

Abstract
Acquired aplastic anemia (AA) is a rare disease with a complex pathogenesis. In most cases, T-cell mediated immune destruction of hematopoietic cells results in peripheral blood pancytopenia and bone marrow hypoplasia. Asubset of the heterodimeric interleukin-23 receptor gene (IL-23R) is significantly associated with autoimmune-mediated diseases. To examine whether IL-23R single nucleotide polymorphisms (SNPs) might contribute to AA, we selected three IL-23R SNPs with amino-acid changes (rs11209026: p.Arg381Gln; rs41313262: p.Val362Ile; rs11465797: p.Thr175Asn), and compared their frequencies in279 AA patients and 184 ethnicallymatched healthy controls. The three SNP prevalences were similar between the AApatients and controls. The Arg381Gln variant, which has a strong protective effect against inflammatory bowel disease, showed no association with AA. Further, IL-23 levels in sera were measured in the AA patients and in controls, and there were no significant differences among them. Our results indicatethat these three IL-23R SNPs and serum IL-23 level have no apparent impact onsusceptibility to AA.
Keywords: aplastic anemia, IL-23R, autoimmune disease, polymorphism
INTRODUCTION
Acquired aplastic anemia (AA) is the paradigm of the human bone marrow failure syndromes [32]. In most cases, peripheral blood and marrow of AApatients contain elevated numbers of activated cytotoxic lymphocytes, whichdecrease with successful immunosuppressive therapy, implicating T-cell mediated immunityin hematopoietic cell destruction. Inalmost all AA patients, a decreased number of regulatory T cells (Tregs) has beenobserved, as in other autoimmune diseases [25]. Increased levels of T-bet (a member of the T-box transcription family) in patients’ T cells are responsible for the increased interferon-γ (IFN-γ) levels and the T helper type 1 (Th1) shift of CD4+ T cells in AA [24]. T-bet mediated IFN-γ elevation would be a trigger for endogenous stimulation in Tcells of AA patients as IFN-γ is a master cytokine of the Th1 immune response.
Interleukin-23 (IL-23), a member ofthe IL-12 family, has proinflammatory and immunoregulatory activities. IL-23 signaling-pathway molecules maybe important candidate genes for autoimmune disease [13,27]. The IL-23 receptor is a heterodimeric complex formed byanIL-12Rβ1 subunit (shared with IL-12) and a unique IL-23R subunit [16]. Although IL-23 and IL-12 as well as their receptors share structural and functional similarities, IL-23 has biological activities in the T-cell mediated immune response similar to but distinct from IL-12. IL-23 promotes the development of T helper cells (Th17) producing IL-17, a proinflammatory cytokine with many targets and multiple downstream mediators [3,12]. Elevated IL-17 levels have been observed in sera and target tissues of various autoimmune diseases and inflammatory conditions, including rheumatoid arthritis (RA), multiple sclerosis (MS), systemic lupus erythematosus(SLE), asthma, the two commonforms of inflammatory bowel disease (IBD) (Crohn’s disease (CD) and ulcerative colitis (UC)), bacterial pneumonia, and in murine experimental autoimmune encephalopathy[32].
A recent genome-wide association studyrevealed that several single nucleotide polymorphisms (SNPs) in the IL-23R gene (chromosome 1p31; GenBank Accession: NM_144701.2, GeneID: 149233) were significantlyassociated with IBD: an uncommon coding variant (rs11209026, c.1227G>A, p.Arg381Gln) conferred strong protection againstCD [8]; several other non-coding variants inthe IL-23R gene also were associated with CD and UC. Subsequent studies also have provided more evidence for anassociation of IL-23R gene withIBD in both adult and child cohorts [4,7,14,15,19,26,29], and further with other immune-mediated disorders[6], such as psoriasis and ankylosing spondylitis [5,21]. Accumulating data indicatethat genetic risk factors may be shared among various autoimmune diseases. This study is the first attempt to assess the possible contribution of IL-23R gene polymorphisms to AA.
