

Abstract
The ability to measure steroid hormone concentrations in blood and urine specimens is central to the diagnosis and proper treatment of adrenal diseases. The traditional approach has been to assay each steroid hormone, precursor, or metabolite using individual aliquots of serum, each with a separate immunoassay. For complex diseases, such as congenital adrenal hyperplasia and adrenocortical cancer, in which the assay of several steroids is essential for management, this approach is time consuming and costly, in addition to using large amounts of serum. Gas chromatography/mass spectrometry profiling of steroid metabolites in urine has been employed for many years but only in a small number of specialized laboratories and suffers from slow throughput. The advent of commercial high-performance liquid chromatography instruments coupled to tandem mass spectrometers offers the potential for medium- to high-throughput profiling of serum steroids using small quantities of sample. Here, we review the physical principles of mass spectrometry, the instrumentation used for these techniques, the terminology used in this field and applications to steroid analysis.
Keywords: Steroid Assay, Mass Spectrometry, Adrenocortical Cancer, Congenital Adrenal Hyperplasia, Multiplex Assay
Introduction
Modern diagnosis of hormone deficiency and excess states, particularly when subtle, relies heavily on static and dynamic tests of hormone production. Consequently, the evolution of clinical endocrinology has been intimately linked to the development of hormone assays, which were often generated by endocrinologists themselves. To this end, a major advance in the endocrinology of human steroid hormone physiology occurred when Abraham developed the radioimmunoassay (RIA) for estradiol (E2) in 1969, replacing the cumbersome and imprecise bioassays of that era [1]. Abraham used an organic solvent extraction and column chromatography to remove cross-reacting compounds, and the assay proved extremely useful. Over time, RIAs were created, improved, and validated for many key steroid hormones and precursors. Two-site immunoradiometric or immunochemiluminometric (ICMA) assays improved sensitivity and specificity, with ICMA assays eliminating the need for radioactivity. Leaving the obscure academic endocrinologists’ laboratories, commercialization of these assays incorporated instrumentation to increase sample throughput, but extraction and chromatography are difficult to automate, and assay performance and reproducibility often suffered. Academic laboratories introduced multiplexed immunoassays, where several different fractions from a single extraction were eluted from a chromatographic column and individually assayed for specific steroids, reducing sample volume, and increasing productivity to some extent. Commercial assays, in contrast, remained mostly single-analyte measurements, largely for economic and quality control reasons.
Meanwhile, analytical chemistry adopted mass spectrometry (MS) as a versatile technique for quantitation of small molecules such as petrochemicals, agricultural chemicals, and pharmaceuticals. Coupled to gas or high-performance liquid chromatography (GC or HPLC), numerous compounds can be separated and measured quickly and accurately, and to some extent these assays can be automated. Steroids are small molecules similar to xenobiotics commonly measured by MS methods, and GC-MS assays for urinary steroid hormones and metabolites appeared in the 1970s. Due to their low volatility and excretion predominantly as sulfate or glucuronide conjugates, urinary steroids require hydrolysis and derivatization following extraction and prior to GC-MS analysis, limiting automation and increasing extraction complexity. Nonetheless, extremely useful procedures have developed to measure up to 40 steroids in a single GC-MS analysis [2]. The mass-production of inexpensive quadrupoles—the heart of the GC-MS instrument—led to the introduction of tandem mass spectrometers in the 1980s. The incorporation of computers for instrument control and data acquisition led to improved sensitivity and speed. Liquid chromatography with tandem mass spectrometry (LC-MS/MS) assays are amenable to measuring several analytes with minimal sample preparation, and this technology has been applied to steroid hormone measurements. The advantages of MS-based assays are their inherent multiplexed nature, measuring many steroids in a small sample. These powerful assays are ideal for evaluating complex adrenal diseases, particularly congenital adrenal hyperplasias (CAH), adrenal adenomas/hyperplasias, and adrenocortical cancer (ACC), as several hormones and precursors are analyzed simultaneously. In this article, we will review principles of MS, its application to adrenal diseases, and pitfalls and promises of these methods.
Basic Concepts in Mass Spectrometry
Physical Principles
The field of MS was developed on the basis of a single physical principle: when a charged particle moves through a magnetic field, the path of the particle is deflected by an amount inversely proportional to the mass of the particle. Consequently, a simple mass spectrometer consists of an ionization apparatus, a vacuum chamber with a magnetic field, and a detector (Fig. 1a). Later, mass spectrometers were devised to measure m/z based on the time required for the ion to fly the length of a vacuum chamber, called time-of-flight or TOF instrument. TOF instruments measure m/z with extreme precision and are very useful for determining the exact mass of a compound, but historically, these instruments were not designed for precise quantitation due in part to limited dynamic range.
Fig. 1.

