2,3 for constructs labeled with 2-AP dimer pairs. Fig. 1 E and F shows equivalent effects in the CD spectra of 2-AP dimer probes incorporated in either the lagging or the leading strand of these constructs. These results are generally consistent with those obtained in our previous study of dsDNA fraying at primer/template and fork junctions on the lagging strand and show that the first noncomplementary base at the fork on both strands is uniquely unstacked and exposed to solvent in the absence of protein.
Monitoring Helicase-Induced dsDNA Unwinding Using 2-AP Probes in the Lagging (5′ → 3′) Strand.
The binding of a helicase at a DNA fork construct could induce structural changes in either or both DNA strands and could also perturb DNA breathing at and near the ss–dsDNA junction. Because of the spectral “transparency” of the canonical DNA bases and the protein components at wavelengths above 300 nm we can use fluorescence and CD changes of 2-AP probes incorporated site specifically to monitor directly the base-pair unstacking and opening events as the active helicase initially binds to the forked constructs, and then follow the reaction as the helicase moves into and through the dsDNA portions of the constructs. To make such measurements possible, a stable halted primosome–DNA construct complex was formed by using a nonhydrolyzable analogue of GTP (GTPγS) to drive the assembly of the hexameric gp41 helicase and (for most experiments) adding primase to form a “GTPγS-locked” tight-binding primosome helicase that binds stably at the fork (or ss–dsDNA junction) of the DNA construct as a “helicase initiation” complex. Nonhydrolyzable NTPs permit formation and initial binding of the primosome helicase but do not allow further unwinding, presumably because NTP hydrolysis is required to trigger the (thermodynamically unfavorable) helicase release step that completes each step of the translocation cycle (2). The T4 helicase or primosome complexes were formed by assembling the components as described in Materials and Methods and ref. 10.
Fig. 2 A and B summarizes as bar graphs the normalized fluorescence intensity changes measured at 370 nm at various probe positions; these changes can be measured at a single wavelength because the fluorescence peaks of the 2-AP probes are not shifted by protein binding to the DNA constructs (Fig. S2). We see in Fig. 2A that the addition of GTPγS-bound gp41 hexameric helicase (blue bar) to free ssDNA (red bar) caused no significant change in the fluorescence intensity of a 2-AP probe located in the middle of the nonpoly(dT) portion of the noncomplementary ssDNA sequence in the lagging strand (see Table S1 for probe positions, construct structures, and nomenclature). This result is in good agreement with the results of our primosome assembly study (10), which showed that the gp41 hexamer helicase alone forms a weak complex with either ssDNA or a replication fork junction, with the binding equilibrium significantly favoring the unbound form.
Fig. 2.

Fluorescence intensity changes caused by helicase binding for forked DNA constructs with 2-AP monomer probes in the lagging (5′ → 3′) strand. (A) Intensities at 370 nm for ssDNA and for constructs with 2-AP monomer probes located directly at the ss–dsDNA fork junction. (B) Intensities for 2-AP monomer probes located deeper within the dsDNA portion of the construct. The first bar (red) in each panel corresponds to the fluorescence intensity of the free construct, the second (blue) shows the fluorescence intensity of that construct in the presence of the hexameric gp41–GTP γS complex, the third (green) shows the intensity in the presence of the primosome helicase, and the fourth (black, when present) corresponds to the intensity in the presence of a gp41 helicase hexamer in the presence of excess GTP. The designation under each group of fluorescent intensities along the x axis identifies the construct and probe position(s) used.
The addition of a single subunit of gp61 to the gp41 hexamer to form the T4 primosome results in a stable helicase–ssDNA (or fork junction construct) complex, which is manifested by an increase in fluorescence intensity (Fig. 2A, green bar). This shows that binding of the primosome helicase to these ssDNA sequences increases somewhat the exposure of the 2-AP probe to the solvent, presumably as a consequence of local base unstacking. Finally, excess GTP was added to the gp41 helicase–ssDNA complex to determine the changes that occur as the helicase continues its translocation along the ssDNA strand. We note that the apparent fluorescent intensity of the probe (Fig. 2A, black bar) decreased slightly, presumably reflecting the translocation to—and binding of—most of the helicase at or near the 3′-end of the ssDNA strand, and thus largely downstream of the central position of the 2-AP probe in the strand.
