

Abstract
The ability to sense and adapt to hypoxic conditions plays a pivotal role in neuronal survival. Hypoxia induces the release of tissue-type plasminogen activator (tPA) from cerebral cortical neurons. We found that the release of neuronal tPA or treatment with recombinant tPA promotes cell survival in cerebral cortical neurons previously exposed to hypoxic conditions in vitro or experimental cerebral ischemia in vivo. Our studies using liquid chromatography and tandem mass spectrometry revealed that tPA activates the mammalian target of rapamycin (mTOR) pathway, which adapts cellular processes to the availability of energy and metabolic resources. We found that mTOR activation leads to accumulation of the hypoxia-inducible factor-1α (HIF-1α) and induction and recruitment to the cell membrane of the HIF-1α-regulated neuronal transporter of glucose GLUT3. Accordingly, in vivo positron emission tomography studies with 18-fluorodeoxyglucose in mice overexpressing tPA in neurons show that neuronal tPA induces the uptake of glucose in the ischemic brain and that this effect is associated with a decrease in the volume of the ischemic lesion and improved neurological outcome following the induction of ischemic stroke. Our data indicate that tPA activates a cell signaling pathway that allows neurons to sense and adapt to oxygen and glucose deprivation.
Introduction
Cerebral cortical neurons require a continuous supply of oxygen and glucose to meet their metabolic demands. Therefore, their survival depends on the activation of mechanisms to detect and adapt to low oxygen and glucose concentrations. The mammalian target of rapamycin (mTOR) is a protein kinase that integrates signals from different pathways to regulate cell survival and growth (Hay and Sonenberg, 2004). Hypoxia and cerebral ischemia cause a rapid inhibition of the mTOR pathway leading to growth arrest and neuronal death (Wouters and Koritzinsky, 2008). Moreover, mTOR activation in neurons has been linked to the development of plasticity (Jaworski and Sheng, 2006) and cell survival and recovery (Shi et al., 2011)
Hypoxia-inducible factor 1 (HIF1) is a transcription factor that plays a central role in hypoxia sensing and adaptation (Sharp and Bernaudin, 2004). HIF1 consists of two subunits, HIF1-α and HIF1-β. HIF1-β is constitutively expressed and does not respond to changes in oxygen tension, whereas HIF1-α is continuously made and rapidly degraded under normoxia (Sharp and Bernaudin, 2004). During hypoxia HIF1-α degradation is inhibited, allowing its rapid accumulation and binding to hypoxia-responsive elements and thereby activating the expression of hypoxia-responsive genes (Shi, 2009), many of which have a neuroprotective effect in the ischemic brain (Sheldon et al., 2009).
A family of membrane transporter proteins known as GLUTs regulates the passage of glucose across cell membranes. GLUT3 is the main transporter of glucose in neurons (Simpson et al., 2008). GLUT3 is found mostly in synaptic contacts in axons and dendrites and its expression is regulated by HIF1-α (Semenza, 2002) and increased by synaptic activity (Ferreira et al., 2011). The onset of cerebral ischemia is followed by a rapid decrease in the extracellular concentration of glucose in the brain accompanied by an induction in neuronal GLUT3 expression in an attempt to increase glucose transport and utilization. However, this rise in GLUT3 expression is transient and its subsequent decline is followed by neuronal death (Vannucci et al., 1996).
The serine proteinase tissue-type plasminogen activator, or tPA, is abundantly expressed in neurons and endothelial cells. Membrane depolarization by hypoxia or cerebral ischemia induces the rapid release of tPA from endothelial cells into the intravascular compartment and from neurons into the synaptic space (Yepes et al., 2000, 2003; Echeverry et al., 2010). In the intravascular space tPA has a beneficial thrombolytic effect associated with its ability to catalyze the conversion of plasminogen into plasmin. In contrast, the role of tPA released from neurons in response to a hypoxic/ischemic injury is still unclear.
The in vitro and in vivo studies presented here indicate that tPA activates a cell signaling pathway in the synaptic space via a plasminogen-independent mechanism that allows neurons to adapt to low oxygen and glucose concentrations. We found that the release of endogenous tPA or treatment with recombinant tPA (rtPA) activates the mTOR pathway in neurons, leading to HIF-1α accumulation and increase in neuronal uptake of glucose via induction and recruitment to the cell membrane of the HIF-1α-regulated neuronal transporter of glucose GLUT3.
Materials and Methods
Animals and reagents.
Murine strains were 8- to 12-week-old males, wild-type (Wt) C57BL/6J, tPA-deficient (tPA−/−), and plasminogen-deficient (Plg−/−) mice backcrossed at least seven generations into C57BL/6J mice and their Wt C57BL/6J controls. We also used mice with a tenfold increase in tPA expression in neurons (Madani et al., 1999) and their Wt littermate controls (T4 mice, kindly provided by Prof. J. D. Vassalli and Dr. R. Mandani, University of Geneva, Geneva, Switzerland). Experiments were approved by the Institutional Animal Care and Use Committee of Emory University, Atlanta, GA. Recombinant murine tPA, inactive tPA (itPA) with an alanine for serine substitution at the active site Ser481 (S481A), human Lys plasmin, and an ELISA kit that detects active tPA were purchased from Molecular Innovations. Other reagents were human recombinant tissue type plasminogen activator (Genentech), the phosphoinositide 3-kinase/Akt inhibitor wortmannin, methanol, methyl salicylate, and triphenyltetrazolium chloride (Sigma-Aldrich), the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (ATCC), the LDH release assay (Roche), the receptor-associated protein (RAP; kindly provided by Dr. Dudley K. Strickland, University of Maryland School of Medicine, Baltimore, MD), the NMDAR antagonist MK-801 (Tocris Bioscience), rapamycin and the TrkB inhibitor K-252a (Calbiochem), HIF-1α shRNA, scramble shRNA lentiviral particles, anti-GLUT3 antibodies, and TRITC-conjugated donkey anti-goat IgG (Santa Cruz Biotechnology), anti-HIF-1α antibodies (Abcam), antibodies against the p70S6 kinase (p70S6K) phosphorylated at Thr389 (Cell Signaling Technology), heparin sodium (Abraxis Pharmaceutical Products), blue latex (Connecticut Valley Biological Supply), ApopTag Plus Fluorescein in Situ Apoptosis Detection Kit (Millipore Bioscience Research Reagents), 4′-6-diamidino-2-phenylindole (DAPI; Invitrogen), triphenyltetrazolium chloride (TTC; Sigma-Aldrich), 2-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG; Invitrogen), and 18-fluorodeoxyglucose (PETNET Solutions).
Animal model of cerebral ischemia.
The dorsal distribution of the middle cerebral artery, the intracerebral circulation, and the presence of communications between the anterior (carotid) and posterior (basilar) circulation were studied in T4 mice and their Wt littermate controls (n = 4) intravenously injected with blue latex and incubated in methyl salicylate as described previously (Murakami et al., 1998). To induce cerebral ischemia, mice were anesthetized with 4% chloral hydrate (400 mg/kg, i.p.) and the middle cerebral artery (MCA) was exposed and occluded for 60 min with a 6-0 silk suture as described previously (Belayev et al., 1999). Cerebral perfusion (CP) was monitored throughout the surgical procedure with a laser Doppler (Perimed), and only animals with a >70% decrease in CP after occlusion and complete recovery after suture removal were included in this study. Heart rate and systolic, diastolic, and mean arterial blood pressure were controlled with an IITC 229 System (IITC Life Science). One hour after reperfusion, mice were treated with human recombinant tPA at 1.0, 4.5, or 9.0 mg/kg, i.v. Arginin was dialyzed from rtPA with SnakeSkin Dialysis Tubing (ThermoScientific) before its administration. Before transient middle cerebral artery occlusion (tMCAO), a subgroup of mice was treated with rapamycin (50 μg, i.p.) or 2 μl of wortmannin at (0.1 mm) injected at the following coordinates (Paxinos and Franklin, 2001): bregma, −1 mm; mediolateral, 2 mm; dorsoventral, 2 mm. Twenty-four hours after tMCAO, the volume of the ischemic lesion was measured in TTC-stained sections as described previously (Swanson et al., 1990). Each observation was repeated 16 times.
Accelerating rotarod and forelimb grip strength test.
