

Abstract
Some antipsychotic drugs are known to cause valvular heart disease by activating serotonin 5-HT2B receptors. We have developed and validated binary classification QSAR models capable of predicting potential 5-HT2B binders. The classification accuracies of the models to discriminate 5-HT2B actives from the inactives were as high as 80% for the external test set. These models were used to screen in silico 59,000 compounds included in the World Drug Index and 122 compounds were predicted as actives with high confidence. Ten of them were tested in radioligand binding assays and nine were found active suggesting a success rate of 90%. All validated binders were then tested in functional assays and one compound was identified as a true 5-HT2B agonist. We suggest that the QSAR models developed in this study could be used as reliable predictors to flag drug candidates that are likely to cause valvulopathy.
Introduction
During the last decade, several drugs have been shown to cause cardiac valvulopathy in humans. The initial discovery of drug-induced valvulopathy occurred when the anorectic drug fenfluramine (approved by the FDA in 1973), one of the active ingredients of the anorectic drug combination fen-phen, was found to increase the risk of developing two potentially serious conditions, pulmonary hypertension and valvular heart disease (VHD), in individuals receiving these medications to treat obesity.1 More recently, a group at the Mayo Clinic reported VHD in patients taking the anti-Parkinson drug pergolide.2 After the initial 2002 report, other cases of VHD associated with pergolide or other dopamine agonists such as cabergoline used as anti-parkinsonian drugs were identified.3-5 In January of 2007, the New England Journal of Medicine published two large European studies that independently verified the association of VHD with pergolide and cabergoline.6;7 Finally, on March 29, 2007, the Food and Drug Administration issued a Public Health Advisory for the voluntary market withdrawal of pergolide. These stunning withdrawals of drugs from the market stressed the importance of elucidating the mechanism by which these drugs induce valvulopathy and of determining the valvulopathic risk that may be associated with new drug candidates or even existing drugs.
To date, all but two of the VHD-associated drugs are ergoline derivatives (dihydroergotamine, methysergide, pergolide and carbergoline) (see Table 1). The two non-ergoline VHD-associated drugs are fenfluramine1 and 3,4-methylenedioxymethamphetamine (MDMA, ecstasy),8;9 both of which are amphetamine analogues (see Table 1). Thus, it appears that compounds from both the ergoline and phenylisopropylamine families can produce VHD.10
Table 1.
Chemical structures of marketed drugs known as 5-HT2B receptor agonists and associated with VHD.
| Compound | PubChem CID | 5-HT2B Agonist | VHD |
|---|---|---|---|

|
54746 | Yes | Yes |
| Carbergoline | |||

|
10531 | Yes | Yes |
| Dihydroergotamine | |||

|
3337 | Yes | Yes |
| Fenfluramine | |||

|
1614 | Yes | ??a |
| MDA | |||

|
1615 | Yes | Yes |
| MDMA | |||

|
8226 | Yes | Yes |
| Methylergonovine | |||

|
47811 | Yes | Yes |
| Pergolide |
Unknown.
There is increasing evidence that activation of serotonin 2B receptors (5-HT2B) may play a significant role in the pathogenesis of drug-induced valvulopathy.11-13 For instance, VHD-associated drugs such as fenfluramine,14 ergotamine,14 pergolide9;15 and cabergoline, and/or selected active metabolites (such as norfenfluramine and methylergonovine),14 all potently activate 5-HT2B receptors. Chemically similar medications that do not activate 5-HT2B receptors (e.g., lisuride) seemingly do not cause valvular heart disease, further implicating the 5-HT2B receptor—but not other receptors that bind ergopeptines/ergolines and phenylisoproylamines with high affinity—in the pathogenesis of heart-valve disease.13
Additionally, valvulopathy-associated drugs have been shown to induce DNA synthesis in cultured interstitial cells from human cardiac valves via 5-HT2B receptor activation.9 It has been suggested that the valvulopathy induced by 5-HT2B receptor agonists is caused by the inappropriate mitogenic stimulation of normally quiescent valve cells, resulting in an overgrowth valvulopathy.9;13 Although the precise signaling pathways underlying drug-induced valvulopathy remain elusive, 5-HT2B receptors are known to activate mitogenic pathways through the phosphorylation of Src kinase and extracellular regulated kinases (ERK), as well as through receptor tyrosine kinase transactivation,16;17 consistent with a role in regulating heart valve interstitial cell proliferation.