Addition of gp41 hexamer (blue bar, {-1} construct) and primosome helicase (green bar) complexes to the forked dsDNA constructs with a 2-AP monomer probe at lagging strand position -1 showed fluorescence intensity changes comparable to those observed with the free ssDNA strand (red bar). The fluorescence intensity for the {-1} construct was essentially unchanged by the binding of the gp41 helicase hexamer, although a small increase in intensity (and thus, presumably, probe exposure) was observed for the binding of the primosome complex, perhaps reflecting a distortion of the sugar-phosphate backbone at the -1 position as a consequence of the tighter binding of this helicase. In contrast, the green bar in Fig. 2A for monomer probes at position 1 (the first dsDNA position beyond the fork in the lagging strand) shows a significant decrease in the large fluorescence intensity at this position upon binding of the primosome helicase, although again the weak binding of the gp41 helicase hexamer alone (blue bar) did not significantly change the fluorescence relative to the free construct (red bar).
This result shows that tight initial binding of the primosome helicase at the fork alters the exposure of the 2-AP probe base at position 1 of the lagging strand, with the relative change in intensity (blue bar to green bar) suggesting that in forming the helicase initiation complex, the primosome traps the first base pair of the forked DNA construct in the open state and drives it towards the spectral behavior of the initial -1 base (red bar) of the free {-1} construct, effectively decreasing its fluorescence intensity. Addition of excess GTP to the gp41 helicase–DNA complex results in a further decrease in intensity (black bar), presumably reflecting the fact that the helicase unwinds the DNA construct to completion under these conditions. This is consistent with the fact that a 2-AP probe at position 1 of a forked {1} DNA construct is more highly fluorescent than a 2-AP probe located elsewhere within partially stacked ssDNA sequences.
If the interactions of the helicase are primarily with the sugar-phosphate backbones of the DNA fork, the binding of the GTPγS-complexed primosome helicase to the DNA construct might be expected to convert the “breathing environment” of the base at lagging strand position {2} to approximately that of position {1} in the free DNA construct, and also to convert the environment of position {3} to approximately that of position {2} in free DNA. If one assumes that the presence nearby of the helicase itself does not significantly change the breathing behavior of the system, the above changes should result in increases in fluorescence intensity at these positions in the lagging strand when the helicase binds, and Fig. 2B shows that this is what is seen, although the absolute magnitudes of the fluorescence intensities in the presence of the helicase may differ somewhat from those of probes at the same positions in the absence of the protein. We note also that the magnitudes of the relative fluorescence intensities for the probes located in the largely base-paired sequences of the duplex construct are all significantly smaller than those for the largely unpaired probes; note differences in y axis scales used in Fig. 2 A and B.
The completion of duplex unwinding that follows the addition of excess GTP results in a significant further increase in fluorescence intensity (Fig. 2B, black bars), confirming that the initial increase in fluorescence intensity observed in Fig. 2B (blue and green bars) must reflect the unwinding/unstacking of the second and third base pairs as a consequence of initial helicase binding. These effects are, of course, weaker for the blue bars than for the green bars because the gp41 hexamer alone binds much more weakly to the construct than does the full primosome helicase. The unchanged fluorescence intensities of 2-AP monomer probes located at lagging strand positions {4} and {6} of the dsDNA portion of the construct (Fig. 2B) show that the initial (GTPγS-locked) complex does not induce conformational changes at these levels within the dsDNA, whereas complete unwinding does leave these probes in an exposed (ssDNA) conformation (black bar for probe in {4} position). Thus, the initial loading of the helicase–primase complex onto the replication fork induces a position-specific conformational change only in the ssDNA sequences and up to the third base pair in the duplex region. A similar study of helicase-induced dsDNA unwinding using 2-AP dimer probes in the lagging (5′) strand yielded results that were largely congruent with those reported for monomer probes here (SI Text and Fig. S3).