Animals were trained on days −3, −2, and −1 on a rotarod apparatus (LE 8500, Harvard Apparatus) for 30 s with no rotation and thereafter for 2 min with a constant low speed rotation (4 rpm). Before surgery and 6, 12, and 24 h after tMCAO mice were tested on accelerating conditions from 4 to 40 rpm over 10 min. Each observation was repeated 3 times with an interval of 5 min between tests. The time each animal was able to walk on the rod before falling was collected before and at each time point after tMCAO. The forelimb grip strength was measured using a grip strength meter (Bioseb) before surgery and 6, 12, and 24 h after tMCAO. Mice were allowed to grasp a smooth metal triangular pull bar with their right forelimb and then pull backward in the horizontal plane. The force applied to the bar at the moment when the grasp was released was recorded as the peak tension (in newtons). Each test was repeated 5 times and the mean of all trials was recorded as the mean grip strength for that animal. For both tests values are given as a percentage compared to results obtained in the baseline evaluation.
Neuronal cultures, exposure to oxygen-glucose deprivation conditions, and determination of cell survival.
Cerebral cortical neurons were cultured from embryonic day 19 Wt and tPA−/− mice as described previously (Echeverry et al., 2010). A subgroup of Wt neurons was infected with lentiviral particles with either scramble or HIF-1α shRNA. The efficiency of HIF-1α downregulation was confirmed by reverse transcriptase (RT)-PCR analysis. To study the effect of hypoxia on neuronal survival/death, neurons were exposed in an anaerobic chamber (Don Whitley Scientific) to oxygen-glucose deprivation (OGD) conditions (<0.1% oxygen) for 55 min followed 24 h later by determination of cell survival and death with MTT and LDH release assays, in accordance with manufacturer's instructions. For the MTT assay, results are expressed as a percentage of cell survival in neurons exposed to OGD conditions compared to cells maintained under normoxic conditions. For the LDH release assay, results are given as percentage of LDH released into the media compared to LDH released from neurons incubated 24 h with Triton X-100 (1%). Wt neurons were exposed to OGD conditions for 55 min, and either kept in the incubator for 24 h or treated 5, 30, 60, 120, 180 or 360 min later for the remainder of the experiment with 5 nm of either active tPA or itPA or 10 nm plasmin. A subgroup of cells was treated with a combination of 5 nm tPA and either 10 μm MK-801, 60 nm RAP, 10 nm rapamycin, 20 nm wortmannin, or 100 nm TrkB inhibitor K-252a. A subset of neurons infected with scramble or HIF-1α shRNA lentiviral particles was exposed to OGD conditions for 55 min, returned to the incubator, and treated 60 min later with 5 nm tPA. To study the protective effect of hypoxic postconditioning, cell survival was determined with the MTT assay in Wt and tPA−/− neurons exposed to 55 min of OGD conditions, returned to the incubator, and then exposed 10 min later to a second episode of hypoxia [10 min OGD, postconditioning event (PCE)]. A subgroup of neurons was incubated with 5 nm tPA, 10 μm MK-801, 60 nm RAP, 20 nm wortmannin, or 10 nm rapamycin during the PCE. Each experiment was performed in cultures from three different animals, and each observation was repeated 10–20 times. Cell survival/death was determined in each experiment 24 h later with the MTT and LDH release assays.
TUNEL, HIF-1α, and GLUT3 staining.
The brains of T4 mice and their Wt littermate controls were harvested 24 h after tMCAO, cut onto 20 μm sections, and stained for TUNEL as described previously (Wu et al., 2010). Observations were made in three previously described areas of interest (AOIs) (Polavarapu et al., 2007) involving the frontoparietal (AOI-1) and parieto-temporal (AOI-2) zones, as well as the necrotic core (AOI-3). Results are given as percentage of TUNEL-positive cells in relation to the total number of cells per field. Each observation was repeated 16 times. To study the expression of HIF-1á in the ischemic brain, we performed immunohistochemical analysis with an antibody against HIF-1á in 20 μm brain sections of T4 mice and their Wt littermate controls (n = 4) 1 h after tMCAO. Observations were made in the three AOI described above. To study the expression of GLUT3, Wt cerebral cortical neurons were incubated with 5 nm tPA for 0–3 h, fixed, and stained with a goat anti-GLUT3 primary antibody (1:200) and a TRITC-conjugated donkey anti-goat IgG (1:500) for 1 h at room temperature. Cells were then stained with DAPI and mounted onto a slide for microscopic view. Each observation was repeated six times.
tPA activity assay.
Wt neurons were exposed to 55 min of OGD conditions, returned to the incubator, and then exposed 10 min later to a postconditioning event (PCE, 10 min OGD). tPA activity was determined in the culture media after 1, 5, and 10 min of the onset of the PCE with an ELISA kit following the manufacturer's instructions (Molecular Innovations). As controls, we measured tPA activity at identical time points in sister cultures exposed to OGD conditions without an ensuing PCE. Each experiment was performed with cultures from three different animals, and each observation was repeated eight times.
Proteomics and Ingenuity pathways analysis.
Wt cerebral cortical neurons were incubated 1 h with 5 nm tPA or vehicle (control). At the end of treatment, neurons were lysed and 50 μg of protein was reduced, resolved on a 10% polyacrylamide SDS gel, and each sample was digested in-gel with trypsin. Extracted peptides were loaded onto a C18 column, eluted, and sequenced using tandem mass spectrometry (MS/MS) on an LTQ-Orbitrap mass spectrometer (Thermo Finnigan) using data-dependent acquisition. All MS/MS files were searched against mouse database downloaded from the National Center for Biotechnology Information (September, 2009) using the SEQUEST Sorcerer algorithm (version 3.11, SAGE-N). Searching parameters included mass tolerance of precursor ions (±20 ppm) and product ion (± 0.5 m/z), semi-tryptic restriction with a dynamic mass shift for oxidized Met (+15.9949), four maximal modification sites, and a maximum of two missed cleavages. To evaluate the false discovery rate (FDR), all original protein sequences were reversed to generate a decoy database that was concatenated to the original database (70,368 total entries). To remove false positive matches, assigned peptides were grouped by a combination of trypticity (fully and partial) and precursor ion-charge state. Each group was first filtered by mass accuracy (10 ppm), and by dynamically increasing correlation coefficient (Xcorr) and ΔCn values to reduce protein FDR to <1%. If peptides were shared by multiple members of a protein family, the matched members were clustered into a single group in which each protein identified by a unique peptide represented a subgroup. Quantitative pairwise comparison of control and tPA-treated samples was performed using the software DQUAN. The log2 ratio averaged over all peptides for a particular protein was used to determine the protein expression ratio and a standard deviation. For Ingenuity Pathway Analysis, log2 (1 h tPA-treated/control) average protein intensity ratios calculated by DQUAN were centered so that the fit gauss curve midpoint (mean) fell at zero. Log2 values 1.63 SDs from the mean (changed with 95% confidence, with absolute value > 0.709) were considered as changing, and these protein identities and quantifications were considered in the analysis. Resulting ingenuity canonical pathways significantly represented in the list of changing proteins were determined by Ingenuity as a p value based upon the fraction of total proteins known to be linked to each pathway.
Western blot analysis.
Wt cerebral cortical neurons were incubated 0–180 min with either active or inactive tPA (5 nm), alone or in combination with 20 nm wortmannin or 60 nm RAP. Wt mice underwent tMCAO and treatment with rtPA 4.5 mg/kg/i.v. or a comparable volume of saline solution as described above. A subgroup of mice was treated before tMCAO with 2 μl of a 0.1 nm solution of wortmannin injected into the third ventricle as the same coordinates described above. Wt and tPA−/− cerebral cortical neurons were exposed to 10 min of OGD (postconditioning event) 10 min after exposure to 55 min of OGD conditions. As controls, cells were maintained under normoxic conditions or exposed to 55 min of OGD conditions without subsequent exposure to a postconditioning event (10 min OGD). In each case, extracts from cells and brain lysates were immunoblotted with antibodies against the p70S6 kinase (p70S6K) phosphorylated at Thr389 (1:1000 dilution). Each observation was repeated 4–6 times. To study the expression of HIF-1α, Wt cerebral cortical neurons were incubated with 5 nm tPA for 0–180 min. A subset of samples was cotreated with 10 nm rapamycin. Wt mice underwent tMCAO and treatment 1 h after reperfusion with rtPA 4.5 mg/kg/i.v. or a comparable volume of saline solution. In each case, extracts from cells and brain lysates were immunoblotted with antibodies against HIF-1α (1:1000 dilution). To study GLUT3 expression, Wt neurons were incubated with 5 nm tPA, followed 1, 3 or 6 h later by membrane extraction with a Mem-PER Mammalian Membrane Protein Extraction Kit (Pierce Chemical). The brains of Wt and T4 mice were harvested 0–3 h after tMCAO. Lysates from membrane extracts and brain tissue were immunoblotted with antibodies against GLUT3 (1:500 dilution). Each experiment was repeated five times.