The discoveries that 5-HT2B receptors were (1) abundantly expressed in heart valves,18 (2) activated by fenfluramine and its metabolite, norfenfluramine,11;18 and (3) activated by other valvulopathy-inducing drugs9;11 suggested that 5-HT2B receptors were involved in the etiology of valvulopathy.11;18 Subsequently, several other 5-HT2B agonists were also found to be valvulopathogenic.9 Since 5-HT2B agonists have the potential of causing valvulopathic side-effects, it has been suggested that all pharmaceuticals should be screened for activity at 5-HT2B receptors prior to further commercial development.13;19
Similar to experimental high throughput screening (HTS), virtual screening (VS) is typically employed as a ‘hit’ identification tool.20 The experimental screening of all molecules against all biological targets is generally cost- and time-prohibitive. Therefore, pre-selection of compounds by VS that have a reasonable probability to act against a given biological target is highly attractive. Typically, VS approaches imply the use of structure based methodologies; nevertheless, we have repeatedly advocated for the use of ligand based cheminformatics approaches such as QSAR models in virtual screening (reviewed in a recent monograph21).
Herein, we report on the development of in silico screening tools for identifying compounds with potentially serious valvulopathic side effects. These tools can be employed as filters to flag and de-select the potentially harmful compounds at the preclinical stage of drug development, thereby potentially avoiding significant economic and human health consequences incurred at later stages of drug discovery. To achieve this goal, validated and externally predictive, binary QSAR models were generated for 5-HT2B active vs. inactive compounds as defined in 5-HT2B functional assays. Similar studies to develop QSAR models for 5-HT2B actives vs. inactives were reported recently by Chekmarev et al.22 However, in our investigations we considered a larger dataset that contained the most complete set of all known valvulopathogens reported by Huang et al,23 and we validated our predictions experimentally in binding assays.
To obtain the most statistically robust and predictive models, we have employed the combinatorial QSAR strategy24;25 implemented as part of our predictive QSAR modeling workflow (reviewed in Tropsha and Golbraikh26). All models were subjected to rigorous internal and external validation. The results confirmed the high external prediction accuracy of our computational models, which led us to conclude that these models can be used reliably to screen chemical databases to identify putative 5-HT2B actives. Screening the World Drug Index (WDI) database using these models led to the identification of 122 possible 5-HT2B actives; 10 of these computational hit compounds were experimentally tested in 5-HT2B radioligand binding assays at the NIMH Psychoactive Drug Screening Program (PDSP), UNC Chapel Hill (http://pdsp.med.unc.edu/). Experiments confirmed that 9 out of 10 compounds were true actives implying a hit rate of 90%. These results indicate the reliability of our computational models as efficient predictors of compounds’ affinity towards 5-HT2B receptors. We suggest that the computational models developed in this study could be used as drug liability predictors similar to commonly used predictors27;28 of other undesired side effects such as carcinogenicity,29-31 mutagenicity,29;32;33 PGP binding,24 or hERG binding.34-37 Our models can be used to flag compounds that are expected to bind to 5-HT2B receptors but they cannot distinguish agonists from antagonists. Nevertheless, as demonstrated in this study, these putative 5-HT2B binders can be tested in functional assays for their potential to activate 5-HT2B receptors to further assess their valvulopathic potential.
Materials and Methods
Dataset
The PDSP recently screened roughly 2,200 FDA-approved drugs and investigational, drug-like molecules against 5-HT2B receptors.23 However, this modeling study was initiated prior to the completion of the screening of the entire compound library. At the time this study began, screening against 5-HT2B receptors had been completed for 800 compounds. This set became the basis for our model development. After preprocessing of the 800-compound dataset and deleting duplicates, the final dataset consisted of a class of 146 ‘actives’, and another class of 608 ‘inactives’. Detailed PDSP protocols are available online (http://pdsp.med.unc.edu/) and in Huang et al.23 All chemical structures were obtained from PubChem38 as SDF files. By the time our modeling studies were completed, functional data for the remainder of the 2,200 compounds (1,400 compounds) had become available. These ‘new’ data became a source for additional, independent validation sets.