Monitoring Helicase-Induced dsDNA Unwinding Using 2-AP Probes in the Leading (3′ → 5′) Strand.
It has been shown previously (8) and confirmed by us in the present study (SI Text and Figs. S4 and S5) that an adequate “dangling” (noncomplementary) 3′-ssDNA sequence on the leading (3′ → 5′) strand is required for effective binding and helicase activity for both the T4 primosome and the gp41 hexamer helicase acting in isolation. This dangling sequence on the leading strand comprises the “loading site” for the helicase, meaning that the ssDNA sequences at the DNA fork are likely to interact differently with a functioning helicase. This prompted us to look for position-specific interactions of the helicase with probes in this strand (for constructs, see Table S1). The results are summarized in Fig. 3.
Fig. 3.

Fluorescence intensity changes caused by helicase binding for forked DNA constructs with 2-AP monomer probes in the leading (3′ → 5′) strand. (A) Intensities at 370 nm for ssDNA and for constructs with 2-AP monomer probes located directly at the ss–dsDNA fork junction. (B) Intensities for 2-AP monomer probes located deeper within the dsDNA portion of the construct. Color coding is the same as in Fig. 2.
The free ssDNA strand showed an increase in fluorescence intensity, suggesting some unstacking of DNA bases at the probe position on primosome helicase binding (green bar relative to the red bar for the ssDNA construct in Fig. 3A). The same effect is seen at leading strand position -6 in the forked [-6] construct, consistent with the position of this probe in the helicase loading site and the suggestion (above) that the helicase binds tightly (and preferentially, relative to ssDNA sequences) to the 3′-ssDNA dangling sequence of the leading strand at distances at least this far removed from the fork junction. In keeping with this interpretation we notice that intensities at comparable ssDNA positions in the lagging strand (see, for example, the {-3} position in Fig. S5 and compare with Fig. 3A), where the primosome presumably does not bind, are not significantly increased relative to the intensities at the same positions in the free constructs. In contrast, the 2-AP probe in the [-1] construct in the leading strand did not show a significant change in fluorescence intensity (Fig. 3A). Fig. 3B shows that DNA duplexes with 2-AP monomer probes at positions 1 and 2 on the leading strand show slight increases in fluorescence intensity with helicase binding (compare red and green bars), whereas the probes in positions 4 and 6 show little or no change ([4] and [6] constructs). We note that the changes observed at all these positions are small and that the addition of excess GTP (Fig. 3B, black bars) did not show the increase in fluorescence intensity as a consequence of construct unwinding that was seen in the lagging strand. This difference may reflect the continued binding of the approximately 20 nucleotides at the 5′-end of the leading strand by the primosome helicase, which could “hold” the 2-AP probes in somewhat stacked configurations even after the helicase has moved through and unwound the duplex region of the construct. Similar results were obtained with dimer probes in the leading strand and are summarized in SI Text and Fig. S6.
Quenching Experiments Reveal Asymmetries in Lagging and Leading Strand Involvement in the Unwinding Reaction Driven by the Binding of the Primosome Helicase.