a Schematic diagram of a simple mass spectrometer with source, quadrupole, and detector, sorting ions by m/z. b Generation of a mass spectrum for a pure compound. The molecular ions fragment, and the charged fragments are separated by the quadrupole and quantitated by the detector, yielding a standard mass spectrum. c Mass spectrum of testosterone. d Schematic diagram of a collision cell. Following ionization, molecular ions are accelerated into the collision cell via electrostatic repulsion where they collide with gas molecules in the collision cell through a process called collision induced dissociation (CID). CID imparts internal energy to the molecular ion, which induces fragmentation
One basic process of ion formation called electron ionization involves exposing the compound of interest to a beam of electrons accelerated via a high-voltage potential, which imparts charge on the molecules by ejecting a core electron. Since the odd electron molecular ion is an unstable, highly energetic species, the ion fragments into smaller ions and neutral species through unimolecular decomposition. The detector then registers the ions, amplifies the signal through an electron cascade, and through computer control converts the electronic signals into a mass spectrum (Fig. 1b). Note that the base peak (largest signal) in the mass spectrum of testosterone is a fragment ion (Fig. 1c), as is often the case in mass spectrometry. Compounds are analyzed in either positive ion mode or negative ion mode, depending on the chemical nature of the analyte, primarily the acid/base characteristics. The detector can measure specific masses individually (selected ion monitoring, SIM) or sum all the masses, affording total ion current.
Analysis of Complex Mixtures: Tandem Mass Spectrometry
An early advance in mass spectrometry came when mass spectrometers were coupled together into tandem or hybrid instruments, introducing the revolutionary ability to both separate mixtures and identify or quantitate the individual components in a single system. In a primitive tandem quadrupole instrument, multiple compounds are ionized in the source, and the first quadrupole is used to select only the molecular “precursor” ion, serving as a mass filter and allowing only the desired analyte to enter the second quadrupole. The molecular ion could simply enter, traverse, and register intact in the second quadrupole, but what if some of these ions are not related to the steroid of interest but rather fragments of other compounds or other isobaric species? To improve specificity, the instrument is normally designed to allow the molecular ion to be selected in the first quadrupole and fragment in the second, depositing a full mass spectrum in the final readout. For quantitation, only one or two abundant fragment or “product” ions specific for the analyte in question need to be measured in the second mass spectrometer to give equivalent accuracy. Finally, many systems contain a third chamber in between the two mass spectrometers, called a “collision cell,” where the molecular ions are accelerated into gas atoms (typically nitrogen or argon), inducing more complete fragmentation (Fig. 1d). Often, this chamber also contains a third quadrupole to steer the ions through, hence completing the triple quadrupole or “triple quad” system most commonly used today (Fig. 2).
Fig. 2.

Schematic diagram of triple-quadrupole LC-MS/MS process and instrumentation. In the upper right, pre-analytical sample cleanup with optional derivatization prepares the sample for injection. Analytes are resolved on the HPLC column, and the effluent is introduced into the mass spectrometer. Steroid molecules are ionized in the source, selected in Q1, fragmented in the collision cell (Q2), and sorted in Q3. The detector monitors the intensity of the chosen (quantifier) Q3 fragments derived from the ions selected in Q1 at the appropriate times in the chromatogram to perform the measurement
Analysis of Complex Mixtures: Gas Chromatography/Mass Spectrometry
Another approach to separation of complex mixtures prior to mass spectrometry is to first employ a chromatographic step such as GC or HPLC, which is called the “front end.” Since ion formation and analysis must occur under high vacuum (10−5 to 10−6 Torr), these systems initially used GC for separation prior to MS (GC/MS), due to the relative ease of coupling these orthogonal techniques with respect to the vacuum considerations. Given the high resolving capability of a GC equipped with a modern capillary column and the superb precision of MS, GC/MS systems have been a mainstay of analytical chemistry for many years in academia and industry, but not without limitations. Many target analytes, such as steroids, are not intrinsically volatile and will decompose during the transition between the liquid and gas phase inside the injection port or during elution from the column at high temperatures. Consequently, several steps, including pre-analytical derivatization, are required for steroid analysis by GC/MS. First, the excreted steroids are separated from salts and concentrated by solid-phase extraction (SPE). Second, since steroids are mostly excreted as glucuronide and sulfate conjugates, these charged moieties are removed chemically or enzymatically by acid or enzyme treatment. Third, the free hydroxyl groups, and in most cases the ketones as well, are modified chemically to render the compounds less polar and more volatile. Most commonly, silylating reagents modify the hydroxyl groups as trimethysilyl or tert-butyldimethylsilyl ethers. The ketone groups are either converted to silyl enol ethers with base and the same silylating reagents or converted into oximes with hydroxylamine before silylating. These derivatives are stable to high temperatures and more hydrophobic and volatile than the parent steroids.
The disadvantages of the derivatization approach includes the potential for isomeric mixtures of derivatives, the substantial time required prior to analysis, and the increased masses of the compounds after derivatization. Acetates or other simpler groups are sometimes used, particularly for carbon isotope ratio instruments, which measure isotopic composition of the carbon atoms in the chemical of interest [3]. Pentafluorobenzyl esters are attractive derivatives for some purposes, because the pentafluorobenzyl group easily forms a very stable negative ion [4]. Thus, these compounds are analyzed in negative ion mode using electron capture negative ionization, yielding highly sensitive spectra with a large molecular ion ([M-H]−·), often with minimal or no fragments.
