

SUMMARY
Monocytes and macrophages (mϕ) are plastic cells whose functions are governed by microenvironmental cues. Wound fluid bathing the wound tissue reflects the wound microenvironment. Current literature on wound inflammation is primarily based on the study of blood monocyte-derived mϕ (MDM), cells that have never been exposed to the wound microenvironment. We sought to pair-match compare MDMs with mϕ isolated from chronic wound of patients. Oncostatin M (OSM) was differentially overexpressed in pair-matched wound mϕ. Both PGE2 and its metabolite 13,14-dihydro-15-keto-PGE2 (PGE-M) were abundant in wound fluid and induced OSM in wound-site mϕ. Consistently, induction of OSM mRNA was observed in mϕ isolated from PGE2–enriched PVA sponges implanted in murine wounds. Treatment of human THP-1 cell-derived mϕ with PGE2 or PGE-M caused dose-dependent induction of OSM. Characterization of the signal transduction pathways demonstrated the involvement of EP4 receptor and cAMP signaling. In human mϕ, PGE2 phosphorylated Axl, a receptor tyrosine kinase (RTK). Axl phosphorylation was also induced by a cAMP analog demonstrating interplay between the cAMP and RTK pathways. PGE2–dependent Axl phosphorylation led to AP-1 transactivation which is directly implicated in inducible expression of OSM. Treatment of human mϕ or mice excisional wounds with recombinant OSM resulted in an anti-inflammatory response as manifested by attenuated expression of endotoxin-induced TNFα and IL-1β. OSM treatment also improved wound closure during the early inflammatory phase of healing. In summary this work recognizes PGE2 in the wound-fluid as a potent inducer of mϕ OSM, a cytokine with anti-inflammatory role in cutaneous wound healing.
INTRODUCTION
In the United States, chronic wounds affect 6.5 million patients posing a major threat to the public health and economy (1). Yet, studies directly investigating chronic wounds as presented in the clinic to develop mechanism-based understanding are scanty. Macrophages (mϕ) play a key role in wound repair such that both inadequate inflammatory responses to wounding as well as unresolved inflammation compromise wound closure (2, 3). Monocytes are highly plastic cells that differentiate into mϕ based on cues at the specific wound microenvironment (4). The functional fate of monocytes recruited to the wound-site is governed by the specific properties of the wound microenvironment (4, 5). We recognize that peripheral blood monocytes differentiated ex vivo using standard laboratory procedures do form mϕ but do not resemble wound mϕ because of the lack of exposure to an elaborate set of microenvironmental cues ex vivo. Wound mϕ can thus only be studied functionally if they can be isolated intact from the actual wound milieu. Characterization of the phenotype of wound mϕ obtained using the PVA sponge approach led to the recognition that wound mϕ possess unique characteristic features (6, 7). At present, evidence supporting the understanding of mϕ directly isolated from the human wound milieu is scanty. Thus, we sought to develop an approach to collect functionally intact mϕ from clinically presented chronic wounds. Outcomes from such cell were compared in a pair-matched manner with the peripheral blood monocyte-derived mϕ (MDM) of the same individual. Such studies identified oncostatin M (OSM) as a key differentially expressed cytokine abundantly produced by human chronic wound mϕ. OSM is a multifunctional cytokine known to be produced by activated mϕ. It is structurally and functionally related to the IL-6-type cytokine family (8–10). In this work, we sought to characterize the mechanism underlying OSM induction in wound mϕ as well as understand the significance of OSM in wound inflammation.
EXPERIMENTAL PROCEDURES
Human subjects and sample collection
Subjects (N=15) participating in the study were chronic wound patients seen at OSU Comprehensive Wound Center (CWC) clinics and have been undergoing NPWT (negative pressure wound therapy) as part of standard clinical care. Demographic characteristics of patients and wound related information are presented in Table 1. The NPWT dressing (sponges) and peripheral blood were collected from each patient. All human studies were approved by The Ohio State University’s (OSU) Institutional Review Board (IRB). Declaration of Helsinki protocols was followed and patients gave their written informed consent.
Table 1.
Demographic characteristics of patients (n=15) and wound size/age.
| Age (y) | 47±8 |
| Females | 8 |
| Race | AA,C |
| Wound size (mm3) | 4.5–729 |
| Wound age | >30d |
| Diabetic | 8 |
| Wound etiology | |
| pressure | 7 |
| surgical | 8 |
| Wound location | |
| abdominal | 5 |
| lower extremity | 10 |
AA, African American; C, Caucasians
Human chronic wound macrophage and fluid collection
Wound fluid and cells were derived from the NPWT dressing by lavaging the wound dressing with saline solution (11). The lavaged fluid was centrifuged to obtain wound cells. Wound mϕ were isolated from NPWT sponge derived wound cells using Ficoll density centrifugation followed by magnet-activated cell sorting (Miltenyi Biotec, Auburn, CA) using CD14 antibody. Isolated cells were seeded in culture dishes for 3h. Non-adherent cells were washed and removed. The phenotype of adherent cells was confirmed by immunofluorescence staining using CD68 antibody.
Peripheral Blood Monocyte Derived Macrophages (MDM)
Blood monocytes from human subjects were isolated using a Ficoll-Hypaque density gradient (GE Healthcare, formerly Amersham Biosciences, Piscataway, NJ). Positive selection for monocytes was performed using using CD14 antibody conjugated to magnetic beads (Miltenyi Biotec, Auburn, CA). Purity of these preparations of monocytes was >90% as determined by fluorescence-activated cell sorting analyses using CD14 antibodies. Differentiation of these cells to mϕ was performed as described (11).
Isolation of murine wound and bone marrow derived macrophages
For wound mϕ, circular (6 mm) sterile polyvinyl alcohol (PVA) sponges were implanted subcutaneously on the back of 8–10 wks old C57BL6 mice. Sponge-infiltrated mϕ were isolated as described (12). To obtain bone marrow derived macrophages (BMDM), the cells from femurs of mice (9 weeks old) were flushed using RPMI 1640 followed by positive selection using CD11b antibody conjugated to magnetic beads. The isolated cells were cultured in RPMI 1640 containing 10% heat-inactivated FBS, 1% antibiotic/antimicotic, Polymyxin B (10 µg/ml) and mouse MCSF (20 ng/ml) at 37°C in a humidified atmosphere containing 5% CO2 for 5 days (13, 14).
“Hunt/Schilling” wire mesh cylinder for wound fluid (WF) collection
The implantation of wire mesh cylinder (stainless steel; 2.5 cm length & 0.8 cm diameter) and wound fluid harvest was performed as described previously {Roy, 2006 #285}. After anesthesia, midline incision (1 cm or smaller) was made on shaved skin with a scalpel. Small subcutaneous pockets were created by blunt dissection. Two wire mesh stainless steel cylinders were inserted into each pocket. Sutures and staples were used to close the incisions. After three days of implantation wound fluid was harvested for analysis.
Secondary-intention excisional dermal splinted murine wound model
Contraction of excisional murine wounds were limited by the application on a split so that the wound could heal through the processes of granulation and re-epithelialization (15). Following anesthesia applied as isoflurane inhalation, two 6 mm full-thickness (skin and panniculus carnosus) excisional wounds were placed on the dorsal skin (shaved and cleaned using betadine), equidistant from the midline and adjacent to the four limbs. A donut-shaped splint with a 8 mm inner diameter created from a 0.5 mm-thick silicone sheet (Grace Bio-Laboratories, Bend, OR) was placed such that the wound was centered within the splint. To affix the splint to the skin, an immediate-bonding adhesive was used followed by interrupted 6–0 nylon sutures (Ethicon, Inc., Somerville, NJ). The wound was covered with semi-occlusive dressing (Tegaderm, 3M, St. Paul, MN). Recombinant mouse OSM was injected under the tegaderm as needed. All animal studies were approved by the OSU Institutional Animal Care and Use Committee (IACUC). Determination of wound area. The imaging of wounds was performed using a digital camera (Canon PowerShot G6). The wound area was determined by planimetry using ImageJ software as described (16).
GeneChip Probe Array Analyses
RNA extraction, target labeling, GeneChip and data analysis were performed as described previously (16–19). In brief, GeneChip IVT Labeling Kit (Affymetrix, Santa Clara, CA) in vitro transcription (IVT) reaction was used to generate biotinylated cRNA from RNA samples. The samples were hybridized to Affymetrix Human Genome U133 Plus 2.0 Array. The arrays were washed, stained with streptavidin-phycoerythrin and scanned with the GeneArray scanner (Affymetrix) in our own facilities as described earlier (17, 18, 20, 21). Data analyses. Data acquisition and image processing was performed using GCOS (Gene Chip Operating Software, Affymetrix). The expression data have been submitted to the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) with the series accession number GSE36995. Raw data were analyzed using Genespring GX (Agilent, Santa Clara CA). Additional processing of data was performed using dChip software (Harvard University) (17, 18, 20, 21).
ELISA
Levels of OSM (R & D Systems, Minneapolis, MN), PGE2 and PGE-M (Cayman Chemicals, Ann Arbor, MI) in wound fluid were measured using commercially available ELISA kits. The levels of OSM, PGE2 and PGE-M in wound fluid were normalized against albumin concentration in the fluid. Albumin levels were determined by ELISA (AssayPro, St. Charles, MO). For measurement of OSM produced by mϕ, cells were seeded in 6-well or 12-well plates and cultured in RPMI 1640 medium containing 10% heat-inactivated bovine serum for 24h under standard culture conditions. After 24h, the culture media was collected and OSM levels were measured using ELISA as described. Phospho-Axl, total Axl and cAMP levels were measured from cell lysates using a sandwich ELISA (R& D Systems, Minneapolis, MN).
Reverse transcription and quantitative RT-PCR (qPCR)
Total RNA was extracted using the mirVana RNA isolation kit (Ambion, Austin, TX), according to manufacturer’s instructions. mRNA was quantified by real-time or quantitative (Q) PCR assay using the double-stranded DNA binding dye SYBR Green-I as described previously (16, 19).
Phospho-RTK array
RTK antibody arrays were purchased from R&D Systems (#ARY-001, Minneapolis, MN). Cell harvest, sample preparation and RTK array assay were performed as recommended by the manufacturer (11).
siRNA delivery
DharmaFECT (Dharmacon RNA technologies, Lafayette, CO) was used to transfect cells with 100nM siRNA pool (Dharmacon RNA technologies, Lafayette, CO) for 48h as described (22). For control, siControl non-targeting siRNA pool (mixture of 4 siRNA, designed to have ≥4 mismatches with the gene) was used.