More insight into the structural and dynamic details of the helicase–DNA complex can be obtained by studying the dynamic quenching of the fluorescence of the 2-AP probes with solvent additives, because changes in the effectiveness of such quenching will track changes in the solvent exposure of the surfaces of the DNA base analogue probes. Acrylamide was used as quencher (1) because it is uncharged and thus should reflect primarily the solvent accessibility of the fluorescent base probes (14). Quenching experiments were performed on forked DNA constructs with monomer 2-AP probes specifically positioned at either the 1 or the -1 position of the leading or the lagging strand in the absence or presence of the bound (GTPγS-locked) primosome helicase. Fig. 4 shows Stern–Vollmer plots with Fo/F plotted against quencher concentration (Q) (Fo is the fluorescence intensity in the absence of quencher and F is the observed fluorescence at a defined acrylamide concentration). Forked DNA constructs with 2-AP incorporated in the lagging strand showed a decrease in quenching when the helicase was bound to the DNA construct, suggesting that these probes become less accessible to the solvent in the presence of the bound helicase (Fig. 4A). In contrast, DNA constructs with 2-AP incorporated in the leading strand showed an increase in quenching, suggesting that at these positions helicase binding renders the probes more accessible to the solvent (Fig. 4B). Structural interpretations of these differences are considered in Discussion. Control experiments were performed with the separate 2-AP–labeled lagging (5′) and leading (3′) ssDNA strands in the absence and presence of helicase. When bound as free sequences both ssDNA strands showed an increase in acrylamide quenching on helicase binding (Fig. 4C).
Fig. 4.
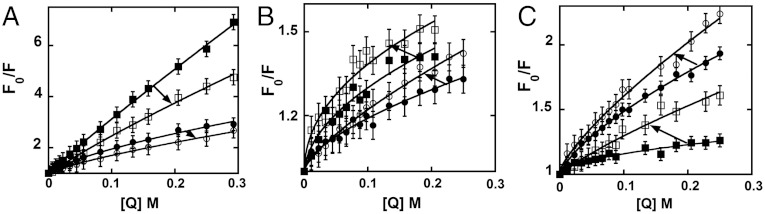
Acrylamide quenching of 2-AP monomer probes in the leading and lagging strands of DNA fork constructs. (A) Forked DNA construct with the 2-AP monomer probe at position -1 on the lagging strand (free DNA, filled circles; helicase-bound DNA, open circles) and at position 1 (free DNA, filled squares; helicase-bound DNA, open squares). (B) Forked DNA constructs with the 2-AP monomer probe at position -1 on the leading strand (free DNA, filled circles; helicase-bound DNA, open circles) and at position 1 (free DNA, filled squares; helicase-bound DNA, open squares). (C) The ssDNA strands with the 2-AP monomer probe on the 5′ strand (free DNA, filled circles; helicase-bound DNA, open circles) and on the 3′ strand (free DNA, filled squares; helicase-bound DNA, open squares). Closed symbols correspond to free DNA alone and open symbols to helicase–DNA complexes. The arrows point from the free to the helicase-bound lines of the Stern–Vollmer plots for the same DNA construct in each case.
Discussion
In contrast to approaches that seek to understand helicase mechanisms primarily by interpreting unwinding rates, our spectroscopic methodology is effectively “model-independent,” in that we monitor effects on specific bases or base pairs as a consequence of helicase binding and ask whether they are perturbed or not perturbed relative to the same bases or base pairs within the free DNA fork construct.
Initial Binding of the T4 Primosome Helicase Opens the First Base Pair at the Replication Fork and Perturbs the Breathing of the Next Three.
We show here that binding of the T4 primosome helicase to a forked construct in the presence of a nonhydrolyzable NTP results in the opening of the first base pair on the duplex side of fork, with the bound primosome translocating into the fork by one base pair. The resultant destabilization is “sensed” over the next three base pairs of the duplex, with the bases beyond the first base pair behaving as if the ds–ssDNA junction had moved one base pair into the duplex, but had otherwise left the base pairs of the fork essentially free to respond to thermal fluctuations as if the helicase were not present. This also confirms that NTP hydrolysis is not required for the initial opening and binding of the first base pair at the fork, but rather that the binding free energy of the NTP-bound helicase subunits and the thermal fluctuations that drive breathing at the ds–ssDNA junction are sufficient to accomplish the initial helicase-induced unwinding step. As suggested previously, these results are consistent with the proposal that hydrolysis of the helicase-bound NTP is only required to release the bound subunit(s) of the helicase complex and thus to “reset” the helicase unwinding cycle (2).