Quantification of glucose uptake.
To quantify the uptake of glucose in the ischemic brain, Wt and T4 mice (n = 4) underwent tMCAO. Thirty minutes after the onset of reperfusion, animals were intravenously treated with 18.5 MBq 18FDG, followed 45 min later by 18FDG PET imaging in a Siemens Inveon micro PET/CT scanner (Siemens). Images with a spatial resolution of 1.8 mm FWHM were acquired during 15 min and reconstructed in with an ordered subset expectation-iterative (OSEM) algorithm. To quantify differences in glucose uptake, the differential uptake ratio images were viewed in coronal sections and regions of interest (ROIs) were placed over the ischemic and nonischemic hemispheres. The uptake of 18FDG in each ROI was recorded as nCi/cc and the differential uptake ratio (DUR) was calculated three separate times in 6 slices per mouse using the following formula:
[(nCi/cc uptake in ischemic hemisphere) − (nCi/cc uptake in nonischemic hemisphere)]/(nCi/cc uptake in ischemic hemisphere).
To quantify the uptake of glucose in vitro, cerebral cortical neurons were cultured in bottom clear black 96-well plates and incubated with tPA (5 nm) and the fluorescent deoxyglucose analog 2-NBDG or with vehicle (control) for 0–80 min. At the end of each time point, cells were washed with PBS and the fluorescence in each well was determined at excitation and emission wavelengths of 485/20 and 528/20 nm, respectively. Values are given as a mean fold increase in glucose uptake in neurons incubated with tPA in relation to neurons incubated with vehicle (control) for an identical period of time. Each experiment was repeated in cultures from three different mice, and each experimental group included 10–12 observations.
Statistical analysis.
Values are expressed as percentage or mean ± SD when appropriate. Statistical tests included the t test followed by the Wilcoxon signed-ranked test and two-way ANOVA for comparisons between groups. p values of <0.05 were considered significant.
Results
Characterization of the cerebrovascular anatomy of mice overexpressing neuronal tPA
First we studied the cerebrovascular anatomy of Wt and T4 mice as described in Materials and Methods. We found no difference in the dorsal and intracerebral distribution of the major blood vessels or in the presence of anastomosis between the carotid and vertebrobasilar systems in both strains of mice (Fig. 1A).
Figure 1.
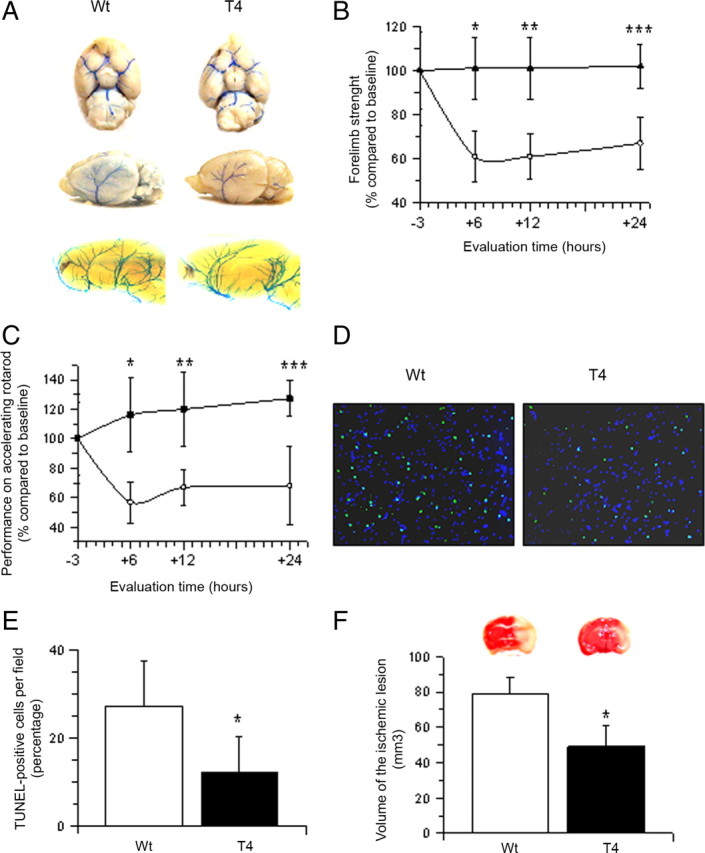
Neuronal tPA has a protective effect in the ischemic brain. A, Circle of Willis (top), dorsal middle cerebral artery (middle), and intracerebral vasculature (bottom) in Wt and T4 mice. B, C, Mean change (percentage) compared to baseline evaluation in right forelimb strength (B) and performance in accelerating rotarod test (C) in Wt (white circles) and T4 mice (black triangles) 6, 12, and 24 h after tMCAO. n = 16; B, *p < 0.05 compared to Wt mice 6 h after tMCAO; **p < 0.05 compared to Wt mice 12 h after tMCAO; ***p < 0.05 compared to Wt mice 24 h after tMCAO. C, *p < 0.05 compared to Wt mice 6 h after tMCAO; **p < 0.05 compared to Wt mice 12 h after tMCAO; ***p < 0.05 compared to Wt mice 24 h after tMCAO. Lines denote SD. D, E. Micrographs of TUNEL staining in area of interest-2 (AOI-2) (D) and mean percentage of TUNEL-positive cells per field in the three AOIs (E) in Wt and T4 mice 24 h after tMCAO. D, Blue, DAPI; green, TUNEL; n = 16. E, *p < 0.05 compared to Wt controls. F, Mean volume of the ischemic lesion and representative TTC-stained sections in T4 and Wt mice 24 h after tMCAO; n = 16. Lines denote SD. *p < 0.05 compared to Wt mice.
Neuronal tPA has a protective effect in the ischemic brain
Because our previous studies indicate that hypoxia and cerebral ischemia induce the release of tPA from cerebral cortical neurons (Echeverry et al., 2010), we decided to investigate whether this release of tPA has an effect on the ischemic brain. Wt and T4 mice (n = 16) underwent tMCAO followed 6–24 h later by assessment of their performance in the accelerating rotarod test and quantification of the grip strength in the right forelimb (affected by the ischemic lesion). At the end of the last evaluation (24 h after tMCAO), we measured the volume of the ischemic lesion and the percentage of apoptotic cells/field in each area of interest defined in the Materials and Methods section. We found that compared to their baseline evaluation, the strength in the right forelimb 6, 12, and 24 h after tMCAO decreased in Wt mice to 61 ± 11.38%, 61 ± 10%, and 67 ± 12%, respectively, and remained unchanged in T4 animals (p < 0.05; Fig. 1B). Likewise, the performance in the accelerating rotarod at 6, 12, and 24 h after tMCAO decreased in Wt mice to 55.75 ± 14%, 66.89 ± 12%, and 67.93 ± 26% of their baseline, respectively, and remained unchanged in T4 mice (Fig. 1C, p < 0.05). Additionally, we found that compared to Wt littermate controls, T4 mice have a 54.91% decrease in the percentage of apoptotic cells/field in the ischemic tissue (27.3 ± 10.31% in Wt and 12.3 ± 8.1% in T4 mice; p < 0.05; Fig. 1D,E) and 38% decrease in the volume of the ischemic lesion (78.90 ± 9.51 mm3 in Wt mice and 48.92 ± 11.9 mm3 in T4 mice, p < 0.05; Fig. 1F).