Analysis of specific binding of AP-1 to DNA
Nuclear protein extracts of cells were prepared using the nuclear extract kit (Active Motif, Carlsbad, CA) according to manufacturer's instructions. Binding of Fos and Jun family of proteins to their consensus sites was determined using a ELISA-based Trans-AM AP-1 kit (Active Motif, Carlsbad, CA).
Statistics
In vitro data are reported as mean ± SD of 3–6 experiments as indicated in respective figure legends. Comparisons among multiple groups were tested using analysis of variance (ANOVA). p<0.05 was considered statistically significant. For animal studies, data are reported as mean ± SD of at least 3–4 animals as indicated. Given the small sample size, Mann–Whitney or Kruskal–Wallis one-way analysis of variance tests was performed to test significance (p<0.05) of difference between means. For wound fluid studies, data from fifteen human subjects (n = 15) have been presented (Table 1).
RESULTS
This work developed a novel approach to isolate and culture functionally intact mϕ from chronic wounds of human subjects (Table 1). To compare the wound mϕ with corresponding mϕ derived from peripheral blood monocytes (MDMs), transcriptome profiling (GeneChip™ Affymetrix) was performed. OSM was among the top-ranked differentially expressed genes that were highly up-regulated in wound mϕ compared to MDMs. A ~9-fold induction in the expression of OSM was observed in wound mϕ compared to pair-matched blood-derived MDM of the same person (Figure 1A). To test the validity of OSM outcomes as evident in profiling data, wound mϕ and MDM were isolated from patients and cultured overnight. Consistent with GeneChip™ data, the level of OSM protein released by wound mϕ was multi-fold higher (~3 fold) compared to that released by peripheral blood MDM of the same individual (Figure 1B). Consistent with elevated production of OSM by cultured wound mϕ, wound-fluid derived from chronic wounds contained elevated levels of OSM compared to pair-matched blood plasma samples from the same patients (Figure 1C). PGE2 is a known inducer of OSM expression in mϕ (23). High levels of PGE2 were noted in wound fluids compared to pair-matched blood plasma obtained from chronic wound patients (Figure 1D). In vivo, PGE2 is rapidly metabolized to 13,14-dihydro-15-keto PGE2 (PGE-M) (24). Higher level of PGE-M was also detected in wound-fluid compared to that in matched plasma samples (Figure 1E). Plasma levels of PGE-M in humans are known to be in the range of 10–100 pg/ml (24). Compared to matched plasma, we detected ~20-fold higher levels of PGE-M in wound-fluid obtained from chronic wounds (Figure 1E). Of interest in this context is the observation that OSM may induce PGE2 (25). In the wound microenvironment, PGE2 may be contributed by a number of cells including mϕ (26). To test whether wound mϕ produce PGE2, levels of this eicosanoid were measured in wound- mϕ culture media. Multifold higher levels of PGE2 were detected in such culture media as compared to that of media hosting MDM (Figure 1F). This observation recognizes wound mϕ as a direct source of PGE2 in the wound microenvironment.
Figure 1. Oncostatin M (OSM) as one of the highly expressed genes in mϕ from human chronic wounds.
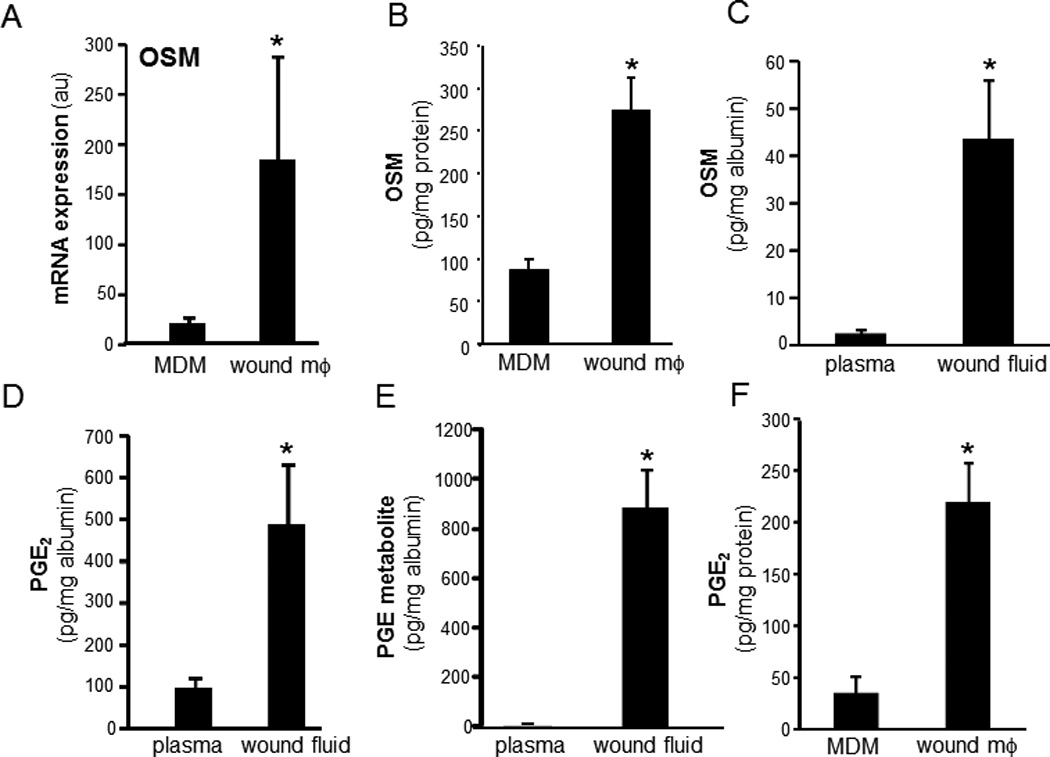
Wound site macrophage (wound mϕ) were isolated from human subjects with chronic wounds. Matching blood monocyte derived macrophages (MDM) were obtained as described in methods from same subjects. A, Expression levels of OSM gene using GeneChip™ analysis. GeneChip™ expression values were normalized using global scaling approach. *, p<0.05 (n=3) compared to matched MDM. B, OSM expression data from GeneChip™ analysis was independently verified using ELISA. Wound site mϕ and matching MDM were cultured for 24h. OSM released by the cells in media was measured by ELISA. p<0.05 (n=3) matched MDM. C–E. OSM, PGE2 and its metabolite 13,14-dihydro-15-keto PGE2 (PGE-M) are abundant in fluid from human chronic wounds. Fluid was obtained from chronic wounds of human subjects. The levels of C, OSM, D, PGE2 and E, PGE-M were determined using ELISA from chronic human wound fluid and matching plasma samples from the patients. The levels were normalized to the total albumin level in the fluid/plasma.* p<0.01 (n=15) compared to plasma. F, wound-site mϕ and matching MDMs were cultured for 24h. PGE2 released by the cells was measured in culture media using ELISA. p<0.05 (n=3) matched MDM.
To characterize the mechanism of PGE2- induced OSM expression, the human monocytic THP-1 cells were used. THP-1 cells were differentiated to mϕ using phorbol-12-myristate-13-acetate (PMA, 20 ng/ml, 48h). PGE2 dose-dependently induced OSM protein expression (Figure 2A). Following PGE2 treatment, OSM gene and protein were significantly unregulated at 6h and 24h post-treatment, respectively (Figure 2B–C). In addition to PGE2, PGE-M also induced OSM expression in a dose-dependent manner (Figure 2D). The expression of OSM mRNA peaked at 48h post-treatment (Figure 2E). Consistent with findings using THP-1, PGE2 potently induced OSM in MDM demonstrating that the finding is applicable to cells of monocytic lineage (Figure 2F). PGE2 is known to signal via G-protein-coupled receptors designated as EP1, EP2, EP3, and EP4 (27). To delineate the PGE2-inducible signaling pathway that causes OSM expression in mature mϕ EP receptors were screened for involvement. In mature mϕ, EP4 represented the most abundant EP receptor (Figure 3A). Next, we addressed the significance of EP4 in PGE2-induced OSM expression. Both inhibition of EP4 using the pharmacologic inhibitor L-161,982 (28) as well as knockdown of EP4 expression inhibited PGE2-induced OSM expression (Figure 3B–C). Transfection of cells with EP4 siRNA was successful in achieving ~70% knock-down of EP4 mRNA expression (Figure 3D). Activation of EP4 is known to induce intracellular cAMP as second messenger (29). We observed that PGE2 rapidly and significantly induced cAMP levels in MDM (Figure 4A). Studies testing the plausible involvement of EP2 in mediating PGE2 signaling demonstrated that selective activation of the EP2 receptor using Butaprost does not elevate cellular cAMP in MDM (Figure 4A) arguing against the potential involvement of EP2 receptors in PGE2-induced cAMP signaling. It may thus be concluded that abundantly expressed EP4 and not EP2 is the key receptor involved in PGE2 signaling in MDM. We observed that the cAMP analog dibutyryl adenosine 3’,5’cyclic monophosphate (db-cAMP) was effective in inducing OSM expression in human mϕ (Figure 4B). Furthermore, the adenylate cyclase agonist forskolin also induced OSM expression (Figure 4B). These data collectively establish that EP4 receptor mediates PGE2-induced OSM expression through adenylate cyclase and cAMP signaling.
Figure 2. PGE2 and PGE-M induced OSM gene and protein expression in human mϕ.
