

Abstract
Reduction of superoxide (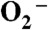 ) by manganese-containing superoxide dismutase occurs through either a “prompt protonation” pathway, or an “inner-sphere” pathway, with the latter leading to formation of an observable Mn-peroxo complex. We recently reported that wild-type (WT) manganese superoxide dismutases (MnSODs) from Saccharomyces cerevisiae and Candida albicans are more gated toward the “prompt protonation” pathway than human and bacterial MnSODs and suggested that this could result from small structural changes in the second coordination sphere of manganese. We report here that substitution of a second-sphere residue, Tyr34, by phenylalanine (Y34F) causes the MnSOD from S. cerevisiae to react exclusively through the “inner-sphere” pathway. At neutral pH, we have a surprising observation that protonation of the Mn-peroxo complex in the mutant yeast enzyme occurs through a fast pathway, leading to a putative six-coordinate Mn3+ species, which actively oxidizes
) by manganese-containing superoxide dismutase occurs through either a “prompt protonation” pathway, or an “inner-sphere” pathway, with the latter leading to formation of an observable Mn-peroxo complex. We recently reported that wild-type (WT) manganese superoxide dismutases (MnSODs) from Saccharomyces cerevisiae and Candida albicans are more gated toward the “prompt protonation” pathway than human and bacterial MnSODs and suggested that this could result from small structural changes in the second coordination sphere of manganese. We report here that substitution of a second-sphere residue, Tyr34, by phenylalanine (Y34F) causes the MnSOD from S. cerevisiae to react exclusively through the “inner-sphere” pathway. At neutral pH, we have a surprising observation that protonation of the Mn-peroxo complex in the mutant yeast enzyme occurs through a fast pathway, leading to a putative six-coordinate Mn3+ species, which actively oxidizes 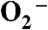 in the catalytic cycle. Upon increasing pH, the fast pathway is gradually replaced by a slow proton-transfer pathway, leading to the well-characterized five-coordinate Mn3+. We here propose and compare two hypothetical mechanisms for the mutant yeast enzyme, differing in the structure of the Mn-peroxo complex yet both involving formation of the active six-coordinate Mn3+ and proton transfer from a second-sphere water molecule, which has substituted for the ─OH of Tyr34, to the Mn-peroxo complex. Because WT and the mutant yeast MnSOD both rest in the 2+ state and become six-coordinate when oxidized up from Mn2+, six-coordinate Mn3+ species could also actively function in the mechanism of WT yeast MnSODs.
in the catalytic cycle. Upon increasing pH, the fast pathway is gradually replaced by a slow proton-transfer pathway, leading to the well-characterized five-coordinate Mn3+. We here propose and compare two hypothetical mechanisms for the mutant yeast enzyme, differing in the structure of the Mn-peroxo complex yet both involving formation of the active six-coordinate Mn3+ and proton transfer from a second-sphere water molecule, which has substituted for the ─OH of Tyr34, to the Mn-peroxo complex. Because WT and the mutant yeast MnSOD both rest in the 2+ state and become six-coordinate when oxidized up from Mn2+, six-coordinate Mn3+ species could also actively function in the mechanism of WT yeast MnSODs.
Keywords: antioxidant enzyme, metalloenzyme, product inhibition, coordination number, proton transfer pathway
Superoxide dismutase (SOD) with manganese at its active site (MnSOD) is remarkable in that it becomes less efficient when levels of its substrate 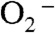 are high, while other subclasses of SOD catalyze the disproportionation of
are high, while other subclasses of SOD catalyze the disproportionation of 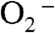 at virtually diffusion-controlled rates independent of
at virtually diffusion-controlled rates independent of 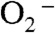 level (1). The degree of depression of MnSOD activity at high levels of
level (1). The degree of depression of MnSOD activity at high levels of 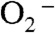 is most prominent in human MnSOD, while the MnSODs from bacteria and yeast are relatively more efficient under the same conditions (2). This unique property of human MnSOD could result from the need for stringent regulation of H2O2 production, due to the complicated roles of H2O2 in mammalian cells, especially as a signaling agent (2).
is most prominent in human MnSOD, while the MnSODs from bacteria and yeast are relatively more efficient under the same conditions (2). This unique property of human MnSOD could result from the need for stringent regulation of H2O2 production, due to the complicated roles of H2O2 in mammalian cells, especially as a signaling agent (2).
The mechanism by which MnSOD removes 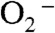 involves product inhibition (1). Specifically, reduction of
involves product inhibition (1). Specifically, reduction of 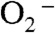 by Mn2+SOD (Scheme 1) occurs either through a pathway (called “prompt protonation” pathway here) (reaction 2), where protonation and dissociation of the peroxo moiety is instantaneous, or through an “inner-sphere” pathway (reaction 3), where a detectable intermediate is formed, which has been suggested to be a side-on Mn(III)-peroxo species (3). (We here refer to this intermediate as a Mn-peroxo complex instead of as a “product-inhibited” complex, the latter term having been used frequently in previous reports.) When protonation of the Mn-peroxo complex (reaction 4) is rate-limiting, the “inner-sphere” pathway has the effect of slowing down MnSOD catalysis.
by Mn2+SOD (Scheme 1) occurs either through a pathway (called “prompt protonation” pathway here) (reaction 2), where protonation and dissociation of the peroxo moiety is instantaneous, or through an “inner-sphere” pathway (reaction 3), where a detectable intermediate is formed, which has been suggested to be a side-on Mn(III)-peroxo species (3). (We here refer to this intermediate as a Mn-peroxo complex instead of as a “product-inhibited” complex, the latter term having been used frequently in previous reports.) When protonation of the Mn-peroxo complex (reaction 4) is rate-limiting, the “inner-sphere” pathway has the effect of slowing down MnSOD catalysis.
Scheme 1.
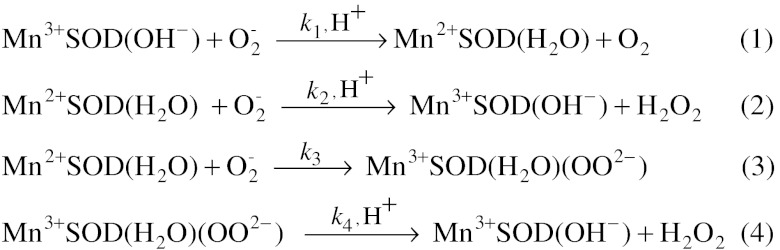
Catalytic mechanism of MnSOD.
Our recent study showed that the two yeast MnSODs, one from Saccharomyce cerevisiae mitochondria (ScMnSOD) and the other from Candida albicans cytosol (CaMnSODc), are both very efficient at high levels of 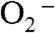 relative to other MnSODs that have been characterized to date (2, 4). Spectroscopic analysis of the oxidized yeast enzymes suggests the presence of a six-coordinate Mn3+ species, in addition to the well-characterized five-coordinate Mn3+ (2). The sixth ligand is likely a hydroxide. Based on these findings, we earlier proposed, for the fast catalysis by yeast MnSODs, a mechanism that involves putative translocation of an outer-sphere solvent molecule (2). More recently we have been pursuing this mechanistic investigation by focusing on mutant yeast MnSOD with substitutions in the second coordination sphere.
relative to other MnSODs that have been characterized to date (2, 4). Spectroscopic analysis of the oxidized yeast enzymes suggests the presence of a six-coordinate Mn3+ species, in addition to the well-characterized five-coordinate Mn3+ (2). The sixth ligand is likely a hydroxide. Based on these findings, we earlier proposed, for the fast catalysis by yeast MnSODs, a mechanism that involves putative translocation of an outer-sphere solvent molecule (2). More recently we have been pursuing this mechanistic investigation by focusing on mutant yeast MnSOD with substitutions in the second coordination sphere.