Treatment with tPA after exposure to lethal hypoxic conditions or induction of cerebral ischemia promotes neuronal survival
Because our data suggest that tPA has a protective effect in the ischemic brain, we decided to use the experimental design depicted in Figure 2A to study cell survival in Wt cerebral cortical neurons treated with 5 nm tPA 5–360 min after exposure to 55 min of OGD conditions. We found that neuronal survival decreases from 100 ± 1.1% in neurons maintained under normoxic conditions to 49.87 ± 3% in cells exposed to OGD conditions without subsequent treatment with tPA. Surprisingly, treatment with tPA 5, 30, 60, 120, 180, or 360 min after exposure to OGD conditions increased cell survival to 70.8 ± 2.2%, 85.3 ± 3%, 82.4 ± 1.8%, 71.6 ± 1.22%, 66.4 ± 2.83%, and 60.02 ± 2.96%, respectively (Fig. 2B, n = 20, p < 0.05). To investigate whether this cell survival-promoting effect of tPA requires its proteolytic activity, we performed similar observations in neurons treated with 5 nm proteolytically inactive tPA or 10 nm plasmin. We found that itPA but not plasmin promotes cell survival in neurons previously exposed to lethal hypoxia (Fig. 2C). To confirm our observations with a different assay, we used the same experimental design to quantify the release of LDH from Wt cerebral cortical neurons treated with proteolytically active or itPA 30–360 min after exposure to lethal hypoxia. We found that treatment with either active tPA or itPA decreases the release of LDH into the media from neurons previously exposed to OGD conditions and, as observed with the MTT assay, this protective effect is maximal when cells are treated 30–60 min after the hypoxic injury (Fig. 2D, n = 20; p < 0.05).
Figure 2.

Treatment with tPA promotes cell survival in cerebral cortical neurons exposed to hypoxic conditions and in the ischemic brain following tMCAO. A, Experimental design used to study the effect of tPA on neuronal survival. Letters denote time of treatment with tPA after exposure to oxygen–glucose deprivation conditions. B–D, Mean cell survival (B, C) and release of LDH into the culture media (D) in Wt cerebral cortical neurons treated with 5 nm proteolytically active (B, D) or inactive tPA (C, D) or 10 nm plasmin (C) 5, 30, 60, 120, 180, or 360 min after exposure to 55 min of OGD conditions; n = 20 in B and 15 in C and D. B, *p < 0.05 compared to neurons exposed to OGD conditions without subsequent treatment with tPA. C, *p < 0.05 compared to neurons left untreated after exposure to OGD conditions; Ns, Nonsignificant. D, *p < 0.05 compared to cells exposed to OGD conditions without subsequent treatment. Lines denote SD. E, Mean cell survival in Wt cerebral cortical neurons exposed to 55 min of OGD conditions and treated 1 h later with 5 nm tPA alone or in combination with 10 μm MK-801, 60 nm RAP, or 100 nm TrkB inhibitor K-252a; n = 10. *p < 0.05 compared to neurons treated with tPA alone or with a combination of tPA and K-252a. Lines denote SD. F, Mean volume of the ischemic lesion in Wt and Plg−/− mice treated 1 h after tMCAO with saline solution (white bars) or rtPA 1 – 9 mg/Kg/IV (gray bars); n = 10 per group. *p < 0.05 compared to Wt mice treated with saline solution; **p < 0.05 compared to mice treated with 1 mg/kg/i.v. of rtPA. ***p < 0.05 compared to Plg−/− mice treated with saline solution. Bars depict mean volume of the ischemic lesion in mm3. Lines denote SD.
Because in the CNS tPA interacts with the NMDAR (Nicole et al., 2001) and the low density lipoprotein receptor-related protein 1 (LRP1; Yepes et al., 2003), we quantified cell survival in Wt neurons exposed to OGD conditions and treated 1 h later with 5 nm tPA alone or in combination with either the NMDAR antagonist MK-801 (10 μm) or the receptor-associated protein RAP (60 nm; an inhibitor of the binding of members of the LDL receptor family to its ligands). We found that the protective effect of treatment with tPA is abrogated by antagonism of either the NMDAR or LRP1. Because experimental evidence indicate that brain-derived neurotrophic factor (BDNF) promotes neuronal survival (Numakawa et al., 2010) and that tPA cleaves BDNF into its active form (Pang et al., 2004), we performed similar experiments in neurons treated with 5 nm tPA alone or in combination with 100 nm K-252a [K-252a is an antagonist of the tyrosine kinase receptor B (TrkB), the cognate receptor for BDNF]. We found that blockade of the BDNF receptor does not abrogate the protective effect of tPA (Fig. 2E; n = 10; p < 0.05). To study whether treatment with rtPA also has a neuroprotective effect in vivo, we measured the volume of the ischemic lesion in Wt mice treated 1 h after tMCAO with either 1.0 or 4.5 or 9 mg/kg rtPA or with a comparable volume of saline solution. We found that compared to animals treated with saline solution, the volume of the ischemic lesion decreases by 13.92 ± 4.4%, 30.37 ± 3.92%, and 31.36 ± 4.6% in mice treated with 1.0, 4.5, or 9.0 mg/kg rtPA, respectively (n = 12; p < 0.05 compared to mice treated with saline solution). To investigate whether this protective effect of rtPA in vivo is mediated by the ability of tPA to cleave plasminogen, we performed similar observations in Plg−/− mice treated 1 h after tMCAO with either rtPA 4.5 mg/kg/i.v. or saline solution. Our data indicate that treatment with rtPA induces a 22.36 ± 6% decrease in the volume of the ischemic lesion in Plg−/− mice (Fig. 2F; n = 12, p < 0.05). Together, these results indicate that treatment with tPA promotes neuronal survival following exposure to lethal hypoxia or ischemia via a mechanism that does not require the conversion of plasminogen into plasmin but needs the engagement of the NMDAR and a member of the LDL receptor gene family.
The release of neuronal tPA promotes cell survival following exposure to hypoxic conditions
Hypoxic postconditioning (HP) is a phenomenon whereby exposure to an episode of hypoxia that does not induce cell death [hereinafter referred to as postconditioning event (PCE)] confers neuroprotection to cells previously exposed to an otherwise lethal hypoxic injury (Zhao, 2009). Because the beneficial effect of HP requires the activation of endogenous neuroprotective pathways, we used an in vitro model of HP previously characterized in our laboratory to study whether the release of endogenous tPA also promotes cell survival in neurons previously exposed to lethal hypoxic conditions. In this model, depicted at the top of Figure 3A, exposure to a postconditioning event (10 min of OGD, PCE in Fig. 3A) promotes neuronal survival when applied 10 min after exposure to an otherwise lethal hypoxic injury (55 min of OGD). First, we investigated whether the PCE has an effect on the release of neuronal tPA. Wt neurons were exposed to 55 min of OGD conditions and then returned to the incubator and either maintained under normoxic conditions for the remaining of the experiment or exposed 10 min later to a PCE. The concentration of tPA in the medium was quantified with an ELISA at 1, 5, and 10 min after the onset of the PCE, or at similar time points in non-postconditioned neurons. We found that the release of tPA from non-postconditioned neurons is negligible. In contrast, the concentration of tPA in the culture medium increased to 10.9 ± 2.6 pg/ml, 21.49 ± 2.16 pg/ml, and 51 ± 7. 2 pg/ml 1, 5, and 10 min after the onset of the PCE, respectively (Fig. 3A). We then used this model to investigate whether the release of endogenous tPA during the PCE mediates the neuroprotective effect of HP. Wt and tPA−/− neurons were incubated 55 min under OGD conditions and either returned to the incubator for the remaining of the experiment or exposed 10 min later to a PCE. A subgroup of neurons was incubated during the PCE with 10 μm MK-801, or 60 nm RAP (Wt neurons) or 5 nm tPA (tPA−/− neurons). Cell survival was determined 24 h later with the MTT assay. We found that neuronal survival decreased from 100 ± 1.8% in control cells to 48 ± 3% in cells exposed to OGD conditions without a subsequent PCE. In contrast, cell survival increased to 72.78 ± 7.8% in Wt neurons exposed to a PCE 10 min after exposure to OGD conditions. However, the protective effect of the PCE was abrogated by genetic deficiency of tPA or by treatment of Wt neurons with RAP or MK-801. Importantly, incubation with 5 nm tPA rescued the protective effect of the PCE in tPA−/− neurons (Fig. 3B, n = 15). Our data indicate that the release of neuronal tPA promotes survival in cells previously exposed to lethal hypoxia and that as observed following treatment with rtPA, this effect also requires the engagement of the NMDAR and a member of the LDL receptor gene family.