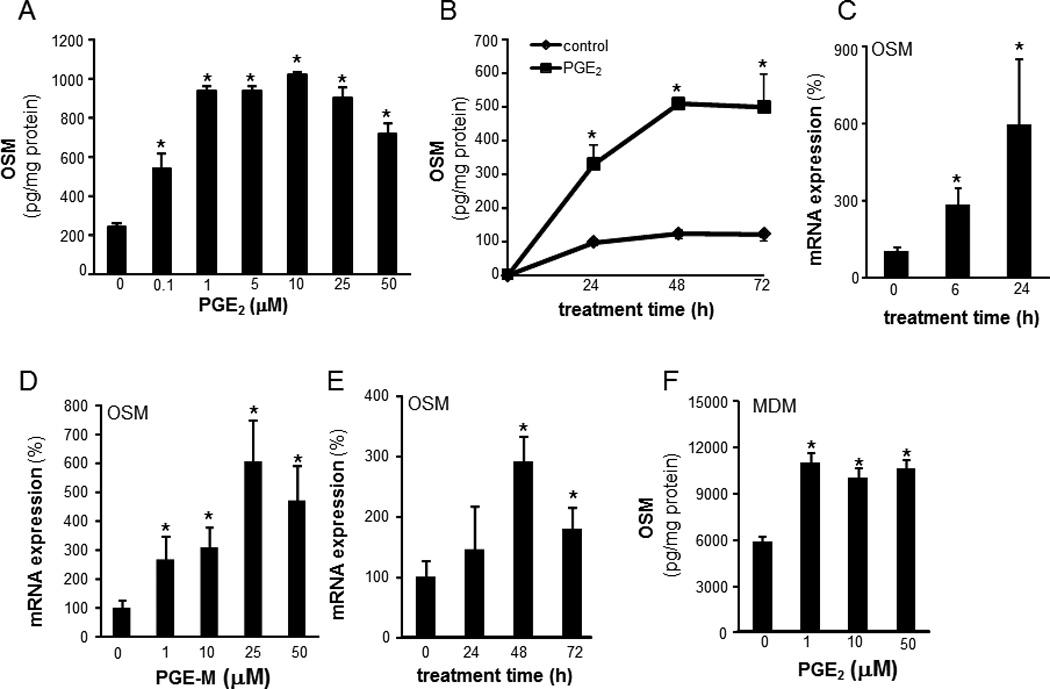
The human monocytic cell line THP-1 cells were differentiated to mϕ with phorbol-12-myristate-13-acetate (PMA, 20 ng/ml, 48h). A, cells were treated with a range of PGE2 concentrations (0.1 –50µM). OSM level in media was measured after following 72h of treatment. Data are mean ± SD (n=5).*, p<0.05 compared to control. B, cells were treated with PGE2 (10 µm) for 0–72h. OSM protein in media was measured using ELISA. OSM levels were normalized to total cellular protein in the culture. Data are mean ± SD (n=4).*, p<0.05 compared to control. C, cells were treated with PGE2 (10 µm) for 0–24h. OSM gene expression was measured using qPCR. Data are mean ± SD (n=4).*, p<0.05 compared to control. D, cells were treated with varying concentrations (1–50 µM) of PGE-M for 72h. OSM gene expression was measured using qPCR. Data are mean ± SD (n=4).*, p<0.05 compared to 0 µM. E, cells were treated with PGE-M (25 µm) for 0–72h. OSM mRNA expression was measured Data are mean ± SD (n=4).*, p<0.05 compared to 0h. F, MDM were treated with a range of PGE2 concentrations (0.1 –50µM). OSM level in media was measured following 72h of treatment. Data are mean ± SD (n=5).*, p<0.05 compared to control.
Figure 3. Role of PGE2 receptor expression and function in PGE2-induced OSM expression.
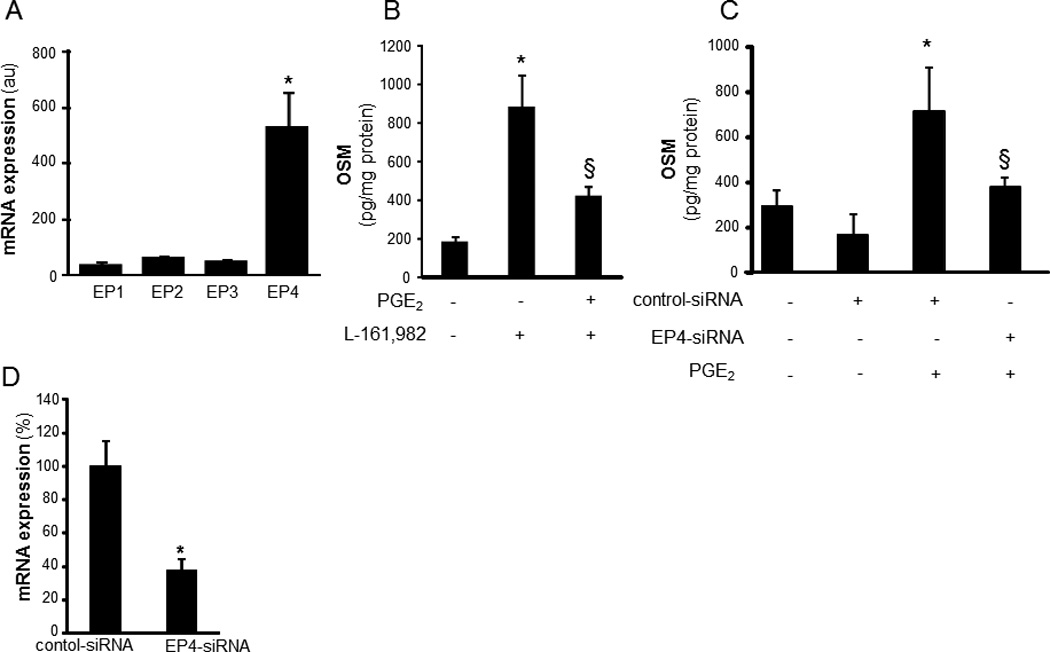
A, relative expression of four PGE2 receptors (EP1-EP4) in differentiated THP-1 mϕ. The mRNA expression of EP receptors was determined using qPCR. Data are mean ±SD (n=4). *, p<0.01 compared to EP1-EP3. B, Cells were pretreated (1h) with L-161,982 (EP4 antagonist, 100 nM) followed by treatment with PGE2 (10 µM, 72h). Level of OSM in culture media was measured using ELISA. Data are mean ± SD (n=6). *, p<0.01 compared to untreated cells; §, p<0.05 compared to PGE2 treated cells. C, cells were subjected to EP4 knockdown (EP4-siRNA) or not (control siRNA, con-siRNA). Following 72h of siRNA transfection, the cells were treated with PGE2 (10 µM, 72h). Level of OSM in culture media was measured using ELISA. Data are mean ± SD *(n=3). p<0.01 compared to PGE2 untreated cells; §, p<0.05 compared to PGE2 treated & con-siRNA treated cells. D, EP4 knockdown was achieved by transfection of cells with EP4 siRNA versus control siRNA. EP4 mRNA levels were determined in cells using qPCR. Data are mean ± SD (n=3). *, p<0.05 compared to control.
Figure 4. Adenylate cyclase, cAMP and receptor tyrosine kinase (RTK) are involved in PGE2 induced OSM expression.
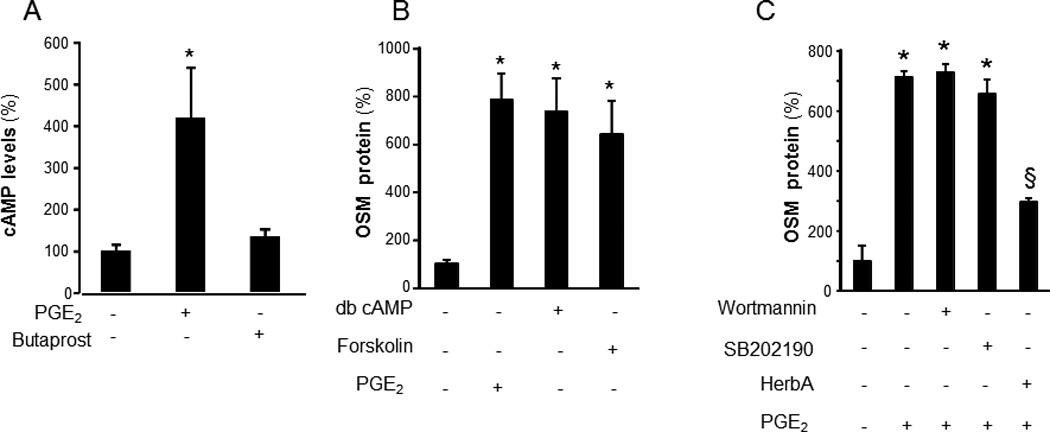
A, Blood monocyte derived macrophages (MDM) were treated with PGE2 (1 µM) or Butaprost (EP2 agonist, 1 µM) for 5 min. Cellular levels of cAMP were determined using ELISA. Data are mean ± SD (n=4). *, p<0.01 compared to untreated cells. B, Differentiated human THP-1 mϕ were treated with an activator of adenylate cyclase, forskolin (100µM), dibutyryl adenosine 3’,5’cyclic monophosphate (db-cAMP, 100µM) or PGE2 (10µM) for 72h. C, cells were pretreated with SB202190 (5 µM), p38 MAPK inhibitor, PI3K inhibitor wortmanin (50 nM) or RTK inhibitors herbimycin A (5 µg/ml, HerbA) for 30 min followed by treatment with PGE2 for 72h. The level of OSM in culture media in both B and C were measured using ELISA. Data are mean ± SD (n=4). *, p<0.01 compared to untreated cells. §, p<0.05 compared to PGE2 treated cells.
Efforts to further characterize the pathways involved in PGE2-induced OSM production led to the observation that the RTK inhibitor herbimycin A potently inhibited PGE2-induced OSM expression (Figure 4C). To investigate which specific RTKs are activated as a result of PGE2 treatment, we employed an antibody array that allows simultaneous assessment of the phosphorylation status of 42 RTKs (human phospho-RTK Array, ARY001, R&D Systems, Minneapolis, MN). This approach led to the observation that PGE2 rapidly (30 min) induces Axl phosphorylation (Figures 5A). This finding was verified by sandwich ELISA using antibodies against phospho-Axl and total Axl. As observed with RTK array screening, potent induction in phosphorylation of Axl was observed following 30min of PGE2 treatment (Figure 5B). Consistently, PGE2 induced Axl phosphorylation in MDM (Figure 5C). Comparable to PGE2, treatment of cells with dbcAMP also resulted in phosphorylation of Axl suggesting that cAMP is sufficient to induce Axl phosphorylation. These data unveil an interesting cross-talk between cAMP and RTK pathways. Knockdown of Axl inhibited PGE2-induced OSM expression demonstrating that Axl is directly implicated in PGE2-induced OSM expression (Figure 5C–E).
Figure 5. Receptor tyrosine kinase Axl is phosphorylated following treatment of mϕ with PGE2: Involvement of Axl in regulation of PGE2 mediated OSM production.