Preprocessing of the Dataset
For the purposes of this work, the data were curated as follows: First, all molecules were “washed” using the Wash Molecules tool in MOE39 (v.2007.09). Using this tool, we processed chemical structures by carrying out several standard operations including 2D depiction layout, hydrogen correction, salt and solvent removal, chirality and bond type normalization (all details are found in the MOE manual39). Second, we used ChemAxon Standardizer40 to harmonize the representation of aromatic rings. Finally, the analysis of the normalized molecular structures resulted in detection of 46 duplicate compounds (i.e., different salts or isomeric states). The functional data for duplicated compounds were found to be identical, so in each case a single example was removed. The curated subset of the original 5-HT2B dataset used in this work contains 754 unique organic compounds (146 actives and 608 inactives). All details about the dataset are available in Supporting Information.
Dataset Division for Model Building and Validation
All QSAR models generated in this study to classify actives vs. inactives were validated by predicting two external validation sets. Each dataset employed in QSAR studies was first randomly divided into a modeling and a validation sets. Additionally, as described above, an independent validation set became available after we completed our modeling studies. Details about this external set are available in Supporting Information, and in Huang et al.23
Another level of internal validation was achieved by comparing model performance for training and test sets. This approach is always employed as a part of our predictive QSAR modeling workflow26 to emphasize the fact that training-set-only modeling is not sufficient to obtain reliable models that are externally predictive.41 Thus, for each collection of descriptors, the modeling sets were further partitioned into multiple chemically diverse training and test sets of different sizes using the Sphere Exclusion method implemented in our laboratory.42 Only models that were highly predictive on the test sets were retained for the consensus prediction of the external validation sets. Finally, only those models that were shown to be highly predictive on both external sets were used in consensus fashion for virtual screening of external compound libraries.
Computational Methods
A combinatorial QSAR approach (Combi-QSAR)24;25 was used to generate classification models for actives vs. inactives (Fig. 1). In this study, four types of descriptors were applied in combination with three types of statistical methods.
Figure 1.

The workflow for QSAR model building and validation as applied to the 5-HT2B dataset (see text for abbreviations).
Molecular Descriptors
Four sets of molecular descriptors were considered in our modeling studies: Dragon,43 MolConnZ (MZ),44 MOE,39 and subgraph descriptors (SG)45 developed in this laboratory. Each type of descriptors was used separately with each of the classification methods in the context of our Combi-QSAR strategy.
DRAGON Descriptors
The Dragon Professional version 5.4 software43 was used to calculate 2D descriptors. These included topological descriptors, constitutional descriptors, walk and path counts, connectivity indices, information indices, 2D autocorrelations, edge adjacency indices, Burden eigenvalues, topological charge indices, eigenvalue-based indices, functional group counts, atom-centered fragments and molecular properties. The initial descriptor set was reduced by eliminating the constant and near-constant variables using built-in functions within the software. The pairwise correlations for all descriptors were examined and one of the two descriptors with the correlation coefficient R2 of 0.95 or higher was excluded. The calculation procedures for these descriptors, with related literature references, are reported by Todeschini and Consonni.46 Finally, the remaining descriptors were normalized by range-scaling so that their values were distributed within the interval 0-1.
MolConnZ Descriptors
The MolConnZ software44 available from EduSoft affords the computation of a wide range of topological indices of molecular structure. These indices include, but are not limited to, the following descriptors: valence, path, cluster, path/cluster and chain molecular connectivity indices,47-49 kappa molecular shape indices,50;51 topological52 and electrotopological state indices,53-56 differential connectivity indices,47;57 graph's radius and diameter,58 Wiener59 and Platt60 indices, Shannon 61 and Bonchev-Trinajstić62 information indices, counts of different vertices, counts of paths and edges between different types of vertices (http://www.edusoftlc.com/molconn/manuals/400). Descriptors with zero values or zero variance were removed; the remaining descriptors were normalized by range-scaling so that their values were distributed within the interval 0-1.
MOE Descriptors
MOE 2007.09 software39 was used to generate 2D MOE descriptors. These included physical properties, subdivided surface areas, atom and bond counts, Kier and Hall connectivity47-49 and kappa shape indices,50;51 adjacency and distance matrix descriptors,58;59;63;64 pharmacophore feature descriptors, and partial charge descriptors. 39 Descriptors with zero values or zero variance were removed; the remaining descriptors were normalized by range-scaling so that their values were distributed within the interval 0-1.