MATERIALS AND METHODS
Patients and controls
Peripheral blood (PB) samples were obtained from 279 unrelated consecutive patients diagnosed with AA (133 females and 146 males), according to the criteria of the International Agranulocytosis and AA Study. These AA patients were treated at a single institution (Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health), and composed of four ethnic groups (159 Caucasians (57.0%), 33African-Americans (11.8%), 49 Hispanics (17.6%), and 33Asians(13.6%)). Blood samples from ethnically matched 184 healthy donors (117 Caucasians (63.6%), 23 African-Americans (12.5%), 22 Hispanics (12.0%), and 22 Asians (12.0%) from SNP-500Cancer) [18] served as controls. Patients or their guardians provided written informed consent for genetic testing, according to protocols approved by the institutional review board of the National Heart, Lung, and Blood Institute.
DNA amplification and sequencing
DNA was extracted from PB mononuclear cells using the Qiagen DNA extraction kit and subjected to PCR amplification using the LA Taq polymerase kit (Takara, Otsu, Japan) and adequate primer sets in order to obtain DNA fragments containing SNPs (rs11209026, rs41313262, and rs11465797). Primers for rs11209026 and rs41313262: 5′-GAGCAGAGTAAAGAGAATAGTAA-3′ (forward), 5′-TGGGCTGAGGACTTAGCCTCTTTAAGCCTC-3′ (reverse); primers for rs11465797: 5′-CAAGTAACTGGGATTACAGGCACATG-3′ (forward), 5′-CTTTACCTATATCATCCAGGT-3′ (reverse). Sequence analysis of the amplified DNA product was performed using the BigDye Terminator version 3.1 ready reactionkit and the ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequencing primers are 5′-GACATTTGTAGAGAGTTTGGCATG -3′ for rs11209026 and rs41313262, and 5′-CTTCCATGCCTAGTGCGTTTGCTG-3′ for rs11465797. Sequencing data were aligned and compared using the SeqMan program (DYNASTER, Madison, WI).
Cytokine enzyme-linked immunosorbent assay (ELISA)
Sera were obtained from 23 AA patients with IL-23R polymorphisms, 37 AA patients without polymorphisms, and 21 healthy controls. IL-23 serum levels were measured using the Human Interleukin 23 (Hu IL-23) Heterodimer ELISA kit (BioSource, Camarillo, CA), following the manufacturer’s recommendations. All samples were assayed in duplicate.
Statistical analysis
Summary statistics, such as proportions and p-values were used to describe the related variables. Statistical analysis was performed by χ2 tests with a significance level of P < 0.05, for SNPs and for ELISA data. All numerical results were computed using Splus 8 (Insightful Corp, Seattle, WA) and NCSS, PASS (Hintze. J, (2006). NCSS, PASS, and GESS. NCSS, Kaysville, Utah) for Windows statistical software.
RESULTS AND DISCUSSION
To assess the contribution of IL-23 SNPs to AA susceptibility and phenotype, we first targeted the non-synonymous rs11209026 SNP (c.1142G>A, p.Arg381Gln) because of its strong association withIBD [10]. In addition, we examined allele frequencies of two non-synonymous SNPs, rs41313262 (c.1084G>A, p.Val362Ile) and rs11465797 (c.524C>A, p.Thr175Asn). Genotype frequencies of the three IL-23R polymorphisms examined in AA patients and healthy controls are shown in Table 1, together with demographic and clinical features. The p.Arg381Gln variant was present in 6.1% ofAA patients and in 6.0% ofethnically matched controls (P = 0.960; β-error =0.16). The carrier frequencyof the p.Val362Ile (rs41313262) variant was low, but similar inAA patients (2.2%) and controls (1.6%) (P = 0.958); the p.Thr175Asn (rs11465797) variant was not observed in patients or controls. No homozygous carriers of the p.Arg381Gln or p.Val362Ile variant were found in either AA patients or controls. All carriers had only a single SNP, except one with two SNPs.