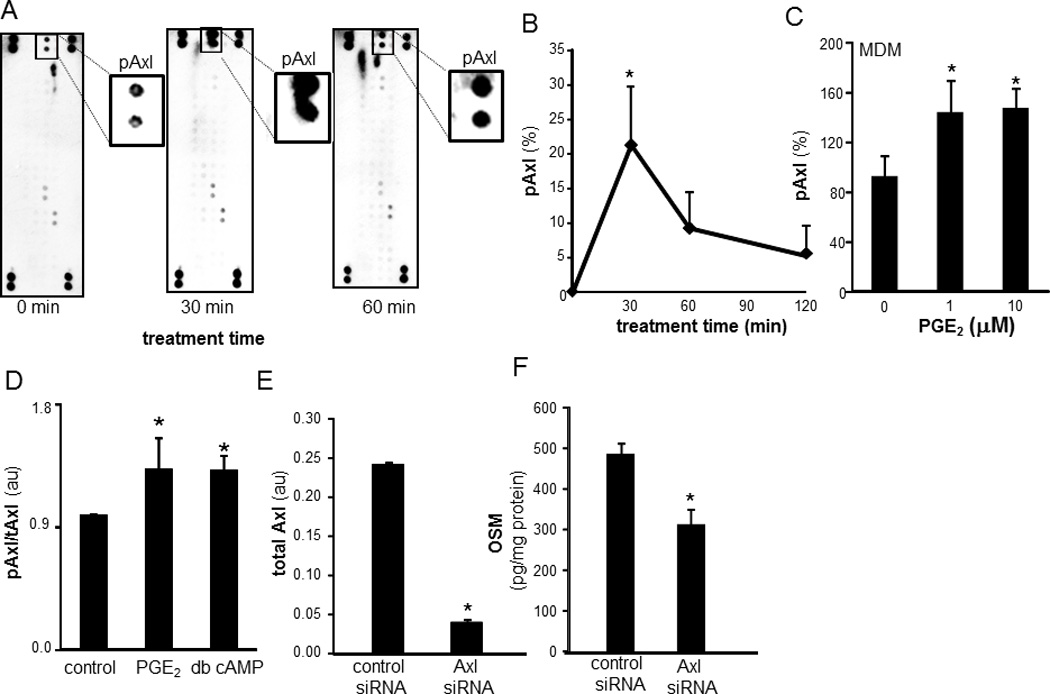
A, Differentiated human THP-1 mϕ were treated with PGE2 (10 µM) for 30 and 60 min. Relative phosphorylation of 42 RTK was screened using phospho-RTK array. The screening revealed PGE2 specifically phosphorylates Axl (pAxl) following 30 min of PGE2 treatment. Insets are zoomed image of pAxl spots in the array. B, The array data was verified using sandwich ELISA to measure tyrosine-phosphorylated Axl in cell lysates. pAxl data was normalized against total Axl (tAxl) present in cell lysates. Data are expressed as % change compared to time 0h. Data are mean ± SD (n=4). *, p<0.05 compared to 0h. C, MDM were treated with PGE2 (1–10 µM) for 30 min. ELISA was used to measure tyrosine-phosphorylated Axl in cell lysates. Data are mean ± SD (n=3). *, p<0.05 compared to control. D, Differentiated human THP-1 mϕ were treated with PGE2 (10 µM) and db-cAMP (100µm) for 30 min. pAxl/tAxl levels were detected using sandwich ELISA. Data are mean ± SD (n=4). *, p<0.05 compared to control. E, Axl knock-down was achieved by transfection of cells with Axl siRNA versus control siRNA. Total Axl levels were determined in cell lysates using ELISA. Data are mean ± SD (n=3). *, p<0.05 compared to control. F, Axl knockdown cells with treated using PGE2 (10 µM) for 72h. The level of OSM in culture media was measured using ELISA. Data are mean ± SD (n=4). *, p<0.05 compared to control siRNA transfected cells.
AP-1 binding sites are present on the human OSM promoter (30). We observed that DNA binding activity of AP-1 was induced in nuclear extracts of PGE2-treated cells. Maximal activation was observed following 1h of PGE2 treatment (Figure 6A). The timeline of induction of RTK phosphorylation (30 min) and AP-1 DNA binding activity (60 min) suggested that phosphorylation of Axl is upstream to induction of AP-1 transactivation. Of the AP-1 proteins, FosB, JunD and Fra-1 were observed to be not sensitive to PGE2 treatment. However, cFos, cJun and Jun B were identified as PGE2-sensitive AP-1 proteins (Figures 6B–D). PGE2-induced DNA binding activity of AP-1 was verified using a lower concentration (1µM) of PGE2 (Figure 7A). PGE2-induced activation of AP-1 was comparable to the activation by a classical phorbol ester inducer (phorbol 12-myristate 13-acetate, PMA) (31) (Figure 7B). Specific inhibition of Axl using R428 (SYN-1131, Synkinase) (32) significantly blunted PGE2-mediated AP-1 activation (Figure 7B). siRNA knockdown studies demonstrated that Axl specifically regulated PGE2-induced activation of Jun (cJun and JunB) proteins (Figure 7C–D), but not that of cFos (data not shown).
Figure 6. Induction of AP-1 DNA binding activity by PGE2.
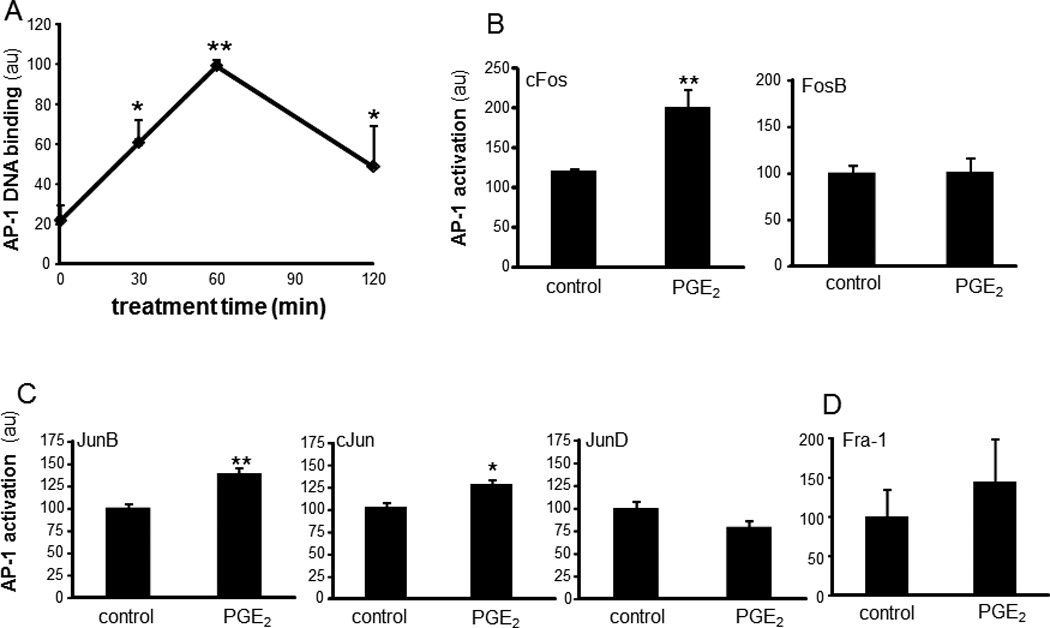
A, An ELISA-based (Trans-AM®) method was used to analyze DNA binding activities of AP-1 from differentiated THP-1 mϕ treated with PGE2 (10 µM) for 0–120 min. B–D, DNA binding activities of AP-1 family of proteins B, Fos (cFos, FosB), C, Jun (JunB, cJun, JunD) and D, Fra-1 in nuclear proteins extracts from cells treated with PGE2 (10 µM) for 60 min. Data are mean ± SD (n=3). *, p<0.05; ** p<0.01 compared to control.
Figure 7. cAMP and Axl regulate cJun and JunB DNA binding activities.
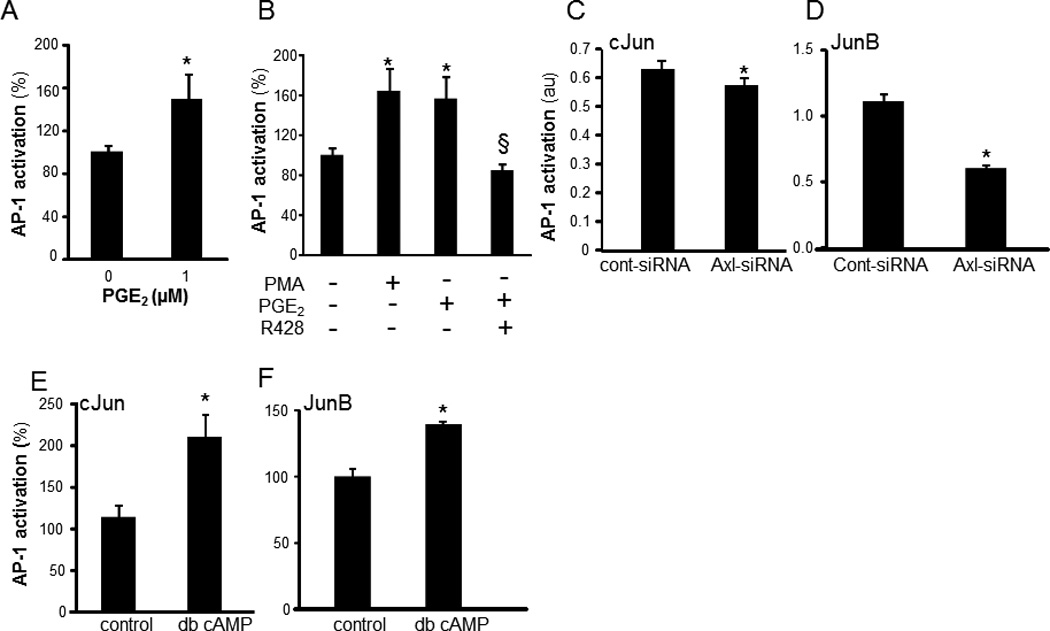
A, DNA binding activity of AP-1 in THP-1 differentiated mϕ treated with PGE2 (1 µM) for 60 min. Data are mean ± SD (n=3). *, p<0.05 compared to control. B, cells were pretreated (1h) with R428 (Axl inhibitor, SYN-1131, 1µM) followed by activation with PGE2 (10 µM, 1h). DNA binding activity of AP-1 was determined. PMA (1 µM, 1h) was used as a classical inducer of AP-1 activity to compare the extent of PGE2-induced AP-1 activation. C–D, Axl knockdown in differentiated human THP-1 mϕ (Axl-siRNA) or control (cont-siRNA) were treated with PGE2 for 1h. C, cJun and D, JunB DNA binding activities were determined from nuclear proteins using Trans-AM® assay. Data are mean ± SD (n=3). * indicated p<0.05 compared to controls. E–F, Cells were treated with db-cAMP (100µM, 1h). E, cJun and F, JunB DNA binding activities were determined from nuclear proteins using Trans-AM® assay. Data are mean ± SD (n=3). * indicate p<0.05 compared to control.