Tyr34 is highly conserved and structurally similar in all known MnSODs, where it participates in a hydrogen-bonding network at the active site. Computational studies suggest that Tyr34 provides one of the two protons needed to form H2O2 in the catalytic cycle (5). Deprotonation of Tyr34 is responsible for the spectroscopic changes, including the blue shift and the decrease in intensity of the optical absorption maximum, that occur for Mn3+SOD with increased pH (6). Wild-type (WT) and Y34F mutant human MnSOD have similar kcat/Km, but kcat is decreased by approximately 10-fold in the mutant protein (7). Tyr34 also plays an interesting role in the “prompt protonation” pathway, as elucidated by the changes in the kinetics of Y34 mutant human MnSOD (8).
To test our previously proposed mechanism and to address questions regarding the role of Tyr34 in yeast MnSOD, we purified and characterized Y34F mutant ScMnSOD. To our surprise, we have found that catalysis by Y34F ScMnSOD is completely “inner-sphere.” More interestingly, our results reveal that Y34F ScMnSOD has a novel mechanism involving an active six-coordinate Mn3+ species, different from any previously characterized WT or mutant enzyme.
Results
An Additional Water Sits in the Active Site of Y34F ScMnSOD.
To aid in our investigation of the role of Tyr34, we determined the X-ray structure of Y34F ScMnSOD (Table S1). The subunit structure and the tetrameric assembly of WT and the mutant ScMnSOD closely resemble each other (Fig. S1). Superimposition of all backbone atoms of the mutant tetramer onto those of the WT protein gives a rmsd of 0.22 Å. Y34F ScMnSOD maintains the strictly conserved five-coordinate Mn at active site, and the substitution of Tyr34 causes only minimal changes to positioning and conformation of active-site residues (Fig. 1A).
Fig. 1.

Active-site structure and oxidation state of Y34F ScMnSOD. (A) Superimposition of the active site of Y34F ScMnSOD (chain A, red) onto that of WT ScMnSOD (chain A, green). Coordination bonds are indicated as solid lines, and hydrogen bonds are shown as dashed lines in WT (black) and Y34F (gray) ScMnSOD, respectively. The graphic was generated using the PyMOL Molecular Graphics System (28). (B) (a, b) Optical spectra of as-isolated (solid line) and reduced (dotted line, by sodium hydrosulfite) Y34F ScMnSOD. Inset: The difference spectrum between as-isolated and reduced Y34F ScMnSOD. (c) Optical spectra of Y34F ScMnSOD oxidized by potassium permanganate ([KMnO4]∶[MnSOD] = 0.75∶1) at pH 7.4 as measured over time. The spectra were recorded after mixing KMnO4 with the enzyme for 0, 10, 20, 30, 45 and 60 min. (d) Reduction (dashed line) of Y34F ScMn3+SOD (oxidized by 0.75 equiv of KMnO4 and allowed to sit for 1 hr) by one equivalent of sodium ascorbate. The sample solutions contained 200 μM (in Mn) enzyme in 25 mM potassium phosphate (pH 7.4).
An additional water molecule occupies the cavity created by the replacement of Tyr34 with phenylalanine in all four subunits in the unit cell of Y34F ScMnSOD. This water has substituted for the ─OH of Tyr34 in the WT enzyme by forming hydrogen bonds to Gln154 and the water molecule hydrogen bonded to His30 (Fig. 1A). Its presence at that location supports the importance of the hydrogen-bonding chain involving Tyr34 (Gln154⋯Tyr34⋯H2O⋯His30) in the WT enzyme (9). This additional water molecule is also present in Y34X (X = A, N, H, or V) mutant human MnSOD (8). However, it was absent in the crystal structure of human Y34F MnSOD, which had been carried out at a resolution similar to that of our study (1.9 Å) (7).
Like the WT enzyme, Y34F ScMn3+SOD Contains Both Five-and Six-Coordinate Mn3+ Centers.
WT MnSODs are usually isolated in the oxidized state, Mn3+SOD (10–12). Yeast MnSODs appear to be an exception, since we showed previously that WT yeast MnSODs from S. cerevisiae and C. albicans rest predominantly in the reduced (2+) state (2, 4). This preference to be reduced is unchanged in Y34F ScMnSOD, the resting form of which absorbs weakly around 480 nm (Fig. 1B).
As in the WT enzyme, the spectrum of Y34F ScMnSOD immediately after oxidation was characteristic of five-coordinate Mn3+SOD (Fig. 1B). Additional absorption intensity appeared at < 400 nm over time (Fig. 1B), which was proposed to result from formation of a six-coordinate Mn3+SOD, with hydroxide as the sixth ligand (2). The intensity is blue-shifted from 390 nm in WT ScMn3+SOD (2) to 370 nm in Y34F ScMn3+SOD (Fig. S2). The time to reach the final spectrum was shorter in the Y34F mutant protein (approximately 45 min) (Fig. 1B) than in WT ScMnSOD (approximately 2 h) (2), indicating faster formation of the putative six-coordinate adduct in Y34F ScMn3+SOD.
Y34F ScMn3+SOD also resembles WT ScMn3+SOD in that its six-coordinate adduct is more prone to reduction by ascorbate than the five-coordinate species (2), as 1.0 equivalent of ascorbate selectively bleached the absorption of Y34F ScMn3+SOD around 370 nm (Fig. 1B). Therefore, the six-coordinate Mn3+ has a higher reduction potential than the five-coordinate Mn3+. Five-coordination is preferred for Mn3+SOD, because six-coordinate Mn3+SOD, with a high-spin d4 center, displays strong Jahn–Teller distortion (13). The slow formation of the six-coordinate adduct and its high reduction potential in both WT and the mutant yeast MnSOD are still not understood in detail, and we will continue exploring their origins in future.
Y34F ScMnSOD is Gated Exclusively Toward “Inner-Sphere” Pathway, yet Still Displays Fast Catalysis.
In pulse radiolysis the disappearance of 2–50 μM 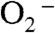 in solutions containing 1–5 μM Y34F ScMnSOD was followed at 260 nm, and the observed traces were compared to previous data collected under identical conditions for WT MnSOD from S. cerevisiae, C. albicans, human, E. coli, and D. radiodurans (Fig. 2). When
in solutions containing 1–5 μM Y34F ScMnSOD was followed at 260 nm, and the observed traces were compared to previous data collected under identical conditions for WT MnSOD from S. cerevisiae, C. albicans, human, E. coli, and D. radiodurans (Fig. 2). When 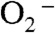 was in 7-fold excess over MnSOD, a relatively uninhibiting condition, human MnSOD, the most inhibited WT MnSOD known, already showed a depressed activity, while the
was in 7-fold excess over MnSOD, a relatively uninhibiting condition, human MnSOD, the most inhibited WT MnSOD known, already showed a depressed activity, while the 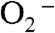 disappearance as a function of time was similar for each of the other four WT enzymes (Fig. 2A). Notably, substitution of Tyr34 caused ScMnSOD to remove
disappearance as a function of time was similar for each of the other four WT enzymes (Fig. 2A). Notably, substitution of Tyr34 caused ScMnSOD to remove 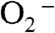 more slowly under this condition than all of the WT enzymes, including WT human MnSOD (Fig. 2A).
more slowly under this condition than all of the WT enzymes, including WT human MnSOD (Fig. 2A).
Fig. 2.