To improve the sensitivity when assaying many compounds from one GC/MS run, specific ions (usually fragments) characteristic of the known compounds are targeted at the time when the compounds elute from the GC into the MS. This method, called SIM, markedly increases sensitivity by reducing the noise inherent in obtaining complete mass spectra. SIM also allows the instrument to focus its power in selected mass regions versus wasting time and resources scanning through masses not relevant to the analysis. Modern instruments can be programmed to measure dozens of ions simultaneously in SIM mode and quantitate target ions at their specified elution times.
Analysis of Complex Mixtures: Liquid Chromatography/Mass Spectrometry
HPLC, unlike GC, separates nonvolatile compounds without derivatization. The HPLC consists of an injector, often configured with a robotic autosampler, and high-pressure pumps capable of mixing two or more solvents or “mobile phases” to gradually change solvent composition. The column is a stainless steel tube filled with silica-based particles or “stationary phase” of <5 μm diameter. Typical column dimensions range between 50 and 250 mm in length, and between 1 and 5 mm in diameter. The smaller the particle, the greater the resolving capacity; however, as particle size decreases, system backpressure increases which requires more force to maintain flow and limits throughput.
The choice of column is critical, since good chromatography is essential to the success of the assay. Systems can be “normal phase,” with a polar stationary phase such as silica and an organic mobile phase. The mobile phase starts with low-polarity solvent, such as hexanes, and the instrument is programmed to mix the gradient to increase mobile phase polarity during the run with a solvent such as ethyl acetate or dichloromethane. The least polar compounds are weakly attracted to the stationary phase and elute first. More polar compounds elute later when the mobile phase polarity is high, overcoming the interaction with the stationary phase. For steroids, “reverse phase” is more effective, since all unconjugated steroids are hydrophobic. The stationary phase contains silica or polymer particles covalently linked to hydrocarbon chains. Typical chain lengths are 8 or 18 carbon atoms, which are “C8” or “C18” resins, respectively. In reverse-phase HPLC, the steroids are injected in a high polarity mobile phase such as 5% methanol in water, and all steroids immediately adhere to the stationary phase in the front of the column, interacting strongly with the hydrophobic resin coating. The gradient is programmed to increase the methanol content in the mobile phase, first eluting the more polar steroids and later the less polar steroids. The order of steroid elution is only partially predictable from their structure, and different solvent mixtures, such as aqueous acetonitrile versus methanol, can change elution characteristics.
Recent advances in HPLC technologies include columns with extremely small particles, <2 μm. Since the back pressures with these columns are so high, up to 1,200 bar (>17,000 psi), special ultrahigh performance pumps, injectors, fittings, and columns, all rated for such higher pressures, are required for these systems (UPLC). A second advance is the development of core–shell particles, where a solid spherical particle (1.2–1.9 μm) is coated with a very thin (<1 μm thick) coating of porous stationary phase. Ordinary silica particles are completely porous, so molecules passing near the particle can transiently interact with the outer layer or take a convoluted path through the center of the particle. These variable paths lead to a phenomenon called “band broadening” where the width of a chromatographic peak increased and its height decreases proportionally. With core–shell particles, there is a defined and limited porous shell on a solid core, limiting the potential paths an analyte can take through the particle and minimizing band broadening. Fewer analyte paths leads to sharper, taller elution peaks, confining all the ions from that compound in a narrow elution time window, which can lead to dramatic improvements in resolution and sensitivity over totally porous particles.
Despite the power of HPLC and UPLC “front end” instruments for separating mixtures prior to MS, marrying an LC to a MS is not exactly a simple technological feat. Taking a typical HPLC flow rate of 0.5 mL/min, ionizing the analytes of interest, and moving them into a vacuum of 10−6 Torr or greater presents an enormous challenge. To introduce the chemical analytes into the MS, the column effluent is directed toward a tiny orifice of the MS, in an interface unit called the “source.” Although this idea might sound easy, it is analogous to spraying water from a fire hose with a 5,000 V potential at a bucket across the street and expecting to get at least some of the water in the bucket! While en route, the solvent must be removed via evaporation, and the chemical must be ionized, requiring a high voltage potential.
One ionization method is electrospray (ESI), in which the column effluent is dispersed into fine droplets on which electric charges are imparted. An opposite charge near the MS orifice attracts these charged droplets, and heated gas evaporates the solvent as the droplets near the MS. As the solvent evaporates, the droplet gets smaller and the ions get closer until the electrostatic repulsion is great enough to break the droplet into smaller droplets. This procedure is iterated until desolvated charged molecules remain. This procedure is similar to the processes used in spray-painting automobile parts and in inkjet printers. The charges are thus deposited on the nonvolatile solute molecules, some of which enter the mass spectrometer. If contaminants or other compounds are present in the droplets, the chemical of interest is still ionized, but a portion of the charge is diverted to these unwanted contaminants, reducing the fraction of the targeted analyte that is successfully ionized and introduced into the MS. This impaired analyte ionization from co-eluting contaminants is called “ion suppression,” a phenomenon that reduces the sensitivity of the assay when ESI is used. An alternative ionization technique, which is less vulnerable to ion suppression, is atmospheric pressure chemical ionization (APCI). In APCI, the gas used for desolvation is ionized first, and charge is transferred from an excess of the ionized gas, usually hydronium ions (H3O+), to the analyte directly without ionization of the droplet first. Regardless of method used, the limitations noted above translate to only a small fraction of the analyte entering the MS as a parent ion, on the order of 1% in most instances.