Subgraph Descriptors (SG)
Frequent subgraph mining of chemical structures is a novel approach to generating fragment descriptors that was developed recently in our group.45 SG descriptors are derived from each dataset, i.e., not pre-defined which gives the advantage of finding important chemical fragments that may have not been defined a priori by other fragment descriptor generating methods. The fragments are derived based on recurring substructures found in at least a subset of molecules (defined by a support value σ) in the dataset. These recurring substructures can implicate chemical features responsible for compounds’ biological activities. First, chemical structures were converted into labeled, undirected graph representations where nodes were labeled by atom types and edges corresponded to chemical bonds. Fast frequent subgraph mining (FFSM) algorithm65 was then used to find common frequent subgraphs for a given support value (σ), which is one of the variables defined by the user that determines the size of the set of subgraphs generated using FFSM. Obviously, the larger is the value of the support, the smaller is the number of subgraphs descriptors. As the support value decreases, the number of subgraphs increases dramatically. Redundant subgraphs were identified and removed leaving only the so called “closed subgraphs”. A subgraph SGi is closed in a database if there exists no supergraph SGj such that SGi ⊆ SGj and σSGi = σSGj. However, subgraph SGi would not be deleted if it also occurs by itself (not as part of the SGj) in the graph database. Removing redundant subgraphs (fragments) reduces the number of subgraphs descriptors drastically and therefore makes the subsequent calculations more efficient. The frequency of individual ‘closed subgraphs’ in each molecule of the dataset is calculated and used as the descriptor value for each molecule. In this study, a support value of 12 % was used, and the upper size limit of the generated subgraphs was 7 atoms.
Balancing the Dataset Using Similarity Searching
The dataset used for model building was imbalanced, consisting of 146 actives vs. 608 inactives. Therefore, only a subset of the larger class of inactives of approximately the same size as the actives was used in model building unless otherwise indicated. This subset was selected to include inactives that were most similar to the actives. Given the vast array of available chemical descriptors and the large number of similarity measures, it is always difficult to decide a priori which combination of descriptors/similarity metrics to use. This problem has been highlighted in several recent publications.66;67 Therefore, similarity searching studies were performed using three types of molecular descriptors: fingerprints (FP), Dragon, and MZ, and applying two similarity metrics, i.e., Euclidean distance and Tanimoto coefficient (Tc). The similarity cutoff was chosen to obtain the most balanced (with roughly equal number of compounds from each class) subset of compounds.
Fingerprints (FP)
166 MACCS68 structural keys implemented in MOE 2007.09 software39 were calculated for all compounds. The similarity searching was performed using an in-house written script applying Tanimoto coefficients for similarity measures.
Dragon Descriptors
Normalized Dragon descriptors of the original dataset were employed to calculate similarities between all compounds in the dataset using Euclidean distance as similarity metric; variable similarity thresholds were used to down-sample the larger class (inactives). Although many schemes could be considered for down-sampling the larger classes, we used the similarity threshold based approach since it restricts the larger class to compounds most similar to the smaller class molecules. This approach makes it more challenging to develop statistically significant models capable of discriminating smaller class compounds from most chemically similar molecules in the larger class. Therefore, it increases the robustness of the binary QSAR models.
MolConnZ Descriptors (MZ)
Similar procedures to those described above for Dragon descriptors were used.
QSAR Methods
k Nearest Neighbors (kNN) QSAR
The kNN QSAR method69 is based on the k nearest neighbors principle and the variable selection procedure. It employs the leave-one-out (LOO) cross-validation (CV) procedure and a simulated-annealing algorithm70;71 to optimize variable selection. The procedure starts with the random selection of a predefined number of descriptors from all descriptors. If the number of nearest neighbors k is higher than one, the estimated activities ŷi of compounds excluded by the LOO procedure are calculated using the following formula:
| (1) |
where yj is the activity of the j-th compound. Weights wij are defined as:
| (2) |
and dij is Euclidean distances between compound i and its j-th nearest neighbor. However, if the number of nearest neighbors k is equal to one, then the estimated activity ŷi of the compound will be equal to the activity of this one nearest neighbor.