Decay of 7 μM (A) and 41 μM (B) 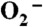 catalyzed by 1 μM (in Mn) human (a, black), E. coli (b, red), D. radiodurans (c, blue) MnSOD, ScMnSOD (d, green), CaMnSODc (e, pink) and Y34F ScMnSOD (f,gray). The samples contained 10 mM potassium phosphate (pH 7), 10 mM sodium formate and 10 μM EDTA. [
catalyzed by 1 μM (in Mn) human (a, black), E. coli (b, red), D. radiodurans (c, blue) MnSOD, ScMnSOD (d, green), CaMnSODc (e, pink) and Y34F ScMnSOD (f,gray). The samples contained 10 mM potassium phosphate (pH 7), 10 mM sodium formate and 10 μM EDTA. [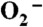 ] in these figures is calculated from the absorbance at 260 nm.
] in these figures is calculated from the absorbance at 260 nm.
We then increased the 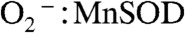 (in concentration) ratio to 41, a condition where MnSOD from different organisms displays dramatically different efficiencies, due to different degrees of product inhibition (10). The degree of product inhibition is defined as the gating ratio k2/k3 (Scheme 1). To our surprise, Y34F ScMnSOD catalyzed the disappearance of
(in concentration) ratio to 41, a condition where MnSOD from different organisms displays dramatically different efficiencies, due to different degrees of product inhibition (10). The degree of product inhibition is defined as the gating ratio k2/k3 (Scheme 1). To our surprise, Y34F ScMnSOD catalyzed the disappearance of 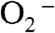 more efficiently than human MnSOD (Fig. 2B), even though the former is gated exclusively toward the “inner-sphere” pathway (k2/k3 < 0.01, see below). The zero-order phase, characteristic of the presence of the Mn-peroxo complex, is dependent on k2/k3 and peroxide dissociation rate (k4) (Scheme 1). The fact that the zero-order phase was not as pronounced in Y34F ScMnSOD as in WT E. coli or human MnSOD (Fig. 2B) suggests faster peroxide dissociation steps in the former.
more efficiently than human MnSOD (Fig. 2B), even though the former is gated exclusively toward the “inner-sphere” pathway (k2/k3 < 0.01, see below). The zero-order phase, characteristic of the presence of the Mn-peroxo complex, is dependent on k2/k3 and peroxide dissociation rate (k4) (Scheme 1). The fact that the zero-order phase was not as pronounced in Y34F ScMnSOD as in WT E. coli or human MnSOD (Fig. 2B) suggests faster peroxide dissociation steps in the former.
To measure individual rate constants, 60 μM of Y34F ScMn2+SOD was pulse-irradiated to react with substoichiometric amounts (2–3 μM) of 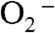 , and the appearance of Mn3+SOD was followed. The absorbance measured at 425 nm within 2 ms after generation of
, and the appearance of Mn3+SOD was followed. The absorbance measured at 425 nm within 2 ms after generation of 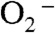 could be divided into two phases, a rapid emergence of absorption within 0.1 ms followed by a relatively slow increase that lasted for approximately 15 ms (Fig. 3A, left). Similar biphasic increases at 425 nm absorption have also been observed for several Y34 mutant human MnSODs (8). The 425 nm absorption of Y34F ScMnSOD slowly decreased at a longer time scale (Fig. 3A, Right), leading to formation of a Mn3+ species (Int3) with an absorption maximum at 480 nm (Fig. 3B), consistent with well-characterized five-coordinate Mn3+SODs (10, 14).
could be divided into two phases, a rapid emergence of absorption within 0.1 ms followed by a relatively slow increase that lasted for approximately 15 ms (Fig. 3A, left). Similar biphasic increases at 425 nm absorption have also been observed for several Y34 mutant human MnSODs (8). The 425 nm absorption of Y34F ScMnSOD slowly decreased at a longer time scale (Fig. 3A, Right), leading to formation of a Mn3+ species (Int3) with an absorption maximum at 480 nm (Fig. 3B), consistent with well-characterized five-coordinate Mn3+SODs (10, 14).
Fig. 3.

Mn3+ species observed from oxidation of Y34F ScMn2+SODc by 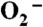 in pulse radiolysis. (A) The absorbance change at 425 nm within 200 ms after pulse radiolysis generated initial
in pulse radiolysis. (A) The absorbance change at 425 nm within 200 ms after pulse radiolysis generated initial 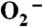 burst of 2.2 μM. (B) The spectra of the Mn3+ species (Int1: hollow circles; Int2: hollow triangles; Int3: solid circles). The sample solutions contained 60 μM (in Mn) Y34F ScMnSOD in 10 mM potassium phosphate (pH 7.4), 10 mM sodium formate, 120 μM H2O2, and 100 μM EDTA.
burst of 2.2 μM. (B) The spectra of the Mn3+ species (Int1: hollow circles; Int2: hollow triangles; Int3: solid circles). The sample solutions contained 60 μM (in Mn) Y34F ScMnSOD in 10 mM potassium phosphate (pH 7.4), 10 mM sodium formate, 120 μM H2O2, and 100 μM EDTA.
We attribute the rapid and slow absorption increase in Fig. 3A (left) to emergence of Intermediate 1 (Int1) and 2 (Int2), respectively (Scheme 2). The data were fitted into two-exponential models, and rate constants and molar-extinction coefficients were calculated. The resulting spectrum of Int1, which has an absorption maximum at 415 nm (Fig. 3B), is identical to those reported for Mn-peroxo complexes (8, 10, 14). Int2 is an intermediate with broad absorption in the visible range and a maximum at 425 nm (Fig. 3B). The structure of this intermediate is unknown, but species with similar spectral features have been observed in similar studies of Y34A, Y34H, and Y34N human MnSOD (8).
Scheme 2.
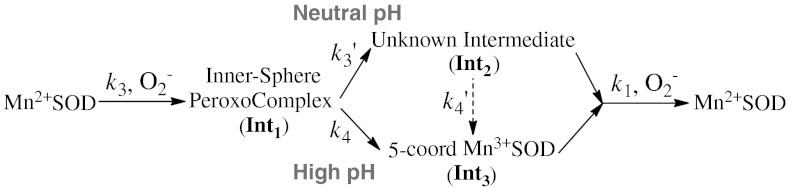
Reduction of 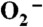 by Y34F ScMn2+SOD.
by Y34F ScMn2+SOD.
The substitution of Tyr34 caused dramatic changes to the kinetics of ScMnSOD. The rate constant for the “prompt protonation” pathway, k2 (Scheme 1), was too low to measure accurately. The rate constant for formation of the Mn-peroxo complex, k3 (Schemes 1 and 2), was dramatically elevated from 0.04–0.05 nM-1 s-1 for WT ScMnSOD to 0.70 nM-1 s-1 for the mutant protein (Table S2), such that the overall rate constant of reduction of 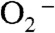 by Mn2+SOD, defined as a sum of k2 and k3, is similar in the WT and the mutant protein. We define the rate constant for formation of Int2 from the Mn-peroxo complex as k3’ (Scheme 2), which was determined to be 310 s-1 for Y34F ScMnSOD and is comparable to that of Y34H human MnSOD but lower than those of Y34A and Y34N human MnSOD (Table S2). The rate constant for formation of the five-coordinate Mn3+ from Int2, k4’ (Scheme 2), was determined to be 20 s-1 for Y34F ScMnSOD and is 3- to 16-fold lower than those determined for Y34 mutant human enzymes (Table S2).
by Mn2+SOD, defined as a sum of k2 and k3, is similar in the WT and the mutant protein. We define the rate constant for formation of Int2 from the Mn-peroxo complex as k3’ (Scheme 2), which was determined to be 310 s-1 for Y34F ScMnSOD and is comparable to that of Y34H human MnSOD but lower than those of Y34A and Y34N human MnSOD (Table S2). The rate constant for formation of the five-coordinate Mn3+ from Int2, k4’ (Scheme 2), was determined to be 20 s-1 for Y34F ScMnSOD and is 3- to 16-fold lower than those determined for Y34 mutant human enzymes (Table S2).