If the HPLC is linked to a standard MS, the LC/MS system can produce full mass spectra or operate in SIM mode for quantitation of many analytes just like GC/MS. Since nonvolatile compounds are analyzed without derivatization, LC/MS is more versatile and amenable to high-throughput formats than GC/MS. Nevertheless, a compromise between speed and sensitivity must be made. For complex biological mixtures such as human serum, some pre-analytical preparation is usually required, particularly for steroid assays, and the method chosen must balance cost, speed, recovery, and efficacy. At a minimum, the sample is deproteinated with acetonitrile or methanol, which frees steroids such as testosterone from their binding proteins and removes some other lipids and salts. “Dirty” samples will clog the vulnerable (and expensive!) HPLC columns, so additional cleanup steps, such as SPE or liquid–liquid extraction, are often employed. SPE can be incorporated as a second small HPLC column prior to the main (resolving) column using additional pumps and switching valves, further automating the assay.
Despite even extensive pre-analytical efforts and excellent chromatography, LC/MS still suffers from noise, limiting assay sensitivity and from isobaric overlap where two compounds of identical mass elute together. To introduce another layer of separation, the LC is interfaced with a triple-quadrupole LC-MS/MS, which is the most versatile and sensitive instrumentation platform for medium- to high-throughput assays. These instruments can detect <1 pg of many steroids such as testosterone injected from a clinical sample or <1 pg “on column,” which equates to a sensitivity of 1 ng/dL (0.03 nmol/L) from 0.1 mL of serum. Sensitivity is even better when using pure steroid standards in a pure solvent, and this discrepancy between results with pure standards and real samples is called the “matrix effect,” a vague term meaning that the biological milieu (matrix) impairs the sensitivity of the assay in multiple, often obscure ways. With LC-MS/MS, assay multiplexing is possible, measuring several steroids simultaneously as done by GC/MS. The enormous dynamic range of these instruments, often spanning 5 orders of magnitude, facilitates multiplex of steroids, whose concentrations vary widely. This feature means that dehydroepiandrosterone sulfate (DHEAS; ∼100 μg/dL) and aldosterone (1–10 ng/dL) can be accurately measured at the same time without sample dilution, whereas the dynamic range of immunoassays is much lower.
In a typical experiment, the individual steroids are chromatographed and fragmented, to determine their elution times, ionization and fragmentation conditions, and optimal product ions. The steroids are then combined to assure that all isobaric steroids and product ion targets are resolved by the HPLC. Finally, a variation of SIM termed multiple reaction monitoring (MRM) is employed. MRM values are the precursor ion selected in Q1 and the best—usually most abundant—product ion measured in Q3. For example, the “MRMs” or “transitions” for testosterone in positive ion mode using ESI are usually m/z 289/97 or 289/109, representing [M + H]+ and high-abundance fragments of 97 and 109 amu, respectively. When two high abundance product ions are identified, one can be the measured or “quantifier” ion (97) and the second tracked as the “qualifier” ion (109), further assuring that the ions derive from the targeted steroid rather than a contaminant and increasing specificity further at the expense of some sensitivity. Many delta-4 steroids give good fragments of 97 or 109 amu, yet these ions can be used repeatedly as quantifier ions in Q3, since the steroids are resolved either by HPLC (elution time) or by precursor ion (Q1 selection; Fig. 3).
Fig. 3.