Figure 3.

Endogenous tPA promotes cell survival in cerebral cortical neurons exposed to hypoxic conditions. A, Top, Model of hypoxic postconditioning. Lethal hypoxia indicates the time interval during which neurons are exposed to 55 min of OGD conditions. R1, Recovery; indicates the moment when neurons are returned to the incubator for 10 min. PCE, Postconditioning event; denotes the time interval during which neurons are exposed to a second episode of OGD conditions (10 min). R2, Recovery; indicates the moment when cells are returned to the incubator before quantification of cell survival. Bottom, Mean concentration of active tPA in the culture media of Wt cerebral cortical neurons during the PCE; n = 8 per time point. *p < 0.05 and **p < 0.05 compared to tPA concentration in the culture media of sister cultures not exposed to a PCE after lethal OGD. Lines denote SD. B, Wt and tPA−/− cerebral cortical neurons were exposed to 55 min of OGD conditions and exposed 10 min later to a PCE in the presence of either 10 mm MK-801, 60 nm RAP, 5 nm tPA, or vehicle (control) followed by determination of cell survival 24 h later. Lines denote SD; n = 15 per experimental group. *p < 0.05 compared to non-postconditioned Wt neurons or with postconditioned in the presence of RAP or MK-801. **p < 0.05 compared to tPA−/− neurons postconditioned in absence of recombinant tPA.
TPA activates the mTOR pathway in neurons
Based on our data, we postulated that tPA activates a neuroprotective cell signaling pathway in cerebral cortical neurons. To test this hypothesis, we used liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) and quantitative analysis to identify protein changes in Wt cerebral cortical neurons incubated for 60 min with 5 nm tPA or vehicle (control) as described in the Materials and Methods section. We found that compared to vehicle (control)-treated neurons, treatment with tPA caused a >50% decrease or increase in abundance in 383 and 206 proteins, respectively (Fig. 4A). Further studies with the Ingenuity Pathway Analysis tool showed that a significant number of these proteins belong to the mammalian target of rapamycin pathway (44 of201 proteins in this pathway were downregulated and 9 proteins were upregulated by treatment with tPA; p < 0.05). Interestingly, we observed that the greatest effect of tPA on this pathway was on the abundance of the transmembrane protein 127 (TMEM127; −4.8 log2).
Figure 4.

tPA induces activation of the mTOR pathway in cerebral cortical neurons. A, Histogram analysis of the population distribution of quantified proteins in cerebral cortical neurons treated with 5 nm tPA or vehicle (control). Thin traces represent theoretical Gaussian distributions (Gauss1) that fit the raw data representing 92% of proteins within the population. The Gaussian 2 (Gauss2) traces represent a unique subset of proteins that decrease in the tPA-treated sample compared to control (shaded area). The sum of the two Gaussian distributions is represented by the thick traces (2 Gauss Fit). B, C, Representative Western blot analysis with an antibody that detects p70S6K phosphorylated at Thr389 [(p) p70S6K] in cell extracts from Wt cerebral cortical neurons left untreated (C, Control) or incubated 5–180 min with 5 nm of either active tPA (atPA) or proteolytically inactive itPA (B) or during 1 h with a combination of 5 nm tPA and either 20 nm wortmannin (Wort) or 60 nm RAP (C). D, Representative Western blot for (p) p70S6K expression in cell extracts from Wt and tPA−/− cerebral cortical neurons exposed to 55 min of OGD conditions and either returned to the incubator for the remaining of the experiment or exposed 10 min later to hypoxic postconditioning (HP, 10 min OGD). E, Representative Western blot analysis for (p) p70S6K expression in brain extracts from Wt mice 1 h after tMCAO and the intravenous administration of rtPA alone or in combination with wortmannin.
Because TMEM127 is a recently described negative regulator of the mTOR pathway (Qin et al., 2010), we decided to investigate the effect of tPA on mTOR pathway activation. Wt cerebral cortical neurons were incubated with 5 nm either active or itPA followed 5–180 min later by a Western blot analysis with antibodies against the p70S6 kinase (p70S6K) phosphorylated at Thr389 (p70S6K phosphorylation is an indicator of mTOR activation). Because Akt mediates the activation of the mTOR pathway and since our earlier studies indicate that tPA induces Akt phosphorylation via LRP1 (An et al., 2008), we performed similar analyses in Wt neurons treated with a combination of tPA and either 20 nm wortmannin or 60 nm RAP. We found that incubation with either active or itPA induces mTOR activation in cerebral cortical neurons (Fig. 4B) and that this effect is mediated by activation of the PI3K/Akt pathway via a member of the LDL receptor family (Fig. 4C). Then we used the model of HP described above to study the effect of endogenous tPA on mTOR activation. We found that the application of a PCE induces mTOR activation in Wt but not in tPA−/− neurons (Fig. 4D). To investigate the effect of treatment with rtPA on mTOR activation in vivo, we studied the expression of (p) p70S6K in the ischemic tissue of Wt mice treated 1 h after tMCAO with rtPA alone or in combination with wortmannin. We found that treatment with rtPA induces mTOR activation in the ischemic brain and that this effect is abrogated by cotreatment with wortmannin (Fig. 4E). Together, our in vitro and in vivo studies indicate that treatment with rtPA or release of endogenous tPA induces mTOR activation in neurons.
mTOR activation mediates the neuroprotective effect of tPA
Because activation of the mTOR signaling pathway plays a pivotal role in the detection, adaptation and survival to hypoxic conditions (Wouters and Koritzinsky, 2008) we used the experimental design described in Figure 5A to investigate whether mTOR mediates the prosurvival effect of tPA in neurons previously exposed to lethal hypoxia. Wt cerebral cortical neurons were exposed to 55 min of OGD conditions followed 30–360 min later by treatment with 5 nm tPA, alone or in combination with 10 nm rapamycin, a selective mTOR inhibitor, and determination of cell survival 24 h later. Our results indicate that the cell survival promoting effect of treatment with tPA is abrogated by mTOR inhibition (Fig. 5A, n = 15, p < 0.05).
Figure 5.

mTOR activation mediates the neuroprotective effect of tPA. A, The top section describes the experimental design used to investigate whether mTOR pathway activation mediates the neuroprotective effect of tPA. The bottom section indicates mean cell survival in Wt cerebral cortical neurons incubated with 5 nm tPA alone or in combination with 10 nm rapamycin 30 (A), 60 (B), 180 (C), or 360 (D) minutes after exposure to 55 min of OGD conditions; n = 15 per experimental condition. *p < 0.05 compared to cultures treated with a combination of tPA and rapamycin at similar time points after OGD, respectively. Lines denote SD. B, Mean cell survival in Wt cerebral cortical neurons subjected to HP alone or in the presence of 10 nm rapamycin 10 min after exposure to 55 min of OGD conditions; n = 12. *p < 0.05 compared to cells preconditioned in absence of rapamycin. Lines denote SD. C, Wt mice underwent tMCAO followed 1 h later by the intravenous administration of either saline solution (black bar), rtPA (white bar), or a combination of rtPA and rapamycin (RAPA; gray bar). The volume of the ischemic lesion was determined 24 h later as described in Materials and Methods; n = 12. *p < 0.05 compared to mice not treated with rtPA after tMCAO; **p = 0.03 compared to mice treated with rtPA alone.
To investigate whether the protective effect of endogenous tPA (Fig. 3B) is also mediated by mTOR, we studied cell survival in Wt neurons exposed to 55 min of OGD conditions followed 10 min later by a PCE applied in the presence of rapamycin 10 nm or vehicle (control). Our results indicate that exposure to OGD conditions decreases neuronal survival from 100 ± 2.3% to 52 ± 3.8%. In contrast, when neurons were exposed to a PCE, cell survival increased to 76 ± 2.8%. However, this protective effect was abrogated by mTOR inhibition with rapamycin (54 ± 4.6% neuronal survival; Fig. 5B, n = 12, p < 0.05). To determine whether the beneficial effect of treatment with rtPA in vivo is also mediated by mTOR activation, we measured the volume of the ischemic lesion in Wt mice treated 1 h after tMCAO with rtPA, alone or in combination with rapamycin 50 μg/i.p. Our results indicate that treatment with rtPA induces a 34 ± 8% decrease in the volume of the ischemic lesion (n = 10, p < 0.05) and that this effect is abrogated by cotreatment with rapamycin (Fig. 5C, n = 12, p < 0.05).