Consistent with the outcome of Axl knockdown studies, dbcAMP resulted in activation of cJun and JunB but not of cFos (Figure 7E–F). Taken together, these findings indicate that PGE2 elicits convergent RTK and cAMP signaling which culminates in OSM expression. To test whether PGE2 may induce OSM production by wound mϕ in vivo, PVA sponges containing either PGE2 or vehicle (ethanol) were subcutaneously implanted on the back of C57BL/6 mice. Potent induction of OSM mRNA expression was observed in mϕ isolated from PVA sponges containing PGE2 (2 nmols/sponge; Figure 8A). Consistently, PGE2 treatment also induced OSM production in human wound mϕ (Figure 8B). Human wound mϕ demonstrated elevated expression of EP4 receptor (Figure 8C). Exposure to PGE2 resulted in increased phosphorylation of Axl in wound mϕ suggesting that PGE2–induced OSM pathway involves EP4-RTK signaling as observed in THP-1 cells (Figure 8 D–E). Next, the significance of wound-fluid PGE2 and cyclooxygenase-derived prostaglandins in induction of wound-mϕ OSM was determined. Treatment of MDM with culture media containing specific dilutions of sterile-filtered wound fluid potently induced OSM expression (Figure 9A). Matching volume of human AB serum (HSA) was added to the culture medium as a control for wound-fluid. Treatment of wound fluid with anti-PGE2 antibody resulted in sequestration of PGE2 from wound fluid. To determine if such sequestration blocks biological activity of PGE2, we determined whether wound fluid may induce cAMP levels and whether anti-PGE2 treatment is able to block such effect. These data demonstrate that level as well as biological activity of PGE2 was effectively blocked using anti-PGE2 antibody (Figure 9B–C). Such blockade of PGE2 resulted in significant inhibition of wound fluid-induced OSM expression in MDM (Figure 9D). Murine wound mϕ obtained from subcutaneously implanted PVA sponges showed higher expression of OSM mRNA compared to bone marrow derived murine macrophages (BMDM) (Figure 9E). Oral supplementation (gavage) of mice with indomethacin (1mg.kg−1.day−1) or saline (control) for 5 consecutive days, resulted in attenuation of OSM mRNA expression in murine wound mϕ as well as attenuated level of PGE2 in wound fluid suggesting that endogenous cyclooxygenase derived prostaglandins are involved in inducing OSM in wound macrophages (Figure 9E–F).
Figure 8. PGE2 mediated induction of OSM and p-Axl in human and mice wound mϕ.
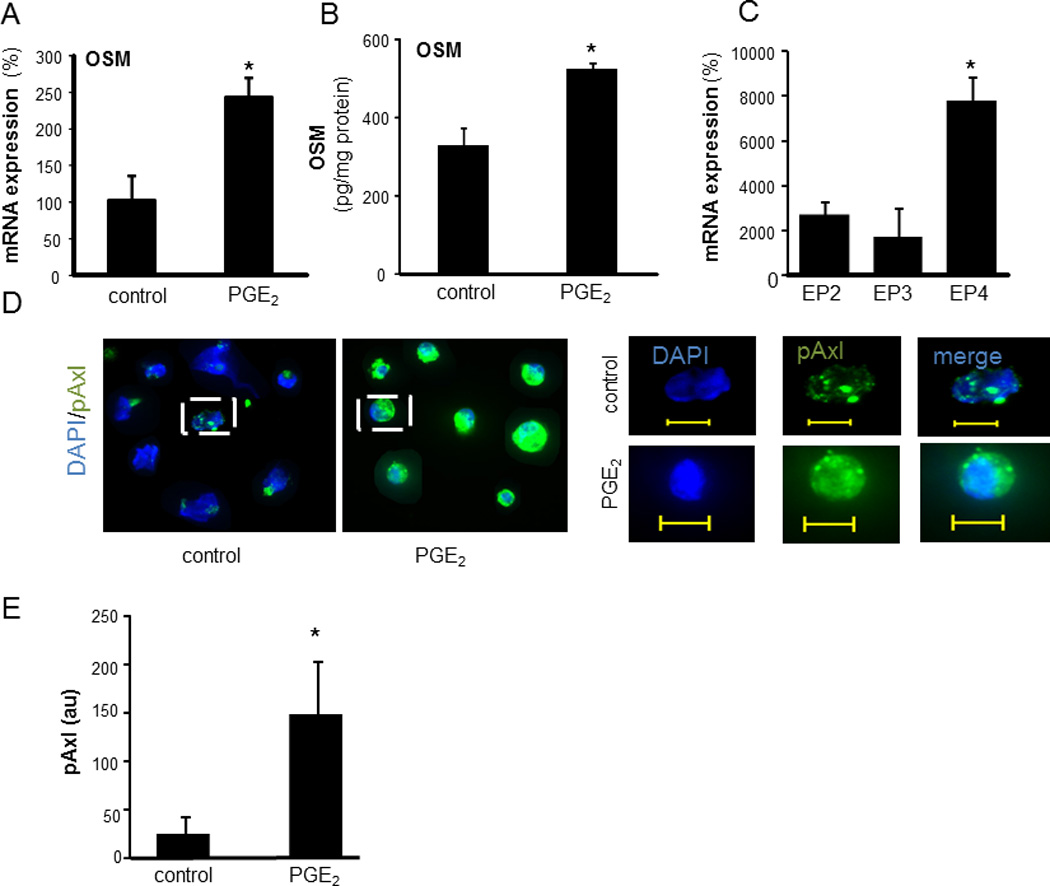
A, PVA sponges containing 20 µl of 0.1 mM PGE2 (2 nmols/sponge) or ethanol (matching volume, 20 µl) were implanted subcutaneously at the back of mice. Wound mϕ were harvested 3 days post implantation and OSM mRNA expression was determined using qPCR. Data are mean ± SD (n=3) *, p<0.05 compared to control. B, The mϕ from human chronic wounds were isolated and treated with PGE2 (10 µM, 24h). Controls were treated with matching volume of ethanol. OSM levels in culture media of wound mϕ was measured using ELISA. Data were normalized to total protein levels. Data are mean ± SD (n=3). *, p<0.05 compared to control. C, relative expression of EP receptors in human wound mϕ. mRNA expression of EP receptors was determined using qPCR. Data are mean ±SD (n=4). *, p<0.01 compared to EP2-EP3. D, Human wound mϕ were stained with anti-pAxl antibody (green) and DAPI (blue nucleus). Increased expression of pAxl in wound mϕ treated with PGE2 (10 µM, 30 min) can be seen. Left: low magnification images; Right: high magnification images of the cell marked in respective left panel. Scale bar = 10 µM. E, Quantification of the fluorescence signal shown in D using Axiovision (Zeiss).
Figure 9. Wound-fluid PGE2 and cyclooxygenase derived prostaglandin induced OSM in human and murine wound mϕ.
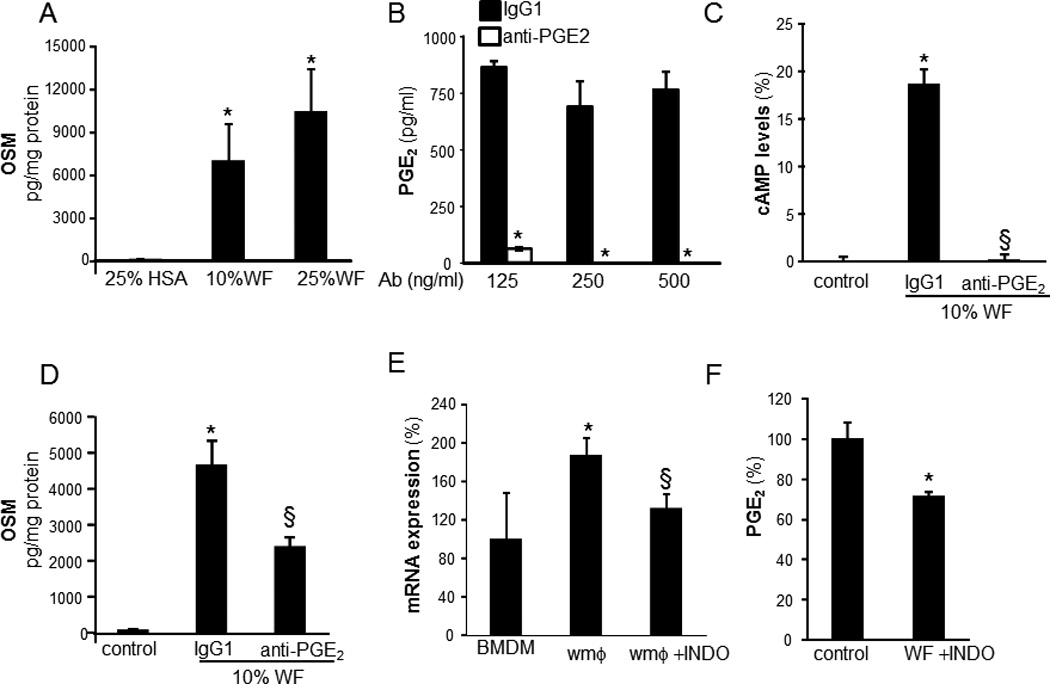
A, human MDM were treated with varying dilutions (0–25%, v/v, 72h) of wound fluid (WF) derived from human chronic wounds. WF was sterile filtered and added directly to the MDM culture medium. For control, matching volume of human AB serum (HSA) was added to the culture medium. OSM levels were measured in culture media 72h following treatment with WF. Data are mean ± SD (n=4).*, p<0.05 compared to control. B, to determine the dose of anti-PGE2 antibody (Ab, clone 2B5) that may effectively sequester PGE2 from WF, varying (125–500 ng/ml) concentrations of anti-PGE2 ab or equivalent amount of IgG1 was added to the WF. PGE2 level in WF was determined using ELISA. Data are mean ± SD (n=4).*, p<0.05 compared to control. C–D, WF (10% v/v) was pre-treated with anti-PGE2 or corresponding IgG1 (250 ng/ml) to sequester PGE2 followed by treatment of MDM with WF (10% v/v) for 72h. C, OSM or D, cAMP level in culture medium was determined using ELISA. Data are mean ± SD (n=3).*, p<0.05 compared to control. D–E, Mice (C56Bl6, 9 week old) were supplemented (intragastric) with indomethacin (INDO, 1mg.kg−1.day−1) or vehicle (saline) for 5 consecutive days. PVA sponges or Hunt-Schilling wire mesh cylinders were subcutaneously implanted at the back of mice on the third day following supplementation. D, Wound mϕ (PVA sponges) and wound fluid (WF, wire mesh cylinders) were harvested 3 days post-implantation and D, OSM mRNA expression (mϕ) or E, PGE2 levels (WF) was determined using qPCR or ELISA, respectively. Bone marrow derived macrophages (BMDM) were obtained from INDO untreated mice. Data are mean ± SD (n=3) *, p<0.05 compared to BMDM. §, p<0.05 compared to wound mϕ.