Int2 is the Active Mn3+ Species in Y34F ScMnSOD.
As we have shown that the mutant yeast enzyme reacts completely through the Mn-peroxo complex, the question arises: What is the Mn3+ species that oxidizes 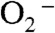 ? Is it Int2 or the five-coordinate Mn3+? Int2 is a product from partial or full protonation of the Mn-peroxo complex. Formation of the five-coordinate Mn3+ from Int2 is so slow (
? Is it Int2 or the five-coordinate Mn3+? Int2 is a product from partial or full protonation of the Mn-peroxo complex. Formation of the five-coordinate Mn3+ from Int2 is so slow (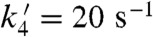 ) that if the former were the oxidant, Y34F ScMnSOD would be much slower than WT human MnSOD (k4 = 120 s-1) under inhibiting conditions. This contradicts our observation (Fig. 2B). Indeed, we were unable to fit our data to a mechanism in which formation of the five-coordinate Mn3+ is the rate limiting step. By contrast, the experimental data (Figs. 2 and 4A) with various enzyme and
) that if the former were the oxidant, Y34F ScMnSOD would be much slower than WT human MnSOD (k4 = 120 s-1) under inhibiting conditions. This contradicts our observation (Fig. 2B). Indeed, we were unable to fit our data to a mechanism in which formation of the five-coordinate Mn3+ is the rate limiting step. By contrast, the experimental data (Figs. 2 and 4A) with various enzyme and 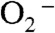 concentrations fit well to a mechanism in which Int2 is the Mn3+ species that oxidizes
concentrations fit well to a mechanism in which Int2 is the Mn3+ species that oxidizes 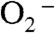 (Fig. S3).
(Fig. S3).
Fig. 4.

pH determines the pathway of protonation of the Mn-peroxo complex in Y34F ScMnSOD. (A) Decay of 41 μM 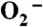 catalyzed by 5 μM (in Mn) Y34F ScMnSOD at pH 7.0 (a) and 8.5 (b). The solutions contained 10 mM potassium phosphate, 10 mM sodium formate, 1 mM Tris-Cl and 10 μM EDTA. Inset: rate constant k3’(solid circles) and k4’ (hollow triangles) as functions of pH. (B) The absorbance changes at 420 nm upon generation of 8.9 μM
catalyzed by 5 μM (in Mn) Y34F ScMnSOD at pH 7.0 (a) and 8.5 (b). The solutions contained 10 mM potassium phosphate, 10 mM sodium formate, 1 mM Tris-Cl and 10 μM EDTA. Inset: rate constant k3’(solid circles) and k4’ (hollow triangles) as functions of pH. (B) The absorbance changes at 420 nm upon generation of 8.9 μM 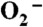 at pH 7.5 (black), 8.0 (red), 8.5 (green), 9.0 (purple), and 9.5 (blue). Fitted curves generated from PRWIN (27) are shown. The solutions contained 80 μM (in Mn) Y34F ScMnSOD in 15 mM potassium phosphate, 10 mM sodium formate, 1 mM Tris-Cl, 160 μM H2O2, and 100 μM EDTA. Inset: percentage of the fast protonation pathway as a function of pH, calculated by dividing the highest 420 nm absorbance at each pH to that at pH 7.5, assuming that at pH 7.5 the enzyme reacts solely through the fast proton transfer pathway.
at pH 7.5 (black), 8.0 (red), 8.5 (green), 9.0 (purple), and 9.5 (blue). Fitted curves generated from PRWIN (27) are shown. The solutions contained 80 μM (in Mn) Y34F ScMnSOD in 15 mM potassium phosphate, 10 mM sodium formate, 1 mM Tris-Cl, 160 μM H2O2, and 100 μM EDTA. Inset: percentage of the fast protonation pathway as a function of pH, calculated by dividing the highest 420 nm absorbance at each pH to that at pH 7.5, assuming that at pH 7.5 the enzyme reacts solely through the fast proton transfer pathway.
Fast and Slow Proton-Transfer Pathways in Y34F ScMnSOD.
Increased pH lowered the overall rate of protonation of the Mn-peroxo complex, which is a combination of k3’, k4, and k4’ (see below), in Y34F ScMnSOD. At pH 7, the overall protonation rate was equal to k3’ (310 s-1); at pH 8.5, it dropped by a factor of three to 100 s-1 (Fig. 4A). In addition, the rapid absorption increase at 420 nm, resulting from emergence of Int2, gradually disappeared at increased pH (Fig. 4B). These facts were not caused by depression of k3’, which, calculated via fitting the data in Fig. 4B, stayed almost the same from pH 7–8.5 (Fig. 4A, inset). The most likely explanation is that Int2 no longer formed at increased pH (Scheme 2). The same experiment recorded at 480 nm clearly indicated generation of the five-coordinate Mn3+ directly from the peroxo complex at pH≥8 (Fig. S4). Protonation of the peroxo complex in Y34F ScMnSOD appears to partition into two pathways—a fast one at neutral pH leading to Int2 and a slow one at high pH leading to the five-coordinate Mn3+. The gating between the two pathways has a pK of about 8, and the enzyme reacts completely through the slow pathway at pH≥9 (Fig. 4B, inset). The rate constant for the slow pathway (k4) was calculated to be 40 s-1, by fitting the absorbance changes at 480 nm to first-order reactions (Fig. S4).
Discussion
Tyr34 is Critical for the “Prompt Protonation” Pathway.
To examine the role of Tyr34 in catalysis by ScMnSOD, we replaced Tyr34 with phenylalanine. WT ScMnSOD reacts predominantly through the “prompt protonation” pathway, with a gating ratio (k2/k3) of 16–20 (Table S2) (2, 4). Like in Y34 mutant human enzymes (8), the “prompt protonation” pathway is completely disrupted in Y34F ScMnSOD. In Y34F ScMnSOD, k3 is close in value (0.70 nM-1 s-1) to k2 of WT ScMnSOD (0.80 nM-1 s-1) (Table S2), indicating that the “prompt protonation” pathway is substituted by the “inner-sphere” pathway in the mutant yeast enzyme.
Putative Six-Coordinate Int2 is Associated with the Additional Second-Sphere Water.
Spectroscopic analysis revealed that resting WT yeast Mn3+SODs consist of both five- and six-coordinate Mn3+ centers (2). In WT yeast enzymes, the absorption intensities around 480 nm and < 400 nm correlate with two effective 55Mn hyperfine couplings in the EPR spectra, consistent with five- and six-coordinate Mn3+ centers, respectively (2). Like the WT enzyme, Y34F ScMn3+SOD also absorbs at < 400 nm in addition to the 480 nm band (Fig. 1B), suggesting that the oxidized mutant protein also contains both five- and six-coordinate Mn3+.