TPA induces mTOR-regulated HIF-1α activation in cerebral cortical neurons
Because HIF-1α accumulation plays a central role in the adaptation of neurons to hypoxic and ischemic conditions (Semenza, 2002), we decided to investigate whether treatment with tPA has an effect on neuronal HIF-1α. Wt cerebral cortical neurons were incubated with 5 nm tPA, followed 0–180 min later by detection of HIF-1α expression with Western blot analysis. We found that treatment with tPA induces a progressive increase in HIF-1α accumulation in neurons (Fig. 6A). To investigate the association between tPA-induced mTOR activation and tPA-induced HIF-1α accumulation in neurons, we performed similar observations in Wt neurons following 1 h of incubation with tPA alone or in combination with rapamycin. Our results indicate that the effect of tPA on neuronal HIF-1α is mediated by the mTOR pathway (Fig. 6B). Then we studied the effect of genetic overexpression of neuronal tPA on cerebral ischemia-induced HIF-1α accumulation in vivo. Our immunohistochemical studies indicate that HIF-1α expression is virtually undetectable in the ischemic brain of Wt mice following reperfusion. In contrast, we detected in T4 mice a significant increase in HIF-1α accumulation in the areas surrounding the necrotic core in the parietal and frontal lobes (areas of interest 1 and 2) and in the center of the ischemic lesion (area of interest 3; Fig. 6C). To investigate the effect of treatment with rtPA on HIF-1α accumulation in the ischemic tissue, we performed a Western blot analysis for HIF-1α expression in brain extracts from Wt mice treated with either rtPA or saline solution 1 h after tMCAO. We found that treatment with rtPA but not saline solution induces HIF-1α accumulation in the ischemic tissue (Fig. 6D).
Figure 6.

TPA promotes neuronal survival via mTOR-regulated HIF-1a activation. A. Representative Western blot analysis for HIF-1a expression in extracts from Wt cerebral cortical neurons incubated 5–180 min with 5 nm tPA. B, Representative Western blot analysis for HIF-1a expression in extracts from Wt cerebral cortical neurons incubated 60 min with 5 nm tPA alone or in combination with 10 nm rapamycin. C, Representative micrographs of HIF-1α expression in the frontoparietal area of the ischemic tissue of T4 mice and their Wt littermate controls 1 h after tMCAO and complete reperfusion. Magnification, 40×. Blue, DAPI; red, HIF-1α. D, Representative Western blot analysis for HIF-1α expression in brain extracts from the ischemic tissue of Wt mice treated with wither rtPA or a comparable volume of saline solution 1 h after tMCAO and complete reperfusion. E, Mean cell survival in Wt cerebral cortical neurons left uninfected or infected with lentiviral particles containing either HIF-1a shRNA or scramble shRNA and treated with 5 nm tPA 1 h after exposure to 55 min of OGD conditions; n = 8 per condition. Lines denote SD. *p < 0.05 compared to cell survival in Wt neurons either uninfected (white bar) or treated with scramble shRNA (gray bar).
HIF-1α mediates the neuroprotective effect of tPA
To investigate whether HIF-1α mediates the neuroprotective effect of tPA, Wt neurons left untreated or infected with lentiviral particles containing either scramble or HIF-1α shRNA were exposed to 55 min of OGD conditions and treated 1 h later with 5 nm tPA. Cell survival was quantified 24 h later with the MTT assay. HIF-1α downregulation was confirmed by RT-PCR analysis (data not shown). We found that cell survival decreased from 100 ± 0.9% in controls neurons to 50 ± 1.2% in cells exposed to OGD conditions without subsequent treatment. Treatment with tPA 1 h after exposure to OGD conditions increased neuronal survival in noninfected (76 ± 2.8%) and scramble-treated neurons (76 ± 2.8%), but not in cells with HIF-1α downregulated with shRNA (51 ± 2.2%; Fig. 6E; n = 10, p < 0.05). Together, our data indicate that mTOR-regulated HIF-1α accumulation mediates the neuroprotective effect of tPA in cerebral cortical neurons.
TPA induces the expression of GLUT3 in cerebral cortical neurons and increases the uptake of glucose in the ischemic brain
Because the expression of the HIF-1α-regulated neuronal transporter of glucose GLUT3 has been linked to neuroprotection following exposure to hypoxic/ischemic conditions (Bergeron et al., 2000; Bernaudin et al., 2002; Sharp and Bernaudin, 2004), we studied the expression GLUT3 in Wt cerebral cortical neurons incubated with 5 nm tPA. Our results indicate that tPA not only increases the expression of GLUT3 in cerebral cortical neurons (Fig. 7A) but also induces its recruitment to the cell membrane (Fig. 7B). To investigate the functional effect of tPA-induced expression of GLUT3, we measured the uptake of glucose in Wt cerebral cortical neurons treated 0–3 h with 5 nm tPA or vehicle control. We found that compared to vehicle-treated neurons treatment with tPA during 1 or 3 h induces a 7.19 ± 3.14-fold and 5.20 ± 3.66-fold increase in glucose uptake, respectively (n = 12; p < 0.05; Fig. 7C).
Figure 7.
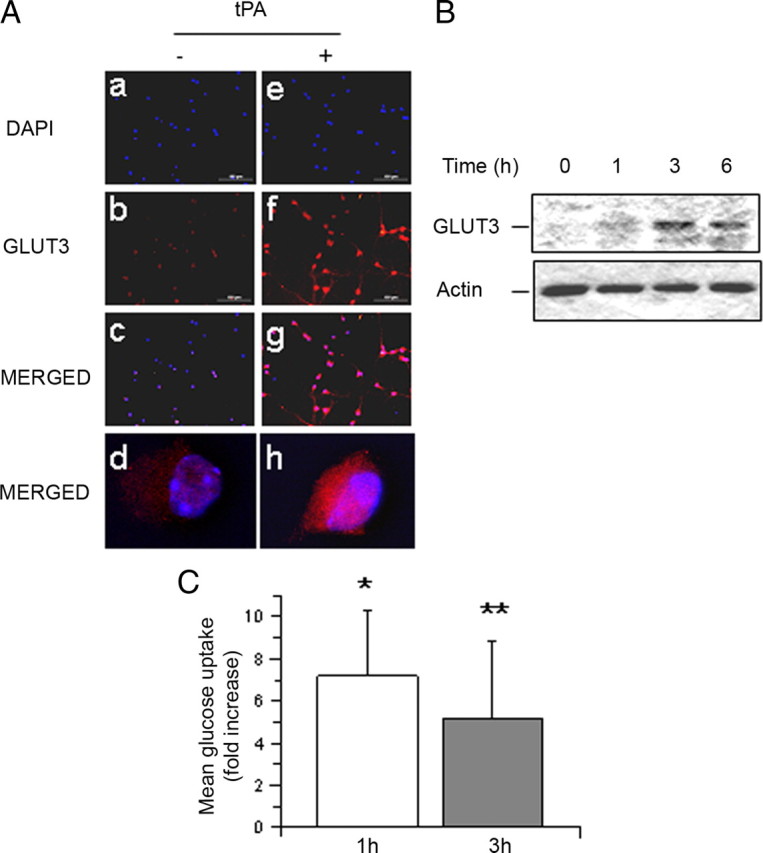
TPA induces the expression of GLUT3 and the uptake of glucose in cerebral cortical neurons. A, Representative micrographs of GLUT3 staining in Wt cerebral cortical neurons incubated for 3 h with 5 nm tPA (tPA +) or vehicle (control; tPA −). Blue, DAPI; red, GLUT3. Magnification: 20× in a–c and e–g; 100 × in d and g. B, Representative Western blot analysis for GLUT3 expression in membrane extracts from cerebral cortical neurons incubated 0–6 h with 5 nm tPA. C, Mean fold increase in the uptake of a fluorescent deoxyglucose analog (2-NBDG) in cerebral cortical neurons following 0–3 h of treatment with 5 nm tPA. Data are presented as mean fold increase in glucose uptake in neurons treated with tPA in relation to glucose uptake in neurons treated with vehicle (control) for similar periods of time; n = 12 per experimental condition. *p < 0.05 compared to neurons treated with vehicle control during 1 h; **p < 0.05 compared to neurons treated with vehicle control for 3 h.