Table 1.
Genotype frequencies of the three IL23R SNPs as well as demographic and clinical features in AA patients and controls
| Risk Factor | AA patients N (%) |
Healthy control N (%) |
AA vs. controls | |
|---|---|---|---|---|
| OR a | P value | |||
| Total | 279 | 184 | ---- | ---- |
|
| ||||
| Diagnosis (%) | ||||
| MAA | 92 (33.0) | ---- | ---- | ---- |
| SAA | 187 (67.0) | ---- | ---- | ---- |
|
| ||||
| Sex (%) | ||||
| Male | 146 (52.3) | NA | ---- | ---- |
| Female | 133 (47.7) | NA | ---- | ---- |
|
| ||||
| Race/Ethnicity (%) | ||||
| Caucasian | 159 (57.0) | 117 (63.6) | ---- | 0.187 |
| African-American | 33 (11.8) | 23 (12.5) | ---- | 0.943 |
| Hispanic | 49 (17.6) | 22 (12.0) | ---- | 0.132 |
| Asian | 33 (13.6) | 22 (12.0) | ---- | 0.967 |
|
| ||||
| SNPs (% yes) | ||||
| rs11209026 | 17 (6.1) | 11 (6.0) | 1.018 | 0.960 |
| rs41313262 | 6 (2.2) | 3 (1.6) | 1.181 | 0.958 |
| rs11465797 | 0 | 0 | ---- | ---- |
rs11209026 (c.1142G>A, p.Arg381Gln); rs41313262 (c.1084G>A, p.Val362Ile);
rs11465797 (c.524C>A, p.Thr175Asn). An amino acid position is relative to the ATG start site; the first nucleotide of the exon 1 start site is designated as position 1 based on the reference sequence GenBank NM_144701.2 for IL-23R. AA patients and controls examined carried only one of the three SNPs.
OR: odds ratio.
We performed ELISA analysis to measure IL-23 levels in sera of 23 AA patients with SNP polymorphisms and 37 AA patients without polymorphisms, and compared results with those of 21 healthy controls (Figure 1). There wereno statistical differences among thethree groups, even after stratifying AA patients according to treatment status.
Figure 1.
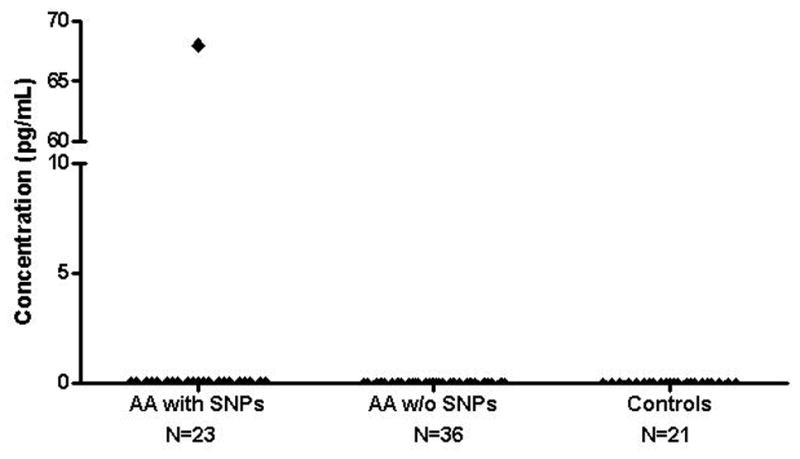
Comparison of IL-23 levels in sera between AA patients and controls by ELISA. Serum samples were obtained from 23 AA patients with SNPs, 37 AA patients without SNPs, and 21 healthy controls. IL-23 levels were measured using the human IL-23 ELISA assay kit. AA patients with and without SNPs vs. controls: P = 0.720; AA patients with SNPs vs. controls: P = 0.344; AA patients without SNPs vs. controls: P = 0.342.