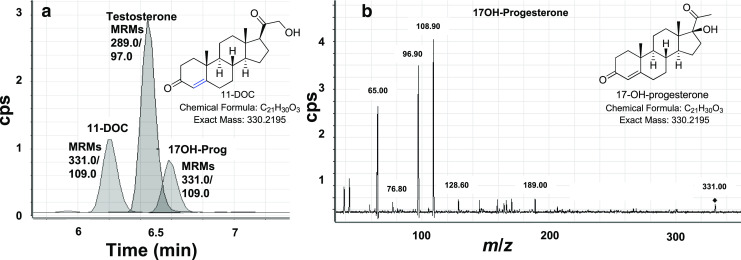
Simultaneous quantitation of 11-deoxycorticosterone (11-DOC), testosterone, and 17-hydroxyprogesterone (17OH-Prog) by LC-MS/MS. a Chromatogram showing tracked MRMs for the three analytes. Note that 11-DOC and 17OH-Prog are isobaric and use identical MRMs; however, chromatography separation allows individual quantitation. In addition, T and 17OH-Prog overlap chromatographically, but the different MRMs allows individual quantitation. b Mass spectrum of 17OH-Prog, showing small molecular ion (331.0, [M + H]+) and fragments, with the 108.9 (109) amu fragment as base peak used for quantitation
Limitations and Caveats to LC-MS/MS Steroid Assays
Like any other technology, LC-MS/MS is not a magical solution to all the difficulties encountered with steroid analysis, and successful application of this method requires a solid background in many aspects of analytical chemistry. First, authentic standards are required to generate standard curves, and these solutions must be prepared accurately and maintained without decomposition. Like any clinical assay, the instrument must be calibrated daily, and a range of sample types must be tested to develop normal ranges and assess limits of detection, interfering substances, and robustness. Second, internal standards are used to correct for ion suppression and recovery during pre-analytical procedures. Ideally, these are isotopically-labeled preparations of each targeted steroid analyte, most commonly with three or more deuterium atoms replacing hydrogens. These compounds are easily distinguished by the tandem mass spectrometer, which selects, sorts, and measures the pairs of molecules simultaneously. Many common steroids are commercially available with deuterium labeling, but the investigator must be prepared to synthesize or have custom-made other less common chemicals if the assay is to be optimized. In truth, not every steroid must have an internal standard, but at a minimum several standards spread throughout the run with proportions comparable to the key analytes is desirable. Third, all steroids do not behave equally in MS, and the sensitivity of the method varies markedly among steroids. Testosterone and most other delta-4 steroids have properties favorable for LC-MS/MS measurements, namely efficient ionization with ESI or APCI and predictable fragmentation to abundant, characteristic product ions. In contrast, assays of delta-5 steroids, 5α-reduced androgens, and estrogens require considerable optimization of instrument conditions. We have found that lowering the gas temperature in the source from the default settings is often beneficial, as many steroids will fragment within the source, particularly with high temperature ESI. Other groups have employed steroid derivatives, such as picolinic acid esters, which efficiently form [M + H]+ ions and fragment predictably [5]. While derivatization certainly improves sensitivity, the additional steps slows throughput, and the reagents might not be ideal for all desired analytes. Some important steroids such as aldosterone and DHEAS are best analyzed in negative ion mode, but most instruments can divide experiments into “periods” and change between positive and negative ion modes during an analysis. Conjugated steroids such as glucuronides are readily analyzed by LC-MS/MS without hydrolysis, but these compounds are best separated with different chromatography protocols than neutral steroids.
Furthermore, although all instruments are similar, specific differences, particularly with source design and performance, means that one assay might not be readily adapted to another instrument without significant method development. Finally, despite all the technology, automation, and sophistication, the accuracy of the assay is completely dependent on accurate construction of the standard curve and pre-analytical processing, before the sample even enters the HPLC. Ironically, with a system costing a half million dollars or more, the success of the LC-MS/MS assay is limited by the accuracy of a $1,000 balance and a $100 pipette.
Applications to Steroid Analysis
GC/MS of Urine Steroid Metabolites
Although individual serum steroid measurements are amenable to GC/MS assays, the requirement for extraction and derivatization renders GC/MS too cumbersome and slow for routine clinical use, particularly when direct immunoassays on serum samples for specific steroids are available. In contrast, the same direct immunoassays are poorly suited for urine steroid analysis for several reasons. First, as discussed above, most steroids are excreted as conjugates. Second, steroids are heavily metabolized to multiple metabolites before excretion, 10 or more for cortisol as an example, all of which can pose problems of cross-reaction in immunoassays. Third, steroid concentrations in urine are more dilute than in plasma, often by an order of magnitude or more and near or below the limits of quantitation. GC/MS is ideally suited to simultaneously quantitate multiple steroid metabolites in urine, although as noted above, urine steroid analysis by GC/MS requires significant time-consuming pre-analytical sample preparation. Because of the technical challenges and more importantly the inability to perform these experiments in a high-throughput format, GC/MS steroid profiling has been limited to a few experienced academic laboratories and has not been adapted to commercial clinical assays. Not only does urinary steroid profiling simultaneously assess all classes of steroids, as well as their precursors and metabolites, but analyses of 24 h urine collections provide integrated measures of steroid production, avoiding the need for precise timing and dynamic testing during sample collection. Consequently, this technology has been a powerful tool for the study of disorders of steroid hormone physiology for several decades [6, 7].