To elucidate the significance of OSM on the inflammatory properties of mϕ, human mϕ were treated with human recombinant OSM. The response of human mϕ to OSM in a setting of LPS-induced inflammation was evaluated using a multiplex cytokine array approach (Figure 10A). OSM clearly modified the inflammatory response of LPS- treated mϕ. Of note, a decline in LPS-induced expression of pro-inflammatory cytokines, TNFα and IL-1β was observed. This array finding was verified independently using ELISA supporting plausible anti-inflammatory properties of OSM (Figure 10B). To test the significance of OSM in wound inflammation in vivo, murine excisional wounds were investigated. Treatment of such wounds with recombinant murine OSM during the early inflammatory phase resulted in increased abundance of tissue OSM in treated wounds demonstrating successful delivery of OSM (Figure 10C). Such treatment proved to be anti-inflammatory by suppressing the expression of pro-inflammatory cytokines TNFα and IL1β (Figure 10D). OSM treated wound also showed improved closure outcomes at day 3 (Figure 10E).
Figure 10. Anti-inflammatory activity of OSM in macrophages and wound inflammation.
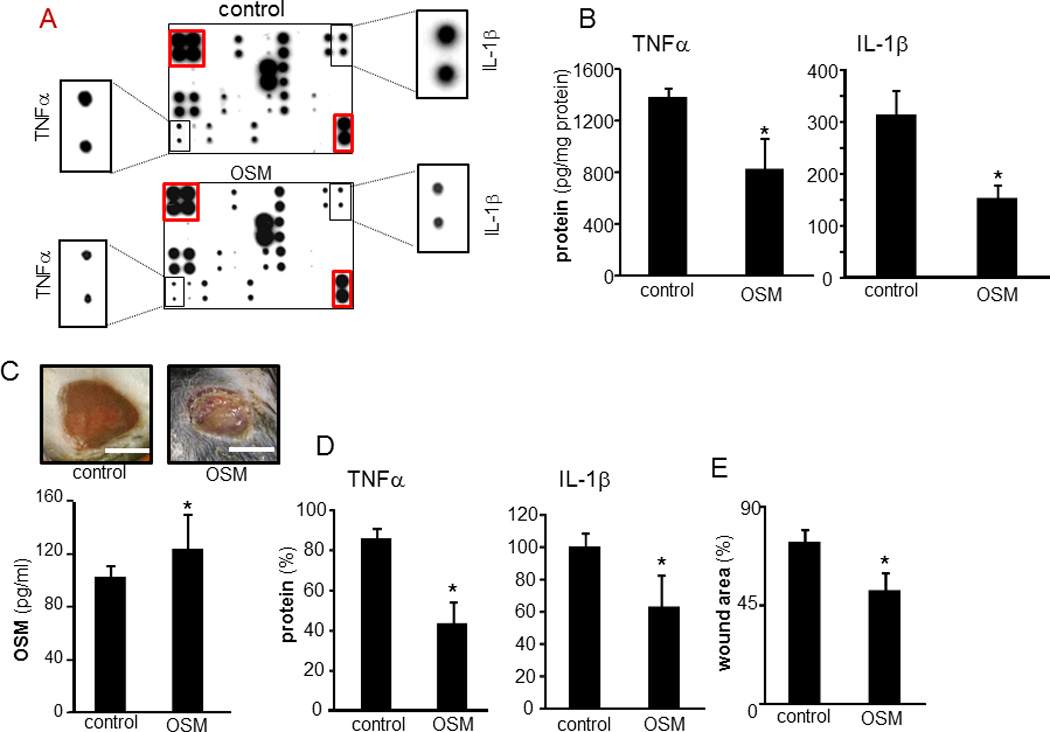
A–B, Human mϕ treated with OSM (25 ng/ml, 72h) followed by treatment with LPS (1 µg/ml, 24h). A, a multiplex cytokine array was performed to evaluate effect of OSM on LPS-induced inflammatory response in human mϕ. The spots marked with red line are housekeeping controls. The zoom of the respective spots for IL-1β and TNFα from each array are shown as insets. B, The levels of LPS-induced TNFα and IL-1β in OSM pretreated mϕ was independently measured using ELISA. Data are mean ± SD (n=3) *, p<0.05 compared to control (con). C–E, Effect of OSM on wound inflammation was evaluated using a murine excisional wound model. C, Representative day 3 post wounding images from excisional wounds treated with recombinant mouse OSM (1.25 µg.15µl−1.wound−1) in early inflammatory phase (0–3d post wounding). Control wounds received vehicle only. OSM levels on day 3 post wounding in wounds tissue treated with recombinant OSM. D, TNFα and IL-1β in OSM pretreated excisional wound on day 3 post wounding. Data are mean ± SD (n=3) *, p<0.05 compared to control. E, wound area in OSM pretreated excisional wound on day 3 post wounding. Wound area is presented as % compared to wound size at day 0 post wounding. Data are mean ± SD (n=3) *, p<0.05 compared to control.
DISCUSSION
The chronic wound microenvironment featuring lower pH, high proteolytic activity, high abundance of lactate and often hosting infection is generally non-conducive to healing (33). Dynamic reciprocity, as an ongoing, bidirectional interaction among cells and their surrounding microenvironment, is viewed as a key contributor to wound healing wherein biochemical, biophysical, and cellular responses to injury play pivotal roles in regulating healing responses to injury (34). Monocyte and mϕ functions are highly responsive to their microenvironment. While this is well characterized in the context of cancer biology (35, 36), there is a void of information on how the human chronic wound microenvironment modifies wound mϕ biology. Current understanding of chronic wound inflammation is primarily based on information from MDM originating from peripheral blood. These cells have never been exposed to the wound microenvironment and therefore are unlikely to be primed by such conditions. Indeed, changes in mϕ phenotype associated with the progression of wound healing do not follow the current mϕ classifications (6, 7). The need to understand unique characteristics of wound mϕ are therefore compelling and would help elucidate novel pathways implicated in determining wound inflammation outcomes.
OSM is an IL-6 family protein discovered in the supernatant of human monocytic cells in 1986 and known to be produced by activated mϕ (9). The anti-inflammatory properties of mϕ-derived OSM are currently unfolding (37). Although the beneficial properties of OSM against cancer is documented (38), this work presents first evidence demonstrating that wound mϕ over-produce OSM and that such OSM displays anti-inflammatory functions. High levels of OSM have been reported in several unrelated inflammatory scenarios (39, 40). This work reports an abundance of OSM in wound fluid. OSM expression in mϕ is known to be induced by factors relevant to the wound-site such as pathogenic bacteria (41), blood coagulation factors such as thrombin (42) and complement component C5a (43). Among lipid mediators, PGE2 is recognized as a potent inducer of OSM expression (23). Prostaglandins are lipid autacoids derived from arachidonic acid. They both sustain homeostatic functions and mediate pathogenic mechanisms, including the inflammatory response (44). PGE2 is the most abundant eicosanoid, lipid mediators generated through oxidative pathways from arachidonic acid. Anti-inflammatory properties of PGE2 are currently unfolding in the context of tumor biology (45). PGE2 helps resolve inflammation by targeting the NF-κB pathway (46). Indeed, administration of esterified PGE2 during the early phase of wound healing showed anti-inflammatory outcomes (47). Abundance of PGE2 at the wound site has been previously reported (48, 49) and findings of the current study support such observations by reporting high levels of PGE2 in the wound fluid. Part of this PGE2 in the wound microenvironment is contributed by wound mϕ. Findings of this study extend that observation to establish a cause and effect relationship between PGE2 at the wound site and induction of OSM in wound mϕ. OSM levels in mϕ derived from human chronic wounds as well as from acute murine wounds were elevated in response to PGE2 treatment. That PGE2 may induce anti-inflammatory responses in mϕ by inhibiting adhesion molecule expression is already reported (50). The current work provides first evidence demonstrating that at the wound site accumulated PGE2 induces OSM which supports wound healing via anti-inflammatory pathways. PGE2 is known to induce OSM expression in microglia, monocytes, and mϕ of human and murine origin via G-protein-coupled receptors, cAMP and PKA (23). PGE2 biosynthesis is noted to have a central role in skin repair. Topically administered PGE2 (dinoprostone) restored normal wound repair. However, the mechanisms underlying the effects of PGE2 on adult dermal wound healing remain unclear. The current study recognizes PGE2 is a potent inducer of OSM expression in mϕ derived from human chronic wounds.
Secreted PGE2 acts in an autocrine or paracrine manner through its four cognate G protein coupled receptors EP1 to EP4 (51). Mϕ predominantly express EP2 and EP4 receptors that are coupled to G-proteins and signal by stimulating adenylyl cyclase (52). That PGE2 mediated OSM expression involves cAMP signaling has been directly demonstrated in microglial cells as well as in human mϕ (23). The current work provides first evidence directly implicating RTK in PGE2-induced signaling directed at OSM expression. Specifically, findings of the study recognized RTK Axl to be phosphorylated in response to PGE2 treatment. Despite 90% knockdown of Axl levels in cells, a modest (~40%) decrease in the level of OSM was noted. Such an effect may be because of the fact that residual Axl function following Axl knockdown is sufficient to transduce signals towards OSM expression. Long turnover time for pre-existing OSM protein could be a contributing factor as well. Axl receptors are known to serve anti-inflammatory functions by suppressing inflammatory cytokine production (53). Also, loss of Axl results in enhanced inflammatory response (54). A striking general observation of the current study is the cross-talk between RTK and cAMP signaling cascades. Exposure of cells to a cAMP analog led to Axl phosphorylation. Both pathways, RTK as well as cAMP, contributed to PGE2-induced expression OSM.