When Y34F ScMn2+SOD was oxidized by 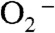 , a Mn3+ intermediate (Int2, Scheme 2) was observed with an absorption maximum at 425 nm (Fig. 3). The blue shifting of its spectrum compared to those of five-coordinate Mn3+SODs leads to a speculation that Int2 is six-coordinate. However, Int2 must differ from the six-coordinate component of resting Y34F ScMn3+SOD, because the two have distinct absorption maxima—i.e., < 400 versus 425 nm (Figs. 1B and 3B). A simple explanation for the different extent of blue shifting is that the sixth ligand is hydroxide in one case and water in the other. Moreover, although we previously proposed that the sixth ligand is hydroxide in resting WT ScMn3+SOD (2), we cannot rule out the possibility that it is instead a water molecule. Alternatively, the coordination geometry could be slightly different in the six-coordinate species present under catalytic conditions from the noncatalytic form.
, a Mn3+ intermediate (Int2, Scheme 2) was observed with an absorption maximum at 425 nm (Fig. 3). The blue shifting of its spectrum compared to those of five-coordinate Mn3+SODs leads to a speculation that Int2 is six-coordinate. However, Int2 must differ from the six-coordinate component of resting Y34F ScMn3+SOD, because the two have distinct absorption maxima—i.e., < 400 versus 425 nm (Figs. 1B and 3B). A simple explanation for the different extent of blue shifting is that the sixth ligand is hydroxide in one case and water in the other. Moreover, although we previously proposed that the sixth ligand is hydroxide in resting WT ScMn3+SOD (2), we cannot rule out the possibility that it is instead a water molecule. Alternatively, the coordination geometry could be slightly different in the six-coordinate species present under catalytic conditions from the noncatalytic form.
An intermediate that is virtually identical in its properties to Int2 observed in Y34F ScMnSOD was previously reported in studies of several Y34 mutant human MnSODs (8). The rate constant for formation of Int2, k3’, in the case of the Y34 mutant human enzymes, was observed to be independent of ionic strength, and it was concluded on this basis that the electrostatic charge had not changed when Int2 was generated and that Int2 could be six-coordinate with the metal ion adding an additional water molecule (8). In the case of Int2 formed by Y34F ScMnSOD, the sixth ligand could likewise be H2O, although we cannot rule out the possibility that it is instead OH- or OOH-.
All Y34 mutant proteins shown to react via Int2 contain an additional second sphere water molecule occupying the cavity created by the replacement of Tyr34 (Fig. 1A) (8), suggesting that the observation of Int2 and the presence of the additional water may be correlated. However, neither the additional water nor Int2 are found in any Y34F mutant protein that has been studied previously (7, 10), suggesting a particularly important role of Tyr34 in WT ScMnSOD. Previously we proposed that fast catalysis by yeast MnSOD could be related to fast proton transfer from an outer-sphere solvent molecule (2). Particularly in Y34 mutant proteins, protonation of the peroxo complex is always fast so long as the additional water molecule is present. One would therefore expect that the additional second-sphere water in these Y34 mutant proteins donates one of the two protons to the bound peroxyl group.
In order to explore the catalytic properties of the six-coordinate species in WT yeast MnSODs, we reduced ScMn3+SOD or CaMn3+SOD with substoichiometric amounts of 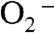 and followed the absorbance changes at 475 nm (Fig. S5). The resting form of oxidized yeast MnSODs is a mixture of five- and six-coordinate Mn3+SOD. In the case of the six-coordinate Mn3+SOD being a “dead-end” complex, k1 would be significantly reduced due to inhibition by the sixth ligand. However, k1 was determined to be 1.0–1.3 × 109 M-1 s-1 for yeast WT MnSODs and is comparable to that of other WT MnSODs (4), whose catalysis apparently only involves five-coordinate Mn3+ species. Therefore, the six-coordinate Mn3+ center is equally as catalytically competent as the well-characterized five-coordinate Mn3+. This, along with the correlation between the additional second-sphere water molecule and fast proton transfer, suggests that the sixth ligand in Int2 is most likely a solvent molecule. Unfortunately, it is difficult to confirm the structure of Int2 using spectroscopic tools, due to short lifetime (approximately 200 ms) of this intermediate.
and followed the absorbance changes at 475 nm (Fig. S5). The resting form of oxidized yeast MnSODs is a mixture of five- and six-coordinate Mn3+SOD. In the case of the six-coordinate Mn3+SOD being a “dead-end” complex, k1 would be significantly reduced due to inhibition by the sixth ligand. However, k1 was determined to be 1.0–1.3 × 109 M-1 s-1 for yeast WT MnSODs and is comparable to that of other WT MnSODs (4), whose catalysis apparently only involves five-coordinate Mn3+ species. Therefore, the six-coordinate Mn3+ center is equally as catalytically competent as the well-characterized five-coordinate Mn3+. This, along with the correlation between the additional second-sphere water molecule and fast proton transfer, suggests that the sixth ligand in Int2 is most likely a solvent molecule. Unfortunately, it is difficult to confirm the structure of Int2 using spectroscopic tools, due to short lifetime (approximately 200 ms) of this intermediate.
Proposed Mechanisms for Y34F ScMnSOD in Which Six-Coordinate Int2 is the Active Species.
Y34F ScMnSOD is even more gated through the Mn-peroxo complex, Int1, than is WT human MnSOD, as indicated by their gating ratios (k2/k3) of < 0.01 and 1, respectively (Table S2). Surprisingly, the mutant ScMnSOD is faster than human MnSOD when 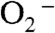 levels are high (Fig. 2B). In the case of human MnSOD, high levels of
levels are high (Fig. 2B). In the case of human MnSOD, high levels of 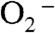 causes the overall reaction to slow because of the relatively low rate of peroxide loss (120 s-1 at pH 7) upon protonation of Int1. In the case of Y34F ScMnSOD, by contrast, protonation and loss of peroxide is much faster (310 s-1 at pH 7). We conclude from curve fitting (Fig. S3) that Int2 is the active Mn3+ species that oxidizes
causes the overall reaction to slow because of the relatively low rate of peroxide loss (120 s-1 at pH 7) upon protonation of Int1. In the case of Y34F ScMnSOD, by contrast, protonation and loss of peroxide is much faster (310 s-1 at pH 7). We conclude from curve fitting (Fig. S3) that Int2 is the active Mn3+ species that oxidizes 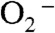 in Y34F ScMnSOD. Although Int2 has been observed in studies of several Y34 mutant human enzymes, the active species that oxidizes
in Y34F ScMnSOD. Although Int2 has been observed in studies of several Y34 mutant human enzymes, the active species that oxidizes 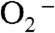 in those systems remains five-coordinate Mn3+ (8). Since Int2 is putatively six-coordinate, Y34F ScMnSOD represents the first MnSOD, whose catalysis may involve a six-coordinate Mn3+.
in those systems remains five-coordinate Mn3+ (8). Since Int2 is putatively six-coordinate, Y34F ScMnSOD represents the first MnSOD, whose catalysis may involve a six-coordinate Mn3+.