GC/MS urine steroid profiling is ideal for diagnosing genetic defects in newborns, such as 21-hydroxylase deficiency (21OHD), the most common form of CAH. In the USA, all newborns are screened for 21OHD using immunoassay of 17-hydroxyprogesterone (17OHP), which characteristically accumulates above the enzymatic block, using dried blood spots [8]. While the sensitivity of this assay is excellent, the specificity is poor, with a positive predictive value of only about 1%, depending on the cutoff value used. The main reason for the low specificity of this screen is that 17OHP is often elevated in premature newborns. In addition, high 17OHP is found in newborns with 11-hydroxylase deficiency and P450-oxidoreductase deficiency, who rarely suffer adrenal crises. Consequently, many states submit all positive screens to a “second tier” confirmatory test, measuring more specific steroids with less routinely available assays, such as 21-deoxycortisol (21dF). Alternatively, the diagnosis can be made by GC/MS analysis of urine steroid metabolites. The major urinary metabolite of 17OHP is pregnanetriol (PT), and the major metabolite of 21dF is pregnanetriolone (PTone). Initially, urinary PT was validated as a diagnostic test for 21OHD in the newborn [9, 10], but not surprisingly, many false-positive tests were observed, primarily in premature babies. Later, PTone was shown to be superior to PT, and the ratio of PTone to the major cortisol metabolites (tetrahydrocortisone and allotetrahydrocortisone, PTone/[THE + aTHE]), yielded the best test performance, with no overlap between cases and controls [11]. The capacity to measure a series of steroids both above and below the enzymatic block simultaneously using a few milliliters of urine illustrates the power of urinary steroid profiling for diagnosing defects in steroid metabolism.
Several reports describing the utility of GC/MS in diagnosing and monitoring ACC have appeared. In ACC, unlike aldosterone- or cortisol-producing adenomas, steroid biosynthesis tends to be inefficient and multidirectional. Consequently, urine steroid profiles from patients with ACC are characterized by abundant metabolites of steroid hormone precursors rather than the metabolites of the active hormones. Each patient’s presurgery profile, which often contains precursors in several biosynthetic pathways [12], thus serves as a “signature” of disease recurrence. A normal postoperative urine profile indicates remission. Subtle changes in these marker steroids, which are not commonly measured in serum immunoassays, herald recurrence, even before clinical or radiographic progression [13]. In particular, tetrahydro-11-deoxycortisol (THS), the major metabolite of 11-deoxycortisol, was found to be the steroid most characteristically elevated in patients with ACC compared to those with adenomas and the single most useful metabolite to distinguish the two conditions [12, 14]. To analyze the data, which typically includes 30 or more steroid measurements, a procedure known as generalized matrix relevance learning vector quantization was employed to identify the most informative steroid metabolites. Using a subset of nine steroids, patients with ACC could be distinguished from adenomas with 90% sensitivity and specificity [14]. The three steroids THS plus the metabolites of androgen precursors 5-pregnenediol and 5-pregnenetriol were the most discriminatory analytes [12, 14].
LC-MS/MS of Serum Steroids
Despite all the advantages of GC/MS-based approaches to urine steroid profiling, the obstacles posed by slow throughput and complex analyses have limited the broader application of steroid GC/MS to clinical endocrinology. Most well-validated diagnostic strategies currently employed in clinical settings rely on serum steroid measurements. For the measurement of circulating steroid hormones, LC-MS/MS has received considerable attention (Table 1) [4, 15, 16]. LC-MS/MS features the potential for high-throughput, since steroids in serum require no derivatization prior to assay, and like GC/MS, many steroids can be measured simultaneously. An example is in the second-tier screening for 21OHD, using LC-MS/MS to measure not only 17OHP and 21dF but also androstenedione, 11-deoxycortisol, and cortisol [17, 18]. Measuring steroids above and below the enzymatic block enables the calculation of sums and/or ratios, which distinguish enzymatic blocks from generalized activation of the hypothalamic–adrenal axis and adrenal prematurity.
Table 1.
Representative parameters for nonderivatized LC-MS/MS steroid measurements
| Steroid | Ionization | Mass transitions | LOD | Normal range (ng/dL) |
|---|---|---|---|---|
| Aldosterone | ESI- | 359→189 | 1 | 2–30 |
| Androstenedione | ESI+ | 287→97, 109 | 40 | 25–250 |
| Corticosterone | ESI+ | 347→121 | 10 | 50–800 |
| Cortisol | ESI+ | 363→121 | 27 | 3,000–25,000 |
| Cortisone | ESI+ | 361→163 | 50 | 1,000–8,000 |
| DHEA | APPI+ | 271→213 | 1 | 30–700 |
| DHEAS | APPI+ | 271→213 | 0.2 | 10,000–500,000 |
| 11-Deoxycorticosterone | ESI+, APCI+ | 331→97, 109 | 75 | 2–20 |
| 11-Deoxycortisol | ESI+ | 347→97, 109 | 60 | 10–80 |
| 21-Deoxycortisol | ESI+ | 347→311 | 35 | <5 |
| Dehydrocorticosterone | ESI+ | 345→121 | 10 | 10–300 |
| Dihydrotestosterone | ESI+ | 291→255 | 85 | 4–85 |
| Estrone | ESI- | 269→145 | 0.2 | 1–15 |
| Estradiol | ESI- | 271→145 | 0.2 | 1–30 |
| 17-Hydroxyprogesterone | ESI+ | 331→97, 109 | 3 | 10–300 |
| Progesterone | APPI+ | 315→97, 109 | 2 | <10–2,500 |
| Testosterone | ESI+ | 289→97, 109 | 5 | 10–60 (female) |
| 350–1,000 (male) |
Just how many steroids can be measured simultaneously using LC-MS/MS with exquisite specificity and sufficient sensitivity for clinical use? The answer to this provocative and clinically pressing question seems to change monthly. Academic groups have published protocols for measuring up to a dozen steroids/sterols at one time on as little as 0.2 mL of serum [19]. Instrument manufactures have recognized the looming demand for serum steroid profiling applications, and method “packages” have been developed and marketed. We caution that any application package is likely to be vulnerable to even slight deviations from the precise protocol, and it is unlikely that a method package developed for one system will function equally well on another system. For LC-MS/MS, in which multiple parameters all contribute unequally to the assay performance for each steroid, profiling methods necessarily reach a compromise amongst the optimal parameters for each analyte. In general, parameters are chosen to favor optimal performance for the least sensitive analytes at the expense of sensitivity for the more abundant steroids.