Collectively, our dataindicatethat the Arg381Gln SNP did not correlate withsusceptibility ofAA.
The IL-23/IL-17-mediated inflammatory axis has been implicated in numerous autoimmune diseases and inflammatory conditions. A series of cytokines, including IL-6, IL-21, TGF-β, and IL-23, sequentially or synergistically induce Th17 lineage cells, together with the cooperative action of transcription factors[11]. Conversely, other cytokines, including IL-2, IL-4, IFN-γ, and IL-27, antagonize differentiation of the Th17 lineage and inhibit IL-17 and IL-23 production. In AA patients, increased productionof type 1 cytokines (IFN-γ, TNF, and IL-2) from T cells has seenobserved, implicating that the hematopoietic cell destruction bya Th1 T-cellresponse [9,23], which includes boththe Fas-mediated cell death and the inhibition of hematopoieticstem cell proliferation [30]. Our previous work has suggestedthat T-bet mediated IFN-γ elevation is responsible for the Th1 shift of CD4+ T cells in AA patients. As CD4+ effecter T cells were shifted to Th1 cells in AA patients, the IL-23/IL-17-mediated inflammatory axis might be suppressed in AA, due to antagonistic effect ofTh1 cytokines onTh17 differentiation and development. Therefore, it is consistent that IL-23R SNPs have no significant association with AA, although it cannot be excluded that other IL-23R SNPs as well as IL-23R isoforms (differentially expressed in distinct cell types) might play protective roles.
In other autoimmune diseases such as MS, RA, and SLE, similar results have beenobtained, IL23R polymorphisms had no significant correlation with susceptibility or severity of disease [2,17,22], in agreement withour results. It has beensuggestedthat the IL23R polymorphisms might play a more important role in regulating local inflammation, as in IBD and psoriasis, rather than in systemic inflammation in RA and SLE, supported by some work in mouse models [28]. Further, the IL-23-pathway molecules have no association with SLE; type I cytokines, which are not induced withIL-23, are relevant in the development and maintenance of this disease [1,20]. AA patients may share a common background of genetic risk affecting disease development and severity, which may be similar to some other autoimmune diseases and completely different from others. Further genetic studies are required to investigate as yet unidentified causal genetic alleles responsible for AA.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) Intramural Research Program. The authors are grateful to Colin Wu for assistance with statistical analysis.
Abbreviations
- AA
aplastic anemia
- SNP
single nucleotide polymorphism
- IL-12, 17 or 23
Interleukin-12, 17 or 23
- IFN-γ
Interferon-γ
- TNF
tumor necrosis factor
- IBD
inflammatory bowel disease
- CD
Crohn’s disease
- UC
ulcerative colitis
- RA
rheumatoid arthritis
- MS
multiple sclerosis
- SLE
systemic lupus erythematosus
- Tregs
regulatory T cells
- Th1
T helper type 1
- Th17
T helper cells type 17
References
- 1.Baechler EC, Gregersen PK, Behrens TW. The emerging role of interferon in human systemic lupus erythematosus. Curr Opin Immunol. 2004;16:801–7. doi: 10.1016/j.coi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 2.Begovich AB, Chang M, Caillier SJ, Lew D, Catanese JJ, Wang J, Hauser SL, Oksenberg JR. The autoimmune disease-associated IL12B and IL23R polymorphisms in multiple sclerosis. Hum Immunol. 2007;68:934–7. doi: 10.1016/j.humimm.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Bettelli E, Oukka M, Kuchroo V. Th-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–50. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 4.Büning C, Schmidt HH, Molnar T, De Jong DJ, Fiedler T, Bühner S, Sturm A, Baumgart DC, Nagy F, Lonovics J, Drenth JP, Landt O, Nickel R, Büttner J, Lochs H, Witt H. Heterozygosity for IL23R p.Arg381Gln confers a protective effect not only against Crohn’s disease but also ulcerative colitis. Aliment Pharmacol Ther. 2007;26:1025–33. doi: 10.1111/j.1365-2036.2007.03446.x. [DOI] [PubMed] [Google Scholar]