Axl (also called UFO, ARK, and Tyro7) receptor tyrosine kinases (RTK) are expressed at abnormally high levels in a variety of malignancies and support mitogensis. Axl is activated by authophosphorylation (55). The mitogenic transcription factor AP-1 is known to promote Axl expression (56, 57). Observations reported herein provide first evidence that Axl supports AP-1 transactivation. Given the key role of AP-1 in executing mitogenesis and an established function of Axl in promoting tumor growth, it is understandable that Axl engages AP-1 for downstream signaling. AP-1 complexes are composed of members of the Jun (cJun, JunB, JunD), and Fos (c-Fos, FosB, Fra-1 and Fra-2) families and bind to specific control elements present in the promoters of genes that regulate cell differentiation and proliferation (58). Members of the Fos family heterodimerize with Jun family members, thus forming the AP-1 complex which transcriptionally regulates numerous genes (58). Findings of this study reveal that members of the Jun family, specifically cJun and JunB but not JunD, were induced by PGE2 treatment. The observation that PGE2 may induce Jun proteins is consistent with a previous observation reported in bone derived fibroblasts (59). Both cJun and JunB contain functional JNK docking sites while JunD lacks such site (60) suggesting a likely involvement of JNK in the Axl-induced cJun and JunB activation pathway.
OSM is a late phase cytokine that elicits an anti-inflammatory response by altering the activities of initiators of the inflammatory response (61). Treatment with OSM attenuated severity of LPS-induced joint inflammation (61). Biological functions of OSM are executed through binding of the cytokine to specific OSM receptor subunit β (OSMRβ) (8). In OSMRβ deficient mice, peritoneal inflammation is associated with enhanced recruitment of monocytic cells suggesting an anti-inflammatory role of OSM signaling in limiting monocyte recruitment (62). Production of OSM during early wound inflammatory phase has been linked to PMN (8). The role of mϕ as an inducible source of OSM in the wound milieu as well as the significance of OSM in wound healing remained unknown. This work address both gaps providing a mechanism for how OSM Is induced at the wound site. Furthermore, findings of the current study support that when made available during the early phase of wound healing, OSM elicits an anti-inflammatory response by suppressing the expression of pro-inflammatory cytokines TNFα and IL-1β by wound-site mϕ. Such anti-inflammatory property of OSM is in concordance with previous reports demonstrating in vivo anti-inflammatory effects of OSM (61).
The wound habitat is isolated and distinguished from its immediate surrounding tissue. The wound fluid bathing the wound tissue reflects the wound microenvironment and shapes the functional response of wound-related cells (63). The approach to study mϕ, differentiated from peripheral blood monocytes in a laboratory setting, is limited in its ability to account for interactions between the wound microenvironment and wound-resident mϕ. This work provides first evidence from a direct pair-matched comparison of the MDM versus wound mϕ identifying OSM as a protein differentially overexpressed in wound mϕ. PGE2, recognized as a growth-promoting autocoid for epidermis, is known to be synthesized in excess at the site of wound healing (64). This work demonstrated that wound-site PGE2 induce OSM in wound mϕ via a pathway that involves both cAMP as well as RTK signaling. Finally, the observation that OSM functions as an anti-inflammatory agent at the wound-site introduces a novel element in the overall biology addressing the control of wound inflammation.
Footnotes
REFERENCES
- 1.Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17:763–771. doi: 10.1111/j.1524-475X.2009.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78:71–100. [PMC free article] [PubMed] [Google Scholar]
- 3.Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Muller W, Roers A, Eming SA. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 4.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stout RD, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. J Leukoc Biol. 2010;87:59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brancato SK, Albina JE. Wound macrophages as key regulators of repair origin, phenotype, and function. Am J Pathol. 2011;178:19–25. doi: 10.1016/j.ajpath.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goren I, Kampfer H, Muller E, Schiefelbein D, Pfeilschifter J, Frank S. Oncostatin M expression is functionally connected to neutrophils in the early inflammatory phase of skin repair: implications for normal and diabetes-impaired wounds. J Invest Dermatol. 2006;126:628–637. doi: 10.1038/sj.jid.5700136. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka M, Miyajima A. Oncostatin M, a multifunctional cytokine. Rev Physiol Biochem Pharmacol. 2003;149:39–52. doi: 10.1007/s10254-003-0013-1. [DOI] [PubMed] [Google Scholar]
- 10.Gomez-Lechon MJ. Oncostatin M: signal transduction and biological activity. Life Sci. 1999;65:2019–2030. doi: 10.1016/s0024-3205(99)00296-9. [DOI] [PubMed] [Google Scholar]
- 11.Roy S, Dickerson R, Khanna S, Collard E, Gnyawali U, Gordillo GM, Sen CK. Particulate b-glucan induces TNFa production in wound macrophages via a redox-sensitive NFkB-dependent pathway. Wound Repair and Regeration. 2011;19:411–419. doi: 10.1111/j.1524-475X.2011.00688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, Bhasker V, Gordillo GM, Sen CK, Roy S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;5:e9539. doi: 10.1371/journal.pone.0009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vairo G, Hamilton JA. CSF-1 stimulates Na+K+-ATPase mediated 86Rb+ uptake in mouse bone marrow-derived macrophages. Biochem Biophys Res Commun. 1985;132:430–437. doi: 10.1016/0006-291x(85)91040-x. [DOI] [PubMed] [Google Scholar]
- 14.Davies JQ, Gordon S. Isolation and Culture of Murine Macrophages. In: Helgason CD, Miller CL, editors. basic cell culture protocols. Totowa: Humana press; 2005. pp. 91–103. [Google Scholar]
- 15.Galiano RD, Michaels J, Dobryansky M, Levine JP, Gurtner GC. Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen. 2004;12:485–492. doi: 10.1111/j.1067-1927.2004.12404.x. [DOI] [PubMed] [Google Scholar]
- 16.Roy S, Khanna S, Rink C, Biswas S, Sen CK. Characterization of the acute temporal changes in excisional murine cutaneous wound inflammation by screening of the wound-edge transcriptome. Physiol Genomics. 2008;34:162–184. doi: 10.1152/physiolgenomics.00045.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy S, Khanna S, Kuhn DE, Rink C, Williams WT, Zweier JL, Sen CK. Transcriptome analysis of the ischemia-reperfused remodeling myocardium: temporal changes in inflammation and extracellular matrix. Physiol Genomics. 2006;25:364–374. doi: 10.1152/physiolgenomics.00013.2006. [DOI] [PubMed] [Google Scholar]
- 18.Roy S, Khanna S, Yeh PE, Rink C, Malarkey WB, Kiecolt-Glaser J, Laskowski B, Glaser R, Sen CK. Wound site neutrophil transcriptome in response to psychological stress in young men. Gene Expr. 2005;12:273–287. doi: 10.3727/000000005783992025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy S, Patel D, Khanna S, Gordillo GM, Biswas S, Friedman A, Sen CK. Transcriptome-wide analysis of blood vessels laser captured from human skin and chronic wound-edge tissue. Proc Natl Acad Sci U S A. 2007;104:14472–14477. doi: 10.1073/pnas.0706793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy S, Khanna S, Wallace WA, Lappalainen J, Rink C, Cardounel AJ, Zweier JL, Sen CK. Characterization of perceived hyperoxia in isolated primary cardiac fibroblasts and in the reoxygenated heart. Journal of Biological Chemistry. 2003;278:47129–47135. doi: 10.1074/jbc.M308703200. [DOI] [PubMed] [Google Scholar]
- 21.Verducci JS, Melfi VF, Lin S, Wang Z, Roy S, Sen CK. Microarray analysis of gene expression: considerations in data mining and statistical treatment. Physiol Genomics. 2006;25:355–363. doi: 10.1152/physiolgenomics.00314.2004. [DOI] [PubMed] [Google Scholar]
- 22.Roy S, Khanna S, Azad A, Schnitt R, He G, Weigert C, Ichijo H, Sen CK. Fra-2 mediates oxygen-sensitive induction of transforming growth factor beta in cardiac fibroblasts. Cardiovasc Res. 2010;87:647–655. doi: 10.1093/cvr/cvq123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Repovic P, Benveniste EN. Prostaglandin E2 is a novel inducer of oncostatin-M expression in macrophages and microglia. J Neurosci. 2002;22:5334–5343. doi: 10.1523/JNEUROSCI.22-13-05334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamberg M, Samuelsson B. On the metabolism of prostaglandins E 1 and E 2 in man. J Biol Chem. 1971;246:6713–6721. [PubMed] [Google Scholar]
- 25.Repovic P, Mi K, Benveniste EN. Oncostatin M enhances the expression of prostaglandin E2 and cyclooxygenase-2 in astrocytes: synergy with interleukin-1beta, tumor necrosis factor-alpha, and bacterial lipopolysaccharide. Glia. 2003;42:433–446. doi: 10.1002/glia.10182. [DOI] [PubMed] [Google Scholar]
- 26.Stenson WF, Parker CW. Prostaglandins, macrophages, and immunity. J Immunol. 1980;125:1–5. [PubMed] [Google Scholar]
- 27.Ushikubi F, Hirata M, Narumiya S. Molecular biology of prostanoid receptors; an overview. J Lipid Mediat Cell Signal. 1995;12:343–359. doi: 10.1016/0929-7855(95)00022-i. [DOI] [PubMed] [Google Scholar]