To determine whether neuronal tPA has an effect on GLUT3 expression in vivo, we performed a Western blot analysis for GLUT3 in brain extracts from Wt and T4 mice 0–3 h after tMCAO. Our results show that neuronal overexpression of tPA is associated with an increase in GLUT3 expression in the ischemic brain (Fig. 8A). We then decided to investigate whether neuronal tPA also has an effect on glucose uptake in the ischemic brain in vivo. Wt and T4 mice underwent tMCAO followed 5 min later by the intravenous administration of 18FDG and quantification of glucose uptake by PET scanning imaging as described in the Materials and Methods section. We found that in Wt mice cerebral ischemia induces a decrease in the DUR of glucose from 1.01 ± 0.02% in nonischemic brains to −18.4 ± 5% indicating a sharp decrease in the uptake of glucose in the ischemic hemisphere (p < 0.05). In contrast, the DUR in T4 mice was +8.0 ± 4.2%, demonstrating not only a lack of decrease but instead an increase in the uptake of glucose in the ischemic hemisphere associated with neuronal overexpression of tPA (Fig. 8B; p < 0.05).
Figure 8.

TPA induces the expression of GLUT3 and the uptake of glucose in cerebral cortical neurons. A, Representative Western blot analysis for GLUT3 expression in the ischemic tissue of Wt and T4 mice 1 h after tMCAO. B, C, Representative transverse (a, c) and coronal (b, d) sections of 18FDG-PET scan imaging (B) and quantification of the differential uptake ratio of glucose (C) in Wt and T4 mice 1 h after tMCAO and the intravenous injection of 18-Fluorodeoxyglucose. Arrows in B denote the area of the brain affected by the ischemic injury; n = 12 observations per animal. *p < 0.05 compared to non ischemic brains; **p < 0.05 compared to nonischemic brains and with brains from Wt mice.
Discussion
A decline in the supply of oxygen and glucose induces the rapid release of tPA from cerebral cortical neurons in vitro (Echeverry et al., 2010) and in vivo (Wang et al., 1998; Yepes et al., 2000). However, a relation between tPA and neuronal survival during hypoxic and ischemic conditions remains controversial. Indeed, whereas some studies have proposed that tPA mediates the harmful effects of hypoxia and cerebral ischemia on neuronal death (Wang et al., 1998; Nagai et al., 1999), more recent evidence using different experimental designs indicates that tPA may instead have a neuroprotective effect (Flavin and Zhao, 2001; Lee et al., 2007; Echeverry et al., 2010; Haile et al., 2012).
Our results show that treatment with rtPA or the induction of the release of endogenous tPA promotes cell survival in neurons previously exposed to lethal hypoxia in vitro or cerebral ischemia in vivo via a mechanism independent of tPA's ability to catalyze the conversion of plasminogen into plasmin. This observation is of significant clinical importance. Indeed, our data suggest that a large number of acute ischemic stroke patients that currently remain untreated due to the existence of contraindications to receive proteolytically active tPA as a thrombolytic may still benefit from the administration of proteolytically inactive tPA as a neuroprotector. Also important, our in vivo data indicate a protective effect with doses of rtPA significantly lower than those used so far in experimental studies with animal models of cerebral ischemia and instead very similar to the doses administered to treat acute ischemic stroke patients (Hacke et al., 2008).
Treatment with rtPA is associated with neurological recovery in a significant number of acute ischemic stroke patients. Interestingly, although it is believed that the recovery observed in rtPA-treated patients is entirely due to plasmin-mediated lysis of the clot, their improvement in neurological function is observed only 90 d later (National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995; Hacke et al., 2008) suggesting that in addition to thrombolysis, other mechanisms may be responsible for rtPA's beneficial effect. These observations are in agreement with our data indicating that tPA has a neuroprotective effect in the ischemic brain independently of its thrombolytic properties.
Our work is in apparent discrepancy with reports by others indicating that tPA has a neurotoxic effect in the ischemic brain (Wang et al., 1998; Nagai et al., 1999). We believe that this disagreement may be explained at least in part by differences between experimental designs. However, our previous work indicates that the concentrations of tPA used in some of these studies are highly unlikely to be found in vitro in neuronal cultures exposed to OGD conditions or in vivo in the ischemic brain following tMCAO, even after treatment with rtPA (Echeverry et al., 2010; Haile et al., 2012). Our finding that mice overexpressing tPA in neurons have a better neurological outcome following tMCAO than their Wt littermate controls not only supports our hypothesis that tPA has a neuroprotective effect in the ischemic brain, but also disagrees with previous studies indicating that mice genetically deficient in tPA in all cell types also have a decrease in the volume of the ischemic lesion following the induction of experimental cerebral ischemia (Wang et al., 1998). We believe that this discrepancy may be explained by a pleiotropic cell type-specific role of tPA in the ischemic brain. Indeed, experimental work in animal models of cerebral ischemia (Yepes et al., 2003) and neuroradiological studies in acute ischemic stroke patients (Kidwell et al., 2008) have demonstrated that tPA also increases the permeability of the BBB with the development of cerebral edema and hemorrhagic transformation. Therefore, it is possible that the decrease in the volume of the ischemic lesion observed in tPA−/− mice is due to preservation of the barrier function of the blood–brain barrier instead of a direct cell survival-promoting effect associated with tPA deficiency. Our data are also in apparent conflict with earlier studies indicating that neuroserpin has a neuroprotective effect in the ischemic brain (Yepes et al., 2000; Cinelli et al., 2001). Despite the fact that neuroserpin has been considered the inhibitor of tPA in the CNS, it has also been shown that tPA activity remains unchanged in the brain of mice overexpressing neuroserpin (Madani et al., 2003) and that neuroserpin also protects neurons genetically deficient in tPA from the deleterious effects of a hypoxic injury (Wu et al., 2010). Together, these observations suggest that neuroserpin has a tPA-independent neuroprotective effect in the CNS.
The mTOR pathway plays a central role adapting cellular processes to the availability of energy and metabolic resources. Our proteomic studies show that tPA causes a significant downregulation of the transmembrane protein 127, which was recently identified as a negative regulator of the mTOR pathway (Qin et al., 2010). In agreement with these observations, we found that the release of endogenous tPA and treatment with rtPA are very efficient activators of the mTOR pathway in neurons via a plasminogen-independent mechanism. The clinical relevance of this finding is underscored by the observation that mTOR inhibition in the ischemic brain has been associated with neuronal death, and mTOR activation in neurons has been linked to recovery after injury (Abe et al., 2010). Our previous studies showed that tPA is an efficient activator of the PI3K/Akt pathway (An et al., 2008). The work presented here shows that Akt activation mediates the effect of tPA on the mTOR pathway.
HIF-1α regulates the expression of genes that facilitate the adaptation to low oxygen concentrations. Despite the fact that HIF-1α is continuously made, it only accumulates in hypoxic cells (Sharp and Bernaudin, 2004). Indeed, in normoxic cells HIF-1α is continuously hydroxylated by the activity of prolyl-4-hydroxylases in the cytoplasm and nucleus, abrogating its intracellular accumulation. In contrast, hypoxia inhibits these hydroxylases, leading to the rapid accumulation of HIF-1α in the cytoplasm. We found that tPA induces mTOR-regulated HIF-1α accumulation in neurons and that this effect does not require hypoxic conditions. This hypoxia-independent effect on HIF-1α accumulation has also been observed with some growth factors and cytokines (Semenza, 2002).
GLUT3 mediates the transport of glucose in neurons and its expression is regulated by HIF-1α and induced by synaptic activity (Simpson et al., 2008) via NMDAR activation (Ferreira et al., 2011). Our results indicate that tPA not only induces the expression of neuronal GLUT3 but also its recruitment to the cell membrane, suggesting a direct effect of tPA on the uptake of glucose. This hypothesis is supported by our observation that either treatment with tPA in vitro or overexpression of neuronal tPA in vivo increases the uptake of glucose in cerebral cortical neurons.