At the moment, commercial clinical laboratories have not embraced steroid profiling, although many reference laboratories now use LC-MS/MS assays for all but the most trivial steroid measurements. The multiplexing of steroid assays, while convenient for the doctor and patient, poses challenges related to quality control documentation and reimbursement for clinical laboratories. Nevertheless, despite the large capital outlay and high technical proficiency required for LC-MS/MS measurements, the superior accuracy to platform immunoassays at low hormone concentrations, the wide dynamic range, and the avoidance of radioactivity and proprietary antibodies make LC-MS/MS assays attractive for steroid assays in reference laboratories. For example, the Center for Disease Control has developed the HoST Program for testosterone assay certification and interlaboratory assay harmonization, as has been done for cholesterol. Many of the laboratories applying for HoST certification are testing their LC-MS/MS assays, recognizing the poor performance of platform immunoasssays for testosterone on samples from women and children [20]. Whether steroid profiling by LC-MS/MS will ever reach “prime time” and routine usage in clinical settings remains to be established.
Conclusions
The era of steroid profiling by GC/MS and LC-MS/MS has arrived, at least for research studies. These technologies provide a dynamic picture of the steroid landscape in an individual, rather than a snapshot or glimpse of one or two guideposts. The endocrinology investigator is no longer limited to available immunoassays offered by a typical test menu or triaging based on volume of precious specimens needed. We have only scratched the surface of deploying steroid profiling for the diagnosis of endocrine diseases like CAH and ACC. We predict that steroid profiling will lead to the elucidation of mild phenotypes, masquerading as primary hypertension or the metabolic syndrome, as seen in “subclinical cushing syndrome” [21]. MS profiling holds the promise of guiding which patients with incidental adrenal adenomas will benefit from surgery and of improved monitoring for CAH and ACC patients on therapy, particularly for adults. No longer exclusively the providence of the analytical chemist, it is now time for the clinical investigator to adopt these methods to solve the difficult problems in the human biology of adrenal diseases. Our intention has been to provide the principles and cautions to inspire and guide these investigators to explore the uncharted waters that lay ahead.
Acknowledgments
This work was supported by a Clinician–Scientist Award in Translational Research from the Burroughs-Wellcome Fund (#1005954 to R.J.A.) and the Michigan Nutrition Obesity Center (DK089503). JGM is supported by National Institute of General Medical Sciences Large Scale Collaborative “Glue” Grant U54 GM069338 (LIPID MAPS).
Disclosures
The authors have no conflicts of interest to disclose.
References
- 1.Abraham GE. Solid-phase radioimmunoassay of estradiol-17β. J Clin Endocrinol Metab. 1969;29:866–870. doi: 10.1210/jcem-29-6-866. [DOI] [PubMed] [Google Scholar]
- 2.Shackleton CHL. Mass spectrometry: application to steroid and peptide research. Endocr Rev. 1985;6:441–486. doi: 10.1210/edrv-6-3-441. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Tobias HJ, Brenna JT. Steroid isotopic standards for gas chromatography–combustion isotope ratio mass spectrometry (GCC-IRMS) Steroids. 2009;74:369–378. doi: 10.1016/j.steroids.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Penning TM, Lee SH, Jin Y, Gutierrez A, Blair IA. Liquid chromatography-mass spectrometry (LC-MS) of steroid hormone metabolites and its applications. J Steroid Biochem Mol Biol. 2010;121:546–555. doi: 10.1016/j.jsbmb.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamashita K, Miyashiro Y, Maekubo H, Okuyama M, Honma S, Takahashi M, Numazawa M. Development of highly sensitive quantification method for testosterone and dihydrotestosterone in human serum and prostate tissue by liquid chromatography-electrospray ionization tandem mass spectrometry. Steroids. 2009;74:920–926. doi: 10.1016/j.steroids.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 6.Shackleton CH. Mass spectrometry in the diagnosis of steroid-related disorders and in hypertension research. J Steroid Biochem Mol Biol. 1993;45:127–140. doi: 10.1016/0960-0760(93)90132-G. [DOI] [PubMed] [Google Scholar]