- 5.Capon F, Di Meglio P, Szaub J, Prescott NJ, Dunster C, Baumber L, Timms K, Gutin A, Abkevic V, Burden AD, Lanchbury J, Barker JN, Trembath RC, Nestle FO. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. 2007;122:201–6. doi: 10.1007/s00439-007-0397-0. [DOI] [PubMed] [Google Scholar]
- 6.Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, Matsunami N, Ardlie KG, Civello D, Catanese JJ, Leong DU, Panko JM, McAllister LB, Hansen CB, Papenfuss J, Prescott SM, White TJ, Leppert MF, Krueger GG, Begovich AB. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–90. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dubinsky MC, Wang D, Picornell Y, Wrobel I, Katzir L, Quiros A, Dutridge D, Wahbeh G, Silber G, Bahar R, Mengesha E, Targan SR, Taylor KD, Rotter JI. IL-23 receptor (IL-23R) gene protects against pediatric Crohn’s disease. Inflamm Bowel Dis. 2007;13:511–5. doi: 10.1002/ibd.20126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giannakoulas NC, Karakantza M, Theodorou GL, Pagoni M, Galanopoulos A, Kakagianni T, Kouraklis-Symeonidis A, Matsouka P, Maniatis A, Zoumbos NC. Clinical relevance of balance between type 1 and type 2 immune responses of lymphocyte subpopulations in aplastic anemia patients. Br J Haematol. 2004;124:97–105. doi: 10.1046/j.1365-2141.2003.04729.x. [DOI] [PubMed] [Google Scholar]
- 10.Glas J, Seiderer J, Wetzke M, Konrad A, Török HP, Schmechel S, Tonenchi L, Grassl C, Dambacher J, Pfennig S, Maier K, Griga T, Klein W, Epplen JT, Schiemann U, Folwaczny C, Lohse P, Göke B, Ochsenkühn T, Müller-Myhsok B, Folwaczny M, Mussack T, Brand S. rs1004819 is the main disease-associated IL23R variant in German Crohn’s disease patients: combined analysis of IL23R, CARD15, and OCTN1/2 variants. PLoS ONE. 2007;2:e819. doi: 10.1371/journal.pone.0000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–17. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;12:467–76. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genetics. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, Vermeire S, Dewit O, de Vos M, Dixon A, Demarche B, Gut I, Heath S, Foglio M, Liang L, Laukens D, Mni M, Zelenika D, Van Gossum A, Rutgeerts P, Belaiche J, Lathrop M, Georges M. Replication of an association between IL23R gene polymorphism with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2007;5:977–81. doi: 10.1016/j.cgh.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 17.Orozco G, Rueda B, Robledo G, García A, Martín J. Investigation of the IL23R gene in a Spanish rheumatoid arthritis cohort. Hum Immunol. 2007;68:681–84. doi: 10.1016/j.humimm.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 18.Packer BR, Yeager M, Burdett L, Welch R, Beerman M, Qi L, Sicotte H, Staats B, Acharya M, Crenshaw A, Eckert A, Puri V, Gerhard DS, Chanock SJ. SNP500-Cancer: a public resource for sequence validation and assay development for genetic variation in candidate genes. Nucleic Acids Res. 2006;32:D528–32. doi: 10.1093/nar/gkh005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts RL, Gearry RB, Hollis-Moffatt JE, Miller AL, Reid J, Abkevich V, Timms KM, Gutin A, Lanchbury JS, Merriman TR, Barclay ML, Kennedy MA. IL23R R381Q and ATG16L1 T300A are strongly associated with crohn’s disease in a study of New Zealand caucasians with inflammatory bowel disease. Am J Gastroenterol. 2007;102:2754–61. doi: 10.1111/j.1572-0241.2007.01525.x. [DOI] [PubMed] [Google Scholar]