In summary, our results indicate that tPA plays a pivotal role in detection of, adaptation to, and survival under hypoxic and ischemic conditions. Based on our data, we here propose a model where either endogenous tPA released in response to a hypoxic injury or rtPA intravenously administered following the onset of acute ischemic stroke promotes neuronal survival via a mTOR-mediated, HIF-1α-regulated increase in the uptake of glucose in neurons. This effect is independent of tPA's ability to cleave plasminogen into plasmin and is observed with doses of tPA not associated with hemorrhagic transformation in the ischemic brain.
Footnotes
This work was supported in part by National Institutes of Health Grants NS-062073 and VA MERIT award BX000474 (to M.Y.) and National Natural Science Foundation of China 81173086 (to J.A.).
The authors declare no competing financial interests.
References
- Abe N, Borson SH, Gambello MJ, Wang F, Cavalli V. Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J Biol Chem. 2010;285:28034–28043. doi: 10.1074/jbc.M110.125336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Zhang C, Polavarapu R, Zhang X, Zhang X, Yepes M. Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein induce Akt phosphorylation in the ischemic brain. Blood. 2008;112:2787–2794. doi: 10.1182/blood-2008-02-141630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Busto R, Zhao W, Fernandez G, Ginsberg MD. Middle cerebral artery occlusion in the mouse by intraluminal suture coated with poly-l-lysine: neurological and histological validation. Brain Res. 1999;833:181–190. doi: 10.1016/s0006-8993(99)01528-0. [DOI] [PubMed] [Google Scholar]
- Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol. 2000;48:285–296. [PubMed] [Google Scholar]
- Bernaudin M, Tang Y, Reilly M, Petit E, Sharp FR. Brain genomic response following hypoxia and re-oxygenation in the neonatal rat. Identification of genes that might contribute to hypoxia-induced ischemic tolerance. J Biol Chem. 2002;277:39728–39738. doi: 10.1074/jbc.M204619200. [DOI] [PubMed] [Google Scholar]
- Cinelli P, Madani R, Tsuzuki N, Vallet P, Arras M, Zhao CN, Osterwalder T, Rülicke T, Sonderegger P. Neuroserpin, a neuroprotective factor in focal ischemic stroke. Mol Cell Neurosci. 2001;18:443–457. doi: 10.1006/mcne.2001.1028. [DOI] [PubMed] [Google Scholar]
- Echeverry R, Wu J, Haile WB, Guzman J, Yepes M. Tissue-type plasminogen activator is a neuroprotectant in the mouse hippocampus. J Clin Invest. 2010;120:2194–2205. doi: 10.1172/JCI41722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira JM, Burnett AL, Rameau GA. Activity-dependent regulation of surface glucose transporter-3. J Neurosci. 2011;31:1991–1999. doi: 10.1523/JNEUROSCI.1850-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavin MP, Zhao G. Tissue plasminogen activator protects hippocampal neurons from oxygen-glucose deprivation injury. J Neurosci Res. 2001;63:388–394. doi: 10.1002/1097-4547(20010301)63:5<388::AID-JNR1033>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- Haile WB, Wu J, Echeverry R, Wu F, An J, Yepes M. Tissue-type plasminogen activator has a neuroprotective effect in the ischemic brain mediated by neuronal TNF-alpha. J Cereb Blood Flow Metab. 2012;32:57–69. doi: 10.1038/jcbfm.2011.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Sheng M. The growing role of mTOR in neuronal development and plasticity. Mol Neurobiol. 2006;34:205–219. doi: 10.1385/MN:34:3:205. [DOI] [PubMed] [Google Scholar]
- Kidwell CS, Latour L, Saver JL, Alger JR, Starkman S, Duckwiler G, Jahan R, Vinuela F, Vinuela F, Kang DW, Warach S. Thrombolytic toxicity: blood brain barrier disruption in human ischemic stroke. Cerebrovasc Dis. 2008;25:338–343. doi: 10.1159/000118379. [DOI] [PubMed] [Google Scholar]
- Lee SR, Lok J, Rosell A, Kim HY, Murata Y, Atochin D, Huang PL, Wang X, Ayata C, Moskowitz MA, Lo EH. Reduction of hippocampal cell death and proteolytic responses in tissue plasminogen activator knockout mice after transient global cerebral ischemia. Neuroscience. 2007;150:50–57. doi: 10.1016/j.neuroscience.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani R, Hulo S, Toni N, Madani H, Steimer T, Muller D, Vassalli JD. Enhanced hippocampal long-term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO J. 1999;18:3007–3012. doi: 10.1093/emboj/18.11.3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani R, Kozlov S, Akhmedov A, Cinelli P, Kinter J, Lipp HP, Sonderegger P, Wolfer DP. Impaired explorative behavior and neophobia in genetically modified mice lacking or overexpressing the extracellular serine protease inhibitor neuroserpin. Mol Cell Neurosci. 2003;23:473–494. doi: 10.1016/s1044-7431(03)00077-0. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Chan PH. The development of a new mouse model of global ischemia: focus on the relationships between ischemia duration, anesthesia, cerebral vasculature, and neuronal injury following global ischemia in mice. Brain Res. 1998;780:304–310. doi: 10.1016/s0006-8993(97)01217-1. [DOI] [PubMed] [Google Scholar]
- Nagai N, De Mol M, Lijnen HR, Carmeliet P, Collen D. Role of plasminogen system components in focal cerebral ischemic infarction: a gene targeting and gene transfer study in mice. Circulation. 1999;99:2440–2444. doi: 10.1161/01.cir.99.18.2440. [DOI] [PubMed] [Google Scholar]
- National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H. BDNF function and intracellular signaling in neurons. Histol Histopathol. 2010;25:237–258. doi: 10.14670/HH-25.237. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Polavarapu R, Gongora MC, Yi H, Ranganthan S, Lawrence DA, Strickland D, Yepes M. Tissue-type plasminogen activator-mediated shedding of astrocytic low-density lipoprotein receptor-related protein increases the permeability of the neurovascular unit. Blood. 2007;109:3270–3278. doi: 10.1182/blood-2006-08-043125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F, Boaretto F, Opocher G, Toledo RA, Toledo SP, Stiles C, Aguiar RC, Dahia PL. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–233. doi: 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64:993–998. doi: 10.1016/s0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5:437–448. doi: 10.1038/nrn1408. [DOI] [PubMed] [Google Scholar]
- Sheldon RA, Osredkar D, Lee CL, Jiang X, Mu D, Ferriero DM. HIF-1 alpha-deficient mice have increased brain injury after neonatal hypoxia-ischemia. Dev Neurosci. 2009;31:452–458. doi: 10.1159/000232563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi GD, OuYang YP, Shi JG, Liu Y, Yuan W, Jia LS. PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem Biophys Res Commun. 2011;404:941–945. doi: 10.1016/j.bbrc.2010.12.085. [DOI] [PubMed] [Google Scholar]
- Shi H. Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr Med Chem. 2009;16:4593–4600. doi: 10.2174/092986709789760779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ. The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab. 2008;295:E242–E253. doi: 10.1152/ajpendo.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Seaman LB, Vannucci RC. Effects of hypoxia-ischemia on GLUT1 and GLUT3 glucose transporters in immature rat brain. J Cereb Blood Flow Metab. 1996;16:77–81. doi: 10.1097/00004647-199601000-00009. [DOI] [PubMed] [Google Scholar]
- Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- Wu J, Echeverry R, Guzman J, Yepes M. Neuroserpin protects neurons from ischemia-induced plasmin-mediated cell death independently of tissue-type plasminogen activator inhibition. Am J Pathol. 2010;177:2576–2584. doi: 10.2353/ajpath.2010.100466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes M, Sandkvist M, Wong MK, Coleman TA, Smith E, Cohan SL, Lawrence DA. Neuroserpin reduces cerebral infarct volume and protects neurons from ischemia-induced apoptosis. Blood. 2000;96:569–576. [PubMed] [Google Scholar]
- Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood–brainbarrier via the LDL receptor-related protein. J Clin Invest. 2003;112:1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J Cereb Blood Flow Metab. 2009;29:873–885. doi: 10.1038/jcbfm.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]