Here we propose a mechanism to describe the catalytic cycle of Y34F ScMnSOD (Mechanism A, Scheme 3, involving Int1-A). It involves binding of 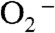 to Mn2+ in the sixth coordination position close to Tyr34 prior to inner-sphere electron transfer, as proposed in the “5–6–5” mechanism (15), referring to increase of coordination number from five to six upon binding of
to Mn2+ in the sixth coordination position close to Tyr34 prior to inner-sphere electron transfer, as proposed in the “5–6–5” mechanism (15), referring to increase of coordination number from five to six upon binding of 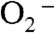 , and decrease of coordination number from six to five upon dissociation of H2O2. The solvent ligand instantaneously transfers the first proton to the distal oxygen of the newly formed
, and decrease of coordination number from six to five upon dissociation of H2O2. The solvent ligand instantaneously transfers the first proton to the distal oxygen of the newly formed 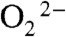 (16), leading to a six-coordinate end-on Mn3+-hydroperoxo species, which in this mechanism corresponds to the Mn-peroxo complex. The additional outer-sphere water molecule then transfers a second proton to the proximal oxygen of HOO-, leading to dissociation of the peroxide ligand. The vacant sixth ligand site is then occupied by a second-sphere solvent molecule. We formulate Int2 as a six-coordinate Mn3+ center binding two solvent molecules. In the other half reaction,
(16), leading to a six-coordinate end-on Mn3+-hydroperoxo species, which in this mechanism corresponds to the Mn-peroxo complex. The additional outer-sphere water molecule then transfers a second proton to the proximal oxygen of HOO-, leading to dissociation of the peroxide ligand. The vacant sixth ligand site is then occupied by a second-sphere solvent molecule. We formulate Int2 as a six-coordinate Mn3+ center binding two solvent molecules. In the other half reaction, 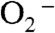 first replaces the solvent molecule in the sixth ligand site (17), before it is oxidized by the Mn3+ ion. Because replacement of the HOO- group by a solvent ligand (
first replaces the solvent molecule in the sixth ligand site (17), before it is oxidized by the Mn3+ ion. Because replacement of the HOO- group by a solvent ligand (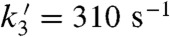 ) is much faster than loss of the HOO- group (k4 = 40 s-1) or the solvent ligand (
) is much faster than loss of the HOO- group (k4 = 40 s-1) or the solvent ligand (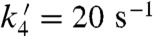 ), fast catalysis can be achieved through formation of the six-coordinate Int2 and bypassing the slow steps leading to five-coordinate Mn3+SOD.
), fast catalysis can be achieved through formation of the six-coordinate Int2 and bypassing the slow steps leading to five-coordinate Mn3+SOD.
Scheme 3.

Proposed mechanisms for Y34F ScMnSOD.
Mechanism A is strongly supported by experimental and computational studies. The “5–6–5” mechanism is supported by the fact that azide, an analog of 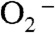 , binds to the metal in the sixth coordination site in Fe and MnSOD (15). Cryotrapped EcMnSOD crystallized at pH 8.5 binds an additional water in the same sixth ligand site (18). In addition, several theoretical calculations suggest that the most stable conformation of a bound peroxyl is end-on (17, 19). The most probable candidate for Mn-peroxo complexes has been proposed as an end-on Mn3+-hydroperoxo complex with HOO- pointing toward the solvent ligand (19).
, binds to the metal in the sixth coordination site in Fe and MnSOD (15). Cryotrapped EcMnSOD crystallized at pH 8.5 binds an additional water in the same sixth ligand site (18). In addition, several theoretical calculations suggest that the most stable conformation of a bound peroxyl is end-on (17, 19). The most probable candidate for Mn-peroxo complexes has been proposed as an end-on Mn3+-hydroperoxo complex with HOO- pointing toward the solvent ligand (19).
The structure of Mn-peroxo complexes is one of the most controversial topics in MnSOD research. To test the feasibility of Mechanism A, we modeled an end-on Mn3+-hydroperoxo species in the active site of Y34F ScMnSOD. The O1 of HOO- in the model was placed at the same (x, y, z) coordinates as the N1 of azide in the six-coordinate azide-MnSOD (Protein Data Bank code: 1MNG). HOO-, sitting in the sixth coordination site, does not have unfavorable steric interactions with the active-site residues (Fig. 5). The only adjustment needed for a good fit was a small displacement of the water molecule that has substituted for the ─OH of Tyr34 to a position 0.85 Å away from the Mn. The proposed new position for this shifted solvent is indicated in cyan in Fig. 5, which should be compared with Fig. 1A. When the Mn–O1(HOO-)–O2(HOO-) angle was modulated between 90° and 180°, the distance between O2(HOO-) and the additional water molecule in Fig. 1A is always < 2.0 Å, suggesting that this water molecule would be in close contact with a bound HOO-, and thus proton transfer from the former to the latter could be quite facile. This is consistent with fast protonation rates observed for Y34F ScMnSOD (Figs. 2B and 4A) and several Y34 mutant human MnSODs (Table S2).
Fig. 5.

A suggestive end-on model of the Mn-peroxo complex built from the crystal structure of Y34F ScMnSOD (chain A). The graphic was generated using the PyMOL Molecular Graphics System (28). Manganese ions are shown as purple spheres. Peroxyl groups are shown as red sticks with hydrogen atoms in gray. Existing and hypothetical solvent molecules are shown as red and cyan spheres, respectively.
There is another mechanism (Mechanism B, Scheme 3, involving Int1-B) that has been proposed by many researchers. The major distinction between Mechanism A and B lies in the structure of the Mn-peroxo complex. In Mechanism B the reaction of 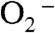 with Mn2+SOD initiates from displacement of the solvent ligand by
with Mn2+SOD initiates from displacement of the solvent ligand by 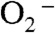 without occurrence of any proton transfer, and the peroxo complex is a six-coordinate Mn3+ binding a side-on
without occurrence of any proton transfer, and the peroxo complex is a six-coordinate Mn3+ binding a side-on 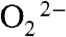 (1, 5). The displaced solvent molecule would become part of the water chain extending from active site to dimer interface (Fig. S6). The side-on peroxo then would need to accept two protons, either through the hydrogen bond chain extending from Gln154 or through direct hydrogen-bonding interactions with second-sphere water molecules. The structure of Int2 and the process of oxidation of
(1, 5). The displaced solvent molecule would become part of the water chain extending from active site to dimer interface (Fig. S6). The side-on peroxo then would need to accept two protons, either through the hydrogen bond chain extending from Gln154 or through direct hydrogen-bonding interactions with second-sphere water molecules. The structure of Int2 and the process of oxidation of 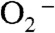 are the same as in Mechanism A.
are the same as in Mechanism A.
Formulation of the Mn-peroxo complex as a side-on Mn3+-peroxo species has been proposed based on computational and spectroscopic studies (3, 5), and on the crystal structure of peroxide-soaked EcMnSOD showing side-on binding of peroxide to active-site Mn (Fig. S7A) (20). The active site structure in Fig. S7A, although determined under cryo conditions, provides a potential pathway for protonation of the side-on peroxo complex through coordination of a water molecule in the sixth ligand site. An end-on Mn-hydroperoxo species is observed in two out of four chains in the same structure (Fig. S7B), and it could be a product of the protonation implied in Fig. S7A.
Here we favor Mechanism A over B, because, in the former, the pathway from the Mn-peroxo complex to Int2 is much simpler. This reaction in Mechanism A requires transfer of one proton and translocation of one solvent molecule, whereas in Mechanism B it requires transfer of two protons and translocation of two solvent molecules. Moreover, one would expect to observe additional intermediates between Int1 and Int2, if Mechanism B in fact occurred. Y34 mutant MnSODs provide ideal systems for studying the reaction mechanism of product inhibition. We will continue our exploration of the mechanism of product inhibition through theoretical calculations in the future.
In Y34F ScMnSOD, pH Controls Proton Transfer Pathways.