- 28.Cherukuri DP, Chen XB, Goulet AC, Young RN, Han Y, Heimark RL, Regan JW, Meuillet E, Nelson MA. The EP4 receptor antagonist, L-161,982, blocks prostaglandin E2-induced signal transduction and cell proliferation in HCA-7 colon cancer cells. Exp Cell Res. 2007;313:2969–2979. doi: 10.1016/j.yexcr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74:143–153. doi: 10.1016/j.lfs.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 30.Ma Y, Streiff RJ, Liu J, Spence MJ, Vestal RE. Cloning and characterization of human oncostatin M promoter. Nucleic Acids Res. 1999;27:4649–4657. doi: 10.1093/nar/27.23.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mollinedo F, Gajate C, Tugores A, Flores I, Naranjo JR. Differences in expression of transcription factor AP-1 in human promyelocytic HL-60 cells during differentiation towards macrophages versus granulocytes. Biochem J. 1993;294(Pt 1):137–144. doi: 10.1042/bj2940137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, Zhang J, Ding P, Apatira A, Chua J, Brandt R, Pine P, Goff D, Singh R, Payan DG, Hitoshi Y. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70:1544–1554. doi: 10.1158/0008-5472.CAN-09-2997. [DOI] [PubMed] [Google Scholar]
- 33.Falanga V. Growth factors and chronic wounds: the need to understand the microenvironment. J Dermatol. 1992;19:667–672. doi: 10.1111/j.1346-8138.1992.tb03756.x. [DOI] [PubMed] [Google Scholar]
- 34.Schultz GS, Davidson JM, Kirsner RS, Bornstein P, Herman IM. Dynamic reciprocity in the wound microenvironment. Wound Repair Regen. 2011;19:134–148. doi: 10.1111/j.1524-475X.2011.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moussai D, Mitsui H, Pettersen JS, Pierson KC, Shah KR, Suarez-Farinas M, Cardinale IR, Bluth MJ, Krueger JG, Carucci JA. The human cutaneous squamous cell carcinoma microenvironment is characterized by increased lymphatic density and enhanced expression of macrophage-derived VEGF-C. J Invest Dermatol. 2011;131:229–236. doi: 10.1038/jid.2010.266. [DOI] [PubMed] [Google Scholar]
- 36.Stearman RS, Dwyer-Nield L, Grady MC, Malkinson AM, Geraci MW. A macrophage gene expression signature defines a field effect in the lung tumor microenvironment. Cancer Res. 2008;68:34–43. doi: 10.1158/0008-5472.CAN-07-0988. [DOI] [PubMed] [Google Scholar]
- 37.Dumas A, Lagarde S, Laflamme C, Pouliot M. Oncostatin M decreases interleukin-1beta secretion by human synovial fibroblasts and attenuates an acute inflammatory reaction in vivo. J Cell Mol Med. 2011 doi: 10.1111/j.1582-4934.2011.01412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.David E, Guihard P, Brounais B, Riet A, Charrier C, Battaglia S, Gouin F, Ponsolle S, Bot RL, Richards CD, Heymann D, Redini F, Blanchard F. Direct anti-cancer effect of oncostatin M on chondrosarcoma. Int J Cancer. 2011;128:1822–1835. doi: 10.1002/ijc.25776. [DOI] [PubMed] [Google Scholar]
- 39.Huang FM, Tsai CH, Yang SF, Chang YC. The upregulation of oncostatin M in inflamed human dental pulps. Int Endod J. 2009;42:627–631. doi: 10.1111/j.1365-2591.2009.01567.x. [DOI] [PubMed] [Google Scholar]
- 40.Albasanz-Puig A, Murray J, Preusch M, Coan D, Namekata M, Patel Y, Dong ZM, Rosenfeld ME, Wijelath ES. Oncostatin M is expressed in atherosclerotic lesions: a role for Oncostatin M in the pathogenesis of atherosclerosis. Atherosclerosis. 2011;216:292–298. doi: 10.1016/j.atherosclerosis.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Zeaiter Z, Diaz H, Stein M, Huynh HQ. Helicobacter pylori induces expression and secretion of oncostatin M in macrophages in vitro. Dig Dis Sci. 2011;56:689–697. doi: 10.1007/s10620-010-1341-z. [DOI] [PubMed] [Google Scholar]
- 42.Kastl SP, Speidl WS, Katsaros KM, Kaun C, Rega G, Assadian A, Hagmueller GW, Hoeth M, de Martin R, Ma Y, Maurer G, Huber K, Wojta J. Thrombin induces the expression of oncostatin M via AP-1 activation in human macrophages: a link between coagulation and inflammation. Blood. 2009;114:2812–2818. doi: 10.1182/blood-2009-01-200915. [DOI] [PubMed] [Google Scholar]
- 43.Kastl SP, Speidl WS, Kaun C, Katsaros KM, Rega G, Afonyushkin T, Bochkov VN, Valent P, Assadian A, Hagmueller GW, Hoeth M, de Martin R, Ma Y, Maurer G, Huber K, Wojta J. In human macrophages the complement component C5a induces the expression of oncostatin M via AP-1 activation. Arterioscler Thromb Vasc Biol. 2008;28:498–503. doi: 10.1161/ATVBAHA.107.160580. [DOI] [PubMed] [Google Scholar]
- 44.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Elssen CH, Vanderlocht J, Oth T, Senden-Gijsbers BL, Germeraad WT, Bos GM. Inflammation restraining effects of prostaglandin E2 on natural killer-dendritic cell (NK-DC) interaction are imprinted during DC maturation. Blood. 2011;118:2473–2482. doi: 10.1182/blood-2010-09-307835. [DOI] [PubMed] [Google Scholar]
- 46.Gomez PF, Pillinger MH, Attur M, Marjanovic N, Dave M, Park J, Bingham CO, 3rd, Al-Mussawir H, Abramson SB. Resolution of inflammation: prostaglandin E2 dissociates nuclear trafficking of individual NF-kappaB subunits (p65, p50) in stimulated rheumatoid synovial fibroblasts. J Immunol. 2005;175:6924–6930. doi: 10.4049/jimmunol.175.10.6924. [DOI] [PubMed] [Google Scholar]
- 47.Talwar M, Moyana TN, Bharadwaj B, Tan LK. The effect of a synthetic analogue of prostaglandin E2 on wound healing in rats. Ann Clin Lab Sci. 1996;26:451–457. [PubMed] [Google Scholar]
- 48.Baxter CR. Immunologic reactions in chronic wounds. Am J Surg. 1994;167:12S–14S. doi: 10.1016/0002-9610(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 49.Egg D. Concentrations of prostaglandins D2, E2, F2 alpha 6-keto-F1 alpha and thromboxane B2 in synovial fluid from patients with inflammatory joint disorders and osteoarthritis. Z Rheumatol. 1984;43:89–96. [PubMed] [Google Scholar]
- 50.Hasegawa S, Ichiyama T, Kohno F, Korenaga Y, Ohsaki A, Hirano R, Haneda Y, Fukano R, Furukawa S. Prostaglandin E2 suppresses beta1-integrin expression via E-prostanoid receptor in human monocytes/macrophages. Cell Immunol. 2010;263:161–165. doi: 10.1016/j.cellimm.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 51.Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141:1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- 52.Nataraj C, Thomas DW, Tilley SL, Nguyen MT, Mannon R, Koller BH, Coffman TM. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–1235. doi: 10.1172/JCI13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharif MN, Sosic D, Rothlin CV, Kelly E, Lemke G, Olson EN, Ivashkiv LB. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. 2006;203:1891–1901. doi: 10.1084/jem.20051725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weinger JG, Brosnan CF, Loudig O, Goldberg MF, Macian F, Arnett HA, Prieto AL, Tsiperson V, Shafit-Zagardo B. Loss of the receptor tyrosine kinase Axl leads to enhanced inflammation in the CNS and delayed removal of myelin debris during Experimental Autoimmune Encephalomyelitis. J Neuroinflammation. 2011;8:49. doi: 10.1186/1742-2094-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Konishi A, Aizawa T, Mohan A, Korshunov VA, Berk BC. Hydrogen peroxide activates the Gas6-Axl pathway in vascular smooth muscle cells. J Biol Chem. 2004;279:28766–28770. doi: 10.1074/jbc.M401977200. [DOI] [PubMed] [Google Scholar]
- 56.Mudduluru G, Leupold JH, Stroebel P, Allgayer H. PMA up-regulates the transcription of Axl by AP-1 transcription factor binding to TRE sequences via the MAPK cascade in leukaemia cells. Biol Cell. 2010;103:21–33. doi: 10.1042/BC20100094. [DOI] [PubMed] [Google Scholar]
- 57.Sayan AE, Stanford R, Vickery R, Grigorenko E, Diesch J, Kulbicki K, Edwards R, Pal R, Greaves P, Jariel-Encontre I, Piechaczyk M, Kriajevska M, Mellon JK, Dhillon AS, Tulchinsky E. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene. 2011 doi: 10.1038/onc.2011.336. [DOI] [PubMed] [Google Scholar]
- 58.Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117:5965–5973. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- 59.Suda M, Tanaka K, Sakuma Y, Yasoda A, Ozasa A, Fukata J, Tanaka I, Narumiya S, Nakao K. Prostaglandin E(2) (PGE(2)) induces the c-fos and c-jun expressions via the EP(1) subtype of PGE receptor in mouse osteoblastic MC3T3-E1 cells. Calcif Tissue Int. 2000;66:217–223. doi: 10.1007/s002230010043. [DOI] [PubMed] [Google Scholar]
- 60.Mechta-Grigoriou F, Gerald D, Yaniv M. The mammalian Jun proteins: redundancy and specificity. Oncogene. 2001;20:2378–2389. doi: 10.1038/sj.onc.1204381. [DOI] [PubMed] [Google Scholar]
- 61.Wahl AF, Wallace PM. Oncostatin M in the anti-inflammatory response. Ann Rheum Dis. 2001;60(Suppl 3):iii75–iii80. doi: 10.1136/ard.60.90003.iii75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hams E, Colmont CS, Dioszeghy V, Hammond VJ, Fielding CA, Williams AS, Tanaka M, Miyajima A, Taylor PR, Topley N, Jones SA. Oncostatin M receptor-beta signaling limits monocytic cell recruitment in acute inflammation. J Immunol. 2008;181:2174–2180. doi: 10.4049/jimmunol.181.3.2174. [DOI] [PubMed] [Google Scholar]
- 63.Drinkwater SL, Smith A, Burnand KG. What can wound fluids tell us about the venous ulcer microenvironment? Int J Low Extrem Wounds. 2002;1:184–190. doi: 10.1177/153473460200100307. [DOI] [PubMed] [Google Scholar]
- 64.Pentland P, Needleman AP. Modulation of keratinocyte proliferation in vitro by endogenous prostaglandin synthesis. J Clin Invest. 1986;77:246–251. doi: 10.1172/JCI112283. [DOI] [PMC free article] [PubMed] [Google Scholar]