Our spectroscopic methodology also permits us to address more detailed structural questions because one can monitor directly what happens at the level of each probe-substituted base or base pair as the helicase advances into the fork construct. Thus, we can ask how the helicase actually binds to and interacts with each strand of DNA at the replication fork, both upstream and downstream of the ss–dsDNA junction itself. We can also ask what the primase subunit contributes to this binding interaction in order to determine how the primosome helicase actually binds and moves during the DNA replication process. To approach these issues we used a GTPγS-locked initiation complex to examine the initial interactions of the helicase complex with base probes in specific positions on both strands of the fork. Further helicase-driven unwinding of the construct could then be induced and monitored by setting up the primosome helicase reaction again, this time using GTP as the NTP helicase substrate*. This permits the NTP hydrolysis and helicase release steps of the unwinding reaction to proceed and thus allows measurement of the spectral properties of individual base positions after the unwinding reaction has passed through.
A Model for the Unwinding of a Forked DNA Duplex by the T4 Primosome.
We recently reexamined the assembly pathway and subunit stoichiometry for the T4 DNA–primosome complex and showed that the binding of a single primase (gp61) subunit to the hexameric (gp41) helicase significantly increases the binding affinity of the resulting complex for the forked helicase unwinding substrate and also significantly increases the helicase activity of the primosome complex relative to that of the gp41 hexameric helicase without primase (10). Here, we ask what happens to individual leading and lagging strand bases and base pairs at the replication fork on initial interaction with the T4 primosome helicase. Some aspects of this process are shown schematically in Fig. 5: In step A the initial binding of the NTP-complexed form of the primosome helicase at a ds–ssDNA fork unwinds the dsDNA region by one base pair. In addition, and consistent with the apparent sequence independence of the helicase unwinding rate, helicase binding at this step appears to involve primarily interactions with the DNA backbones, because the bases on both strands appear to retain the position-specific breathing behavior of the free fork, but translated by one base pair into the duplex DNA as a consequence of the opening of the first base pair of the fork by the advancing helicase. As also shown in Fig. 5, the 5′-end of the ssDNA lagging strand threads through the hole in the center of the gp41 helicase ring and, although the bases do not appear to be constrained, the acrylamide quenching experiments suggest that solvent access to their surfaces is reduced, perhaps as a consequence of the partial shielding of these lagging strand bases from solvent by their position within the ring. The 3′-ssDNA portion of the leading strand, on the other hand, is involved in binding the primosome to the replication fork, likely by interacting with sites on the surfaces of two to three subunits of the hexamer, based on an estimated binding site size of approximately 20 nt (6, 15). The presence of the primase subunit significantly tightens the binding interaction of the fork construct with the helicase, although the exact position of the single primase subunit in the initial complex is presently undefined. This binding seems to leave the bases free, but apparently with somewhat modified stacking, because access of the acrylamide quencher to the surfaces of the leading strand bases is increased. Furthermore, the CD spectra of the leading strand 2-AP dimer probes also suggest that the stacking interactions of these bases are somewhat altered, whereas the CD spectra of lagging strand probes at the same positions appear unperturbed.
Fig. 5.