For classification kNN, the predicted ŷi values (see expression (1)) are rounded to the closest whole numbers (which are, in fact, the class numbers), and the prediction accuracy (correct classification rate, CCRtrain) is calculated as follows:
| (3) |
where and are the number of correctly classified and total number of compounds of class j (j=1, 2). Then, a predefined small number of descriptors are randomly replaced by other descriptors from the original pool, and the new value of CCRtrain is obtained. If CCRtrain (new) > CCRtrain (old), the new set of descriptors is accepted. If CCRtrain (new) ≤ CCRtrain (old), the new set of descriptors is accepted with probability p = exp (CCR (new) - CCR (old))/T, or rejected with probability (1-p), where T is a simulated annealing (SA) “temperature” parameter. During this process, T is decreasing until a predefined threshold. Thus, the optimal (highest) CCRtrain is achieved. For the prediction, the final set of selected descriptors is used, and expressions (1) and (2) are applied to predict activities of compounds of the test sets. Then the activities are rounded to the closest whole numbers, and the correct classification rate for the test set is calculated using formula (3).
In the case when compounds belong to two classes (e.g., active and inactive compounds), a 2 × 2 confusion matrix can be defined, where N(1) and N(0) are the number of compounds in the data set that belongs to classes (1) and (0) respectively. TP, TN, FP, and FN are the number of true positives (actives predicted as actives), true negatives (inactives predicted as inactives), false positives (inactives predicted as actives), and false negatives (actives predicted as inactives), respectively. The following classification accuracy characteristics associated with confusion matrices are widely used in QSAR studies: sensitivity (SE=TP/N(1)), specificity (SP=TN/N(0)), and enrichment E = TP*N/[(TP+FP)*N(1)]. In this study, we have employed normalized confusion matrices. A normalized confusion matrix can be obtained from the non-normalized one by dividing the first column by N(1) and the second column by N(0). Normalized enrichment is defined in the same way as E but is calculated using a normalized confusion matrix: En = 2TP*N(0)/[TP*N(0)+FP*N(1)]. En takes values within the interval of [0, 2].25;72
Classification Based on Association (CBA)
This method integrates both classification rule mining,73;74 which aims to discover a small set of rules in the database that forms an accurate classifier, and association rule mining,75 which finds all the rules existing in the database that satisfy some minimum support and minimum confidence constraints. For association rule mining, the target of discovery is not pre-determined, while for classification rule mining there is one and only one predetermined target. The integration is done by focusing on mining a special subset of association rules, called class association rules (CARs). An efficient algorithm is also used for building a classifier based on the set of discovered CARs.
The CBA algorithm76;77 consists of two parts, a rule generator, which is based on the a priori algorithm for finding association rules, and a classifier builder. The candidate rule generator is similar to the a priori one. The difference is that CBA updates the support value in each step while the a priori algorithm only updates this value once. This allows us to compute the confidence of the ruleitem. A ruleitem is of the form: <condset, y> where condset is a set of items, y ∈ Y is a class label. The support count of the condset (called condsupCount) is the number of cases in the dataset (D) that contain the condset.
Next, a classifier is built from CARs. To produce the best classifier out of the whole set of rules would involve evaluating all the possible subsets of it on the training data and selecting the subset with the right rule sequence that gives the least number of errors. There are 2m such subsets, where m is the number of rules. It is a heuristic algorithm. Given two rules, ri and rj, ri precedes rj if (1) the confidence of ri is greater than that of rj, or (2) their confidences are the same, but the support of ri is greater than that of rj, or (3) both the confidences and the supports of ri and rj are the same, but ri is generated earlier than rj. If R is a set of generated rules (i.e. CARs) and D the training data, the basic idea of the algorithm is to choose a set of high precedence rules in R to cover D. The classifier follows this format: <r1, r2, . . ., rn, default_class>, where ri ∈ R. In classifying an unseen case, the first rule that satisfies the case will classify it. If there is no rule that applies to the case, it takes on the default class.