- 20.Ronnblom L, Eloranta ML, Alm GV. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 2006;54:408–20. doi: 10.1002/art.21571. [DOI] [PubMed] [Google Scholar]
- 21.Rueda B, Orozco G, Raya E, Fernandez-Sueiro JL, Mulero J, Blanco FJ, Vilches C, González-Gay MA, Martin J. The IL23R Arg381Gln non-synonymous polymorphism confers susceptibility to ankylosing spondylitis. Ann Rheum Dis. 2008 Jan 16; doi: 10.1136/ard.2007.080283. [DOI] [PubMed] [Google Scholar]
- 22.Sánchez E, Rueda B, Callejas JL, Sabio JM, Ortego-Centeno N, Jimenez-Alonso J, López-Nevot MA, Martín J. Analysis of interleukin-23 receptor (IL23R) gene polymorphisms in systemic lupus erythematosus. Tissue Antigens. 2007;70:233–37. doi: 10.1111/j.1399-0039.2007.00881.x. [DOI] [PubMed] [Google Scholar]
- 23.Sloand E, Kim S, Maciejewski JP, Tisdale J, Follmann D, Young NS. Intracellular interferon-gamma in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemia. Blood. 2002;100:1185–91. doi: 10.1182/blood-2002-01-0035. [DOI] [PubMed] [Google Scholar]
- 24.Solomou EE, Keyvanfar K, Young NS. T-bet, a Th1 transcription factor, is up-regulated in T cells from patients with aplastic anemia. Blood. 2006;107:3983–91. doi: 10.1182/blood-2005-10-4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solomou EE, Rezvani K, Mielke S, Malide D, Keyvanfar K, Visconte V, Kajigaya S, Barrett AJ, Young NS. Deficient CD4+ CD25+ FOXP3+ T regulatory cells in acquired aplastic anemia. Blood. 2007;110:1603–06. doi: 10.1182/blood-2007-01-066258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tremelling M, Cummings F, Fisher SA, Mansfield J, Gwilliam R, Keniry A, Nimmo ER, Drummond H, Onnie CM, Prescott NJ, Sanderson J, Bredin F, Berzuini C, Forbes A, Lewis CM, Cardon L, Deloukas P, Jewell D, Mathew CG, Parkes M, Satsangi J. IL23R variation determines susceptibility but not disease phenotype in inflammatory bowel disease. Gastroenterology. 2007;132:1657–64. doi: 10.1053/j.gastro.2007.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimetric cytokines: new players in the regulation of T-cell responses. Immunity. 2003;19:641–4. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 28.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, Powrie F. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–18. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 29.Van Limbergen J, Russell RK, Nimmo ER, Drummond HE, Smith L, Davies G, Anderson NH, Gillett PM, McGrogan P, Hassan K, Weaver L, Bisset WM, Mahdi G, Wilson DC, Satsangi J. IL23R Arg381Gln is associated with childhood onset inflammatory bowel disease in Scotland. Gut. 2007;56:1173–74. doi: 10.1136/gut.2007.122069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young NS, Maciejewski J. The pathophysiology of acquired aplastic anemia. N Engl J Med. 1997;336:1365–72. doi: 10.1056/NEJM199705083361906. [DOI] [PubMed] [Google Scholar]
- 31.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–19. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, Hinrichs DJ, Lu H, Chen H, Zhong W, Kolls JK. After interleukin-12p40, are interleukin-23 and interleukin17 the next therapeutic targets for inflammatory bowel disease? Int Immunopharmacol. 2007;7:409–16. doi: 10.1016/j.intimp.2006.09.024. [DOI] [PubMed] [Google Scholar]