Proposed unwinding mechanism of the T4 primosome helicase. The constituents of the primosome are gp41 subunits (blue ellipses), gp61 subunit (red ellipses), GTP (yellow rectangles), and GDP (red rectangles). The “degree of openness” of the base pairs adjacent to the ss–dsDNA junction is indicated in each panel, and the numbers below each DNA construct represent the numbering of the various base pairs prior to initial helicase binding. Step A: The GTP-bound gp41 hexameric helicase loads onto the free DNA fork construct and the gp61 primase subunit binds and stabilizes the complex at the fork junction. Positioning is facilitated by the uniquely unstacked conformation of the -1 bases. As a result of this initial binding the first duplex (breathing) base pair at position 1 is fully unwound and the breathing of the base pairs at initial positions 2, 3, and 4 are all enhanced. Step B: GTP hydrolysis occurs at the gp41–gp41 interface positioned adjacent to the bound gp61 subunit, destabilizing that subunit interface and permitting the primosome to “capture” the now unwound first base pair of the original duplex. This base pair becomes the new -1 position, thereby moving the breathing properties of each base pair at the fork one position further into the duplex sequence. The gp41 hexamer rotates by one subunit (approximately 60°) and the primase translocates to the next gp41–gp41 interface. Step C: The GDP (and Pi) hydrolysis products formed in step B dissociate, a new GTP binds and stabilizes the previously destabilized gp41–gp41 subunit interface, and the primosome helicase is ready to begin a new unwinding-rotation-hydrolysis cycle.
Additional binding via the primase subunit may be responsible for at least a part of the observed stronger binding (relative to the binding of the T4 helicase in the absence of primase) of the primosome to the DNA fork. If movement of the helicase through the construct entails rotation of the helicase subunits relative to the fork, the presence of only a single tightly bound primase subunit on the hexameric helicase requires that the primase subunit must also move from one helicase subunit–subunit interface to the next as a consequence of the coupled translocation of the hexamer along the helicase and the unwinding of the next base pair at the fork (10). Hydrolysis of the NTP located between the helicase subunits at the fork (modeled in Fig. 5, step B) likely leads to destabilization of the helicase subunit–subunit interface at the fork, increasing the exchange rate of the NDP and Pi hydrolysis products into the solvent (16) and permitting a new NTP to bind, as well as facilitating the rotation of the helicase ring to which the leading strand backbone is bound and thus moving the helicase one base pair further into the fork (Fig. 5, step C). Fig. 5 illustrates these interpretations of our spectroscopic data in schematic form, though we stress that only step A has been molecularly defined in this study, and that steps B and C represent the simplest model that is consistent with the subunit stoichiometry of the system and our primosome helicase assembly studies (10).
Materials and Methods
GTP and GTPγS were purchased from Sigma-Aldrich and [γ-32P] GTP and [γ-32P] ATP were obtained from NEN. Unless stated otherwise, all experiments were performed at 20 °C in buffer containing 20 mM Hepes (pH 7.5), 150 mM potassium acetate, 10 mM Mg(OAc)2, 0.1 mM EDTA, and 1 mM DTT. DNA constructs were assembled and characterized as described previously (1, 10) and construct sequences and nomenclature are shown in Tables S1 and S2. The purification, properties, and characterization of the T4 helicase and primase used have also been previously described (10, 16) and are summarized in SI Text. Fluorescence and CD measurements were performed as previously described (1) and are also summarized in SI Text. The procedures used to assemble the T4 primosome from its components are described in ref. 10 and summarized in SI Text.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Walt Baase, Sandra Greive, Kausiki Datta, and other members of our laboratory for suggestions and good advice. This work was supported by National Institutes of Health Grant GM-15792 (to P.H.v.H.). P.H.v.H. is an American Cancer Society Research Professor of Chemistry.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1212929109/-/DCSupplemental.
*We note that in most of the present studies we did not add excess GTP directly to the helicase initiation complex because we have shown that the “buried-between-subunits” positions of the bound NTPs make direct exchange of the initially bound GTPγS ligands with added GTP very slow. Hydrolysis of the natural NTP ligand destabilizes the adjacent subunit interactions, thus allowing the NDP and Pi hydrolysis reaction products to escape into solution and a new NTP to bind more rapidly.