MnSOD is well known for the gating between the “prompt protonation” and “inner-sphere” pathways (Scheme 1), based on the likelihood of reduction of 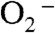 going through the Mn-peroxo complex. We report here that protonation of the peroxo complex in Y34F ScMnSOD is also gated between a fast and a slow pathway (Fig. 4), with the rate constant differing by approximately 7-fold. The fast pathway goes through the putative six-coordinate Int2, while the slow pathway leads directly to the five-coordinate Mn3+SOD and occurs in all MnSODs (Scheme 2, 3).
going through the Mn-peroxo complex. We report here that protonation of the peroxo complex in Y34F ScMnSOD is also gated between a fast and a slow pathway (Fig. 4), with the rate constant differing by approximately 7-fold. The fast pathway goes through the putative six-coordinate Int2, while the slow pathway leads directly to the five-coordinate Mn3+SOD and occurs in all MnSODs (Scheme 2, 3).
Previous studies on bacterial reaction centers (RCs) elucidated mechanisms for electron transfer coupled with multiple proton transfer pathways, and the gating is controlled by conformational changes of the protein (21). In Y34F ScMnSOD, by contrast, the gating is pH-dependent. The fast pathway dominates at neutral pH and is replaced by the slow pathway at increased pH with a pK of about 8 (Fig. 4B, inset). Examples of “fast” and “slow” proton transfer being pH-dependent and pH-independent, respectively, have been reported for metalloenzymes (22). The pH effects to Y34F ScMnSOD could result from deprotonation of active-site water molecules, especially the one that has substituted the ─OH of Tyr34, at high pH. If the additional solvent molecule stays as a H2O, it could donate a proton quickly to the Mn-peroxo complex. When it is deprotonated at increased pH and forms a OH-, the fast proton transfer would be shut down, and the bound peroxyl would need to get a proton through the slow pathway that occurs in all MnSODs.
Formation of the six-coordinate Int2, which is coupled with the fast proton transfer pathway, is no longer observed at high pH. We conclude, therefore, that the five-coordinate Mn3+ generated through the slow proton transfer pathway becomes the active species at high pH (Scheme 2, 3). This is the first time for pH controlling proton transfer pathways and determining the active Mn3+ species to be reported in any WT or mutant MnSOD.
A Six-Coordinate Mn3+ could be an Active Species in WT Yeast MnSOD.
Two unique biophysical properties distinguish yeast MnSODs from any previously characterized MnSOD. First, yeast MnSODs rest predominantly in the reduced 2+ form (2, 4), while all other MnSODs rest ≥50% in the oxidized 3+ form (10–12). The positioning of Gln154 has been proposed to control the resting oxidation state of MnSOD (3, 23–25). Previously we showed that the increased separation of the Gln154 side chain from the metal ion in ScMnSOD leads to a smaller stabilization of OH- vs. H2O and thus a relative stabilization of the 2+ vs. 3+ state (2). Second, yeast Mn3+SODs contain both five- and six-coordinate Mn3+ centers (2), while only five-coordinate Mn3+ centers are present in oxidized MnSODs from other organisms (26). Y34F ScMnSOD resembles the WT enzyme in that it rests as Mn2+SOD and is oxidized to a mixture of five- and six-coordinate Mn3+ centers (Fig. 1B).
Although Y34F ScMnSOD and several Y34 mutant human enzymes share similar active sites and formation of the six-coordinate Int2, only in the mutant yeast MnSOD does Int2 react with 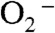 . This may result from the fact that Y34F ScMn3+SOD is inclined to form a six-coordinate Mn3+ adduct. Because WT and the mutant yeast Mn3+SODs are both composed of five- and six-coordinate species (2), the active Mn3+ species in WT yeast MnSODs could also be six-coordinate. Indeed, our recent work has revealed that catalysis by WT yeast MnSODs involves fast proton transfer (2). Compared to that of human MnSOD, the catalytic reactivity of WT yeast enzymes is significantly more decreased at high pH with indications of depression of the protonation rate (2). Therefore, the mechanism proposed for Y34F ScMnSOD could well apply to WT yeast enzymes. Decay of the Mn-peroxo complex and the six-coordinate Int2 may be too fast to be detectible in catalysis by WT yeast enzymes under the conditions of our experiments.
. This may result from the fact that Y34F ScMn3+SOD is inclined to form a six-coordinate Mn3+ adduct. Because WT and the mutant yeast Mn3+SODs are both composed of five- and six-coordinate species (2), the active Mn3+ species in WT yeast MnSODs could also be six-coordinate. Indeed, our recent work has revealed that catalysis by WT yeast MnSODs involves fast proton transfer (2). Compared to that of human MnSOD, the catalytic reactivity of WT yeast enzymes is significantly more decreased at high pH with indications of depression of the protonation rate (2). Therefore, the mechanism proposed for Y34F ScMnSOD could well apply to WT yeast enzymes. Decay of the Mn-peroxo complex and the six-coordinate Int2 may be too fast to be detectible in catalysis by WT yeast enzymes under the conditions of our experiments.
In conclusion, Y34F ScMnSOD has a unique mechanism in which a putative six-coordinate Mn3+ species reacts with 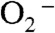 at neutral pH. Fast catalysis can be achieved through formation of this six-coordinate Mn3+ species and bypassing the slow steps leading to five-coordinate Mn3+SOD. Protonation of the Mn-peroxo complex has two distinct pathways with the gating controlled by pH. Because both WT and Y34F yeast Mn3+SOD have the tendency to become six-coordinate, six-coordinate Mn3+ species could also function in the catalysis by WT yeast MnSODs.
at neutral pH. Fast catalysis can be achieved through formation of this six-coordinate Mn3+ species and bypassing the slow steps leading to five-coordinate Mn3+SOD. Protonation of the Mn-peroxo complex has two distinct pathways with the gating controlled by pH. Because both WT and Y34F yeast Mn3+SOD have the tendency to become six-coordinate, six-coordinate Mn3+ species could also function in the catalysis by WT yeast MnSODs.
Materials and Methods
DNA Manipulation, Expression, and Purification of Y34F ScMnSOD.
Strain, plasmid construction, and yeast transformation is detailed in SI Text. Y34F ScMnSOD was expressed and purified as described (2).
Pulse Radiolysis.
The experiments were carried out using the 2 MeV Van de Graaff accelerator at Brookhaven National Laboratory. 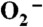 is generated in air-saturated aqueous solution containing sodium formate through the following reactions:
is generated in air-saturated aqueous solution containing sodium formate through the following reactions: 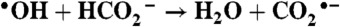 ,
,  ,
, 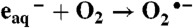 ,
, 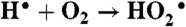 .
.
The experiments to measure catalytic rates were carried out using two methodologies. One set of studies involved either oxidizing an resting enzyme or reducing an oxidized enzyme (40–120 μM) with substoichiometric quantities of 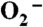 and following the appearance or disappearance of Mn3+SOD, respectively, via following absorbance change in the visible range (370–550 nm). The other method involved following the decay of various concentrations of
and following the appearance or disappearance of Mn3+SOD, respectively, via following absorbance change in the visible range (370–550 nm). The other method involved following the decay of various concentrations of 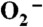 at 260 nm using a 1∶1 to 1∶50 ratio of
at 260 nm using a 1∶1 to 1∶50 ratio of 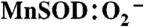 . Here, rate constants were calculated by fitting the data obtained to a mechanism using the Chemical Kinetics program in PRWIN (27).
. Here, rate constants were calculated by fitting the data obtained to a mechanism using the Chemical Kinetics program in PRWIN (27).
All MnSOD were taken as the ICP-measured concentration of manganese in the sample. The pH of the buffer was adjusted using ultrapure (Baker Ultrex) sodium hydroxide and sulphuric acid.
Crystallography: Crystallization, Data Collection, and Refinement.
Reductive methylation (Fig. S8) and crystallization of Y34F ScMnSOD, crystallography data collection, and refinement was performed as described in SI Text.