- 7.Krone N, Hughes BA, Lavery GG, Stewart PM, Arlt W, Shackleton CH. Gas chromatography/mass spectrometry (GC/MS) remains a pre-eminent discovery tool in clinical steroid investigations even in the era of fast liquid chromatography tandem mass spectrometry (LC/MS/MS) J Steroid Biochem Mol Biol. 2010;121:496–504. doi: 10.1016/j.jsbmb.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Therrell BLJ, Berenbaum SA, Manter-Kapanke V, Simmank J, Korman K, Prentice L, Gonzalez J, Gunn S. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101:583–590. doi: 10.1542/peds.101.4.583. [DOI] [PubMed] [Google Scholar]
- 9.Caulfield MP, Lynn T, Gottschalk ME, Jones KL, Taylor NF, Malunowicz EM, Shackleton CH, Reitz RE, Fisher DA. The diagnosis of congenital adrenal hyperplasia in the newborn by gas chromatography/mass spectrometry analysis of random urine specimens. J Clin Endocrinol Metab. 2002;87:3682–3690. doi: 10.1210/jc.87.8.3682. [DOI] [PubMed] [Google Scholar]
- 10.Wudy SA, Hartmann M, Homoki J. Hormonal diagnosis of 21-hydroxylase deficiency in plasma and urine of neonates using benchtop gas chromatography–mass spectrometry. J Endocrinol. 2000;165:679–683. doi: 10.1677/joe.0.1650679. [DOI] [PubMed] [Google Scholar]
- 11.Homma K, Hasegawa T, Takeshita E, Watanabe K, Anzo M, Toyoura T, Jinno K, Ohashi T, Hamajima T, Takahashi Y, Takahashi T, Matsuo N. Elevated urine pregnanetriolone definitively establishes the diagnosis of classical 21-hydroxylase deficiency in term and preterm neonates. J Clin Endocrinol Metab. 2004;89:6087–6091. doi: 10.1210/jc.2004-0473. [DOI] [PubMed] [Google Scholar]
- 12.Tiu SC, Chan AO, Taylor NF, Lee CY, Loung PY, Choi CH, Shek CC. Use of urinary steroid profiling for diagnosing and monitoring adrenocortical tumours. Hong Kong Med J. 2009;15:463–470. [PubMed] [Google Scholar]
- 13.Khorram-Manesh A, Ahlman H, Jansson S, Wangberg B, Nilsson O, Jakobsson CE, Eliasson B, Lindstedt S, Tisell LE. Adrenocortical carcinoma: surgery and mitotane for treatment and steroid profiles for follow-up. World J Surg. 1998;22:605–611. doi: 10.1007/s002689900442. [DOI] [PubMed] [Google Scholar]
- 14.Arlt W, Biehl M, Taylor AE, Hahner S, Libe R, Hughes BA, Schneider P, Smith DJ, Stiekema H, Krone N, Porfiri E, Opocher G, Bertherat J, Mantero F, Allolio B, Terzolo M, Nightingale P, Shackleton CH, Bertagna X, Fassnacht M, Stewart PM. Urine steroid metabolomics as a biomarker tool for detecting malignancy in adrenal tumors. J Clin Endocrinol Metab. 2011;96:3775–3784. doi: 10.1210/jc.2011-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kushnir MM, Rockwood AL, Roberts WL, Yue B, Bergquist J, Meikle AW. Liquid chromatography tandem mass spectrometry for analysis of steroids in clinical laboratories. Clin Biochem. 2011;44:77–88. doi: 10.1016/j.clinbiochem.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Rauh M. Steroid measurement with LC-MS/MS. Application examples in pediatrics. J Steroid Biochem Mol Biol. 2010;121:520–527. doi: 10.1016/j.jsbmb.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Janzen N, Peter M, Sander S, Steuerwald U, Terhardt M, Holtkamp U, Sander J. Newborn screening for congenital adrenal hyperplasia: additional steroid profile using liquid chromatography-tandem mass spectrometry. J Clin Endocrinol Metab. 2007;92:2581–2589. doi: 10.1210/jc.2006-2890. [DOI] [PubMed] [Google Scholar]
- 18.Minutti CZ, Lacey JM, Magera MJ, Hahn SH, McCann M, Schulze A, Cheillan D, Dorche C, Chace DH, Lymp JF, Zimmerman D, Rinaldo P, Matern D. Steroid profiling by tandem mass spectrometry improves the positive predictive value of newborn screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2004;89:3687–3693. doi: 10.1210/jc.2003-032235. [DOI] [PubMed] [Google Scholar]
- 19.Guo T, Taylor RL, Singh RJ, Soldin SJ. Simultaneous determination of 12 steroids by isotope dilution liquid chromatography-photospray ionization tandem mass spectrometry. Clin Chim Acta. 2006;372:76–82. doi: 10.1016/j.cca.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 20.Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an endocrine society position statement. J Clin Endocrinol Metab. 2007;92:405–413. doi: 10.1210/jc.2006-1864. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell IC, Auchus RJ, Juneja K, Chang AY, Holt SA, Snyder WH, 3rd, Nwariaku FE. “Subclinical Cushing’s syndrome” is not subclinical: improvement after adrenalectomy in 9 patients. Surgery. 2007;142:900–905. doi: 10.1016/j.surg.2007.10.001. [DOI] [PubMed] [Google Scholar]