References
- 1.Jose D, Datta K, Johnson NP, von Hippel PH. Spectroscopic studies of position-specific DNA “breathing” fluctuations at replication forks and primer-template junctions. Proc Natl Acad Sci USA. 2009;106:4231–4236. doi: 10.1073/pnas.0900803106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.von Hippel PH, Delagoutte E. A general model for nucleic acid helicases and their “coupling” within macromolecular machines. Cell. 2001;104:177–190. doi: 10.1016/s0092-8674(01)00203-3. [DOI] [PubMed] [Google Scholar]
- 3.Lohman TM. Escherichia coli DNA helicases: Mechanisms of DNA unwinding. Mol Microbiol. 1992;6:5–14. doi: 10.1111/j.1365-2958.1992.tb00831.x. [DOI] [PubMed] [Google Scholar]
- 4.Lohman TM, Bjornson KP. Mechanisms of helicase-catalyzed DNA unwinding. Annu Rev Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 5.Donmez I, Patel SS. Mechanisms of a ring shaped helicase. Nucleic Acids Res. 2006;34:4216–4224. doi: 10.1093/nar/gkl508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young MC, Schultz DE, Ring D, von Hippel PH. Kinetic parameters of the translocation of bacteriophage T4 gene 41 protein helicase on single-stranded DNA. J Mol Biol. 1994;235:1447–1458. doi: 10.1006/jmbi.1994.1100. [DOI] [PubMed] [Google Scholar]
- 7.Delagoutte E, von Hippel PH. Molecular mechanisms of the functional coupling of the helicase (gp41) and polymerase (gp43) of bacteriophage T4 within the DNA replication fork. Biochemistry. 2001;40:4459–4477. doi: 10.1021/bi001306l. [DOI] [PubMed] [Google Scholar]
- 8.Richardson RW, Nossal NG. Characterization of the bacteriophage T4 gene 41 DNA helicase. J Biol Chem. 1989;264:4725–4731. [PubMed] [Google Scholar]
- 9.Dong F, von Hippel PH. The ATP-activated hexameric helicase of bacteriophage T4 (gp41) forms a stable primosome with a single subunit of T4-coded primase (gp61) J Biol Chem. 1996;271:19625–19631. doi: 10.1074/jbc.271.32.19625. [DOI] [PubMed] [Google Scholar]
- 10.Jose D, Weitzel SE, Jing D, von Hippel PH. Assembly and subunit stoichiometry of the functional helicase–primase (primosome) complex of bacteriophage T4 DNA replication system. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1210040109. 10.1073/pnas.1210040109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cha TA, Alberts BM. Studies of the DNA helicase–RNA primase unit from bacteriophage T4. A trinucleotide sequence on the DNA template starts RNA primer synthesis. J Biol Chem. 1986;261:7001–7010. [PubMed] [Google Scholar]
- 12.Johnson NP, Baase WA, von Hippel PH. Low-energy circular dichroism of 2-aminopurine dinucleotide as a probe of local conformation of DNA and RNA. Proc Natl Acad Sci USA. 2004;101:3426–3431. doi: 10.1073/pnas.0400591101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datta K, Johnson NP, von Hippel PH. Mapping the conformation of the nucleic acid framework of the T7 RNA polymerase elongation complex in solution using low-energy CD and fluorescence spectroscopy. J Mol Biol. 2006;360:800–813. doi: 10.1016/j.jmb.2006.05.053. [DOI] [PubMed] [Google Scholar]
- 14.Lakowicz J. Principles of Fluorescence Spectroscopy. New York: Kluwer/Plenum; 1999. [Google Scholar]
- 15.Bujalowski W, Jezewska MJ. Interactions of Escherichia coli primary replicative helicase DnaB protein with single-stranded DNA. The nucleic acid does not wrap around the protein hexamer. Biochemistry. 1995;34:8513–8519. doi: 10.1021/bi00027a001. [DOI] [PubMed] [Google Scholar]
- 16.Dong F, Gogol EP, von Hippel PH. The phage T4-coded DNA replication helicase (gp41) forms a hexamer upon activation by nucleoside triphosphate. J Biol Chem. 1995;270:7462–7473. doi: 10.1074/jbc.270.13.7462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.