Supplementary Material
ACKNOWLEDGMENTS.
This work was supported by Grant DK46828 to J.S.V. This work was supported by KOSEF/MEST through WCU project (R31-2008-000-10010-0) to J.S.V. Radiolysis studies were carried out at Center for Radiation Chemistry Research at Brookhaven National Laboratory, which is funded under contract DE-AC02-98CH10886 with the United States Department of Energy and supported by its Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences. The authors thank Malcolm Capel, Frank Murphy, Jonathan Schuermann, and Igor Kourinov at NECAT beamline 24-ID-C at APS for help with data collection, and Jason Navarro for help with crystallization screens.
Footnotes
The authors declare no conflict of interest.
Data deposition: Crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4E4E).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1212367109/-/DCSupplemental.
References
- 1.Abreu IA, Cabelli DE. Superoxide dismutases: A review of the metal-associated mechanistic variations. Biochim Biophys Acta. 2010;1804:263–274. doi: 10.1016/j.bbapap.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Sheng Y, et al. Comparison of two yeast MnSODs: Mitochondrial Saccharomyces cerevisiae versus cytosolic Candida albicans. J Am Chem Soc. 2011;133:20878–20889. doi: 10.1021/ja2077476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson TA, Brunold TC. Combined spectroscopic/computational studies on Fe-and Mn-dependent superoxide dismutases: Insights into second-sphere tuning of active site properties. Acc Chem Res. 2004;37:461–470. doi: 10.1021/ar030272h. [DOI] [PubMed] [Google Scholar]
- 4.Barnese K, et al. Investigation of the highly active manganese superoxide dismutase from Saccharomyces cerevisiae. J Am Chem Soc. 2010;132:12525–12527. doi: 10.1021/ja104179r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson TA, Karapetian A, Miller AF, Brunold TC. Probing the geometric and electronic structures of the low-temperature azide adduct and the product-inhibited form of oxidized manganese superoxide dismutase. Biochemistry. 2005;44:1504–1520. doi: 10.1021/bi048639t. [DOI] [PubMed] [Google Scholar]
- 6.Maliekal J, et al. Comparison and contrasts between the active site PKs of Mn-superoxide dismutase and those of Fe-superoxide dismutase. J Am Chem Soc. 2002;124:15064–15075. doi: 10.1021/ja027319z. [DOI] [PubMed] [Google Scholar]
- 7.Guan Y, et al. Crystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34. Biochemistry. 1998;37:4722–4730. doi: 10.1021/bi972394l. [DOI] [PubMed] [Google Scholar]
- 8.Perry JJ, et al. Contribution of human manganese superoxide dismutase tyrosine 34 to structure and catalysis. Biochemistry. 2009;48:3417–3424. doi: 10.1021/bi8023288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perry JJ, Shin DS, Getzoff ED, Tainer JA. The structural biochemistry of the superoxide dismutases. Biochim Biophys Acta. 2010;1804:245–262. doi: 10.1016/j.bbapap.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abreu IA, et al. The kinetic mechanism of manganese-containing superoxide dismutase from Deinococcus radiodurans: A specialized enzyme for the elimination of high superoxide concentrations. Biochemistry. 2008;47:2350–2356. doi: 10.1021/bi7016206. [DOI] [PubMed] [Google Scholar]
- 11.Whittaker JW, Whittaker MM. Active-site spectral studies on manganese superoxide-dismutase. J Am Chem Soc. 1991;113:5528–5540. [Google Scholar]
- 12.Hsu JL, et al. Catalytic properties of human manganese superoxide dismutase. J Biol Chem. 1996;271:17687–17691. doi: 10.1074/jbc.271.30.17687. [DOI] [PubMed] [Google Scholar]
- 13.Siegbahn PE. Quantum chemical studies of manganese centers in biology. Curr Opin Chem Biol. 2002;6:227–235. doi: 10.1016/s1367-5931(02)00312-5. [DOI] [PubMed] [Google Scholar]
- 14.Zheng J, Domsic JF, Cabelli D, McKenna R, Silverman DN. Structural and kinetic study of differences between human and Escherichia coli manganese superoxide dismutases. Biochemistry. 2007;46:14830–14837. doi: 10.1021/bi7014103. [DOI] [PubMed] [Google Scholar]
- 15.Lah MS, et al. Structure-function in Escherichia coli iron superoxide dismutase: Comparisons with the manganese enzyme from Thermus thermophilus. Biochemistry. 1995;34:1646–1660. doi: 10.1021/bi00005a021. [DOI] [PubMed] [Google Scholar]
- 16.Rulisek L, Jensen KP, Lundgren K, Ryde U. The reaction mechanism of iron and manganese superoxide dismutases studied by theoretical calculations. J Comput Chem. 2006;27:1398–1414. doi: 10.1002/jcc.20450. [DOI] [PubMed] [Google Scholar]
- 17.Abreu IA, Rodriguez JA, Cabelli DE. Theoretical studies of manganese and iron superoxide dismutases: Superoxide binding and superoxide oxidation. J Phys Chem B. 2005;109:24502–24509. doi: 10.1021/jp052368u. [DOI] [PubMed] [Google Scholar]
- 18.Borgstahl GEO, Pokross M, Chehab R, Sekher A, Snell EH. Cryotrapping the six-coordinate, distorted-octahedral active site of manganese superoxide dismutase. J Mol Biol. 2000;296:951–959. doi: 10.1006/jmbi.1999.3506. [DOI] [PubMed] [Google Scholar]
- 19.Carrasco R, Morgenstern-Badarau I, Cano J. Two proton-one electron coupled transfer in iron and manganese superoxide dismutases: A density functional study. Inorg Chim Acta. 2007;360:91–101. [Google Scholar]
- 20.Porta J, Vahedi-Faridi A, Borgstahl GE. Structural analysis of peroxide-soaked MnSOD crystals reveals side-on binding of peroxide to active-site manganese. J Mol Biol. 2010;399:377–384. doi: 10.1016/j.jmb.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 21.Okamura MY, Paddock ML, Graige MS, Feher G. Proton and electron transfer in bacterial reaction centers. Biochim Biophys Acta. 2000;1458:148–163. doi: 10.1016/s0005-2728(00)00065-7. [DOI] [PubMed] [Google Scholar]
- 22.Chen K, et al. Atomically defined mechanism for proton transfer to a buried redox centre in a protein. Nature. 2000;405:814–817. doi: 10.1038/35015610. [DOI] [PubMed] [Google Scholar]
- 23.Miller AF, Vance CK. Novel insights into the basis for Escherichia coli superoxide dismutase’s metal ion specificity from Mn-substituted FeSOD and its very high E(m) Biochemistry. 2001;40:13079–13087. doi: 10.1021/bi0113317. [DOI] [PubMed] [Google Scholar]
- 24.Yikilmaz E, et al. How can a single second sphere amino acid substitution cause reduction midpoint potential changes of hundreds of millivolts? J Am Chem Soc. 2007;129:9927–9940. doi: 10.1021/ja069224t. [DOI] [PubMed] [Google Scholar]
- 25.Miller AF. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012;586:585–595. doi: 10.1016/j.febslet.2011.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campbell KA, et al. Parallel polarization EPR characterization of the Mn(III) center of oxidized manganese superoxide dismutase. J Am Chem Soc. 1999;121:4714–4715. [Google Scholar]
- 27.Schwarz H. BNL Pulse Radiolysis Program. Upton, NY: Brookhaven National Laboratory; [Google Scholar]
- 28.Schrodinger LLC. The PyMOL Molecular Graphics System. 2010. Version 1.3r1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.