The descriptors used with CBA need to be discrete in nature76 as is the case with SG descriptors but not Dragon, MolConnZ or MOE. Hence, this method was only used with SG descriptors using CBA (v2.1) software.78
Distance Weighted Discrimination (DWD)
This method was initially proposed by Marron and Todd79 with the goal of improving the performance of SVM80;81 in high dimensional low sample size (HDLSS) contexts. The main idea is to improve upon the criterion used for “separation of classes” in SVM. SVM has data piling problems along the margin, because it is maximizing the minimum distance to the separating plane, and there are many data points that achieve the minimum. A natural improvement is to replace the minimum distance by a criterion that allows all the data to have an influence on the result. DWD does this by maximizing the sum of the inverse distances. This results in directions that are less adversely affected by spurious sampling artifacts. The major contribution of this new discrimination method is that it avoids the data piling problem, to give the anticipated improved generality. Like SVM, DWD is based on computationally intensive optimization; however, while SVM uses well known quadratic programming algorithms, DWD uses interior-point methods for so-called Second-Order Cone Programming (SOCP) problems.82 Detailed discussion of these issues may be found in Marron and Todd (2007),79 which is available with the supporting information at https://genome.unc.edu/pubsup/dwd/. All DWD computations were performed using the DWD software83 written in Matlab84 and kindly provided by Dr. Marron.
Robustness of QSAR Models
Y-randomization test is a widely used validation technique to ensure the robustness of a QSAR model.85 This test includes (i) randomly shuffling the dependent-variable vector, Y-vector of training sets (class labels in this study) and (ii) rebuilding models with the randomized activities (class labels) of the training set. All calculations are repeated several times using the original independent-variable matrix. It is expected that the resulting QSAR classification models, built with randomized activities for the training set, should generally have low CCRs for training, test, and external validation sets. It is likely that sometimes, though infrequently, high CCR values may be obtained due to a chance correlation or structural redundancy of the training set. However, if some QSAR classification models obtained in the Y-randomization test have relatively high CCR it implies that an acceptable QSAR classification model cannot be obtained for the given dataset by the particular modeling method. Y-randomization test was applied to all datasets considered in this study, and the test was repeated five times in each case.
Applicability Domain of kNN QSAR Models
Formally, a QSAR model can predict the target property for any compound for which chemical descriptors can be calculated. However, since the training set models are developed in kNN QSAR modeling by interpolating activities of the nearest neighbor compounds, a special applicability domain (i.e., similarity threshold) should be introduced to avoid making predictions for compounds that differ substantially from the training set molecules.86
The similarity was estimated using Euclidean distances in high-dimensional descriptors space. Compounds with the smallest distance between them have the highest similarity. The distribution of distances (pairwise similarities) of compounds in our training set is computed to produce an applicability domain threshold, DT, calculated as follows:
| (4) |
Here, ȳ is the average Euclidean distance of the k nearest neighbors of each compound within the training set, σ is the standard deviation of these Euclidean distances, and Z is an arbitrary parameter to control the significance level. Based on previous studies, we set the default value of this parameter as 0.5, which formally places the boundary for the applicability domain at one-half of the standard deviation. Thus, if the distance of the external compound from at least one of its nearest neighbors in the training set exceeds this threshold, no prediction is made.86 In this study two types of applicability domains were employed. First, a global applicability domain that ensures some level of global similarity (using all descriptors for similarity calculations) between the predicted compounds and the compounds in the modeling set. The second is a local domain which is the applicability domain of each of the individual models using only descriptors used for the model building.
Consensus Prediction
Our experience suggests that consensus prediction of the target property for external compounds, i.e., when the compound activity is calculated by averaging values predicted by all individual models that satisfy our acceptability criteria always provides the most stable and accurate solution87. In general, consensus prediction implies averaging the predictions for each compound by majority voting for classification QSAR models, using all models passing the validation criteria (e.g., CCRtrain ≥ 0.70 and CCRtest ≥ 0.70). In order to determine the confidence in the obtained predictions we need to define a consensus score. The consensus scores employed in this study take into account the total number of models used to predict the compound's activity, and the number of models that predicted the compound to belong to a specific class. Since we define two classes of compounds, i.e., class 1 (actives) and class 0 (inactives), some models may predict a compound to belong to class 0 and others may predict it to belong to class 1. As a result, a consensus score between 0 and 1 will be obtained for each of the predicted compounds. As an additional measure of confidence (and an additional applicability domain criterion) we only accepted those predictions that had an average predicted value (consensus score) above 0.7 (for actives) or below 0.3 (for inactives).
Virtual Screening and Compound Selection for Experimental Validation
To identify putative actives, validated consensus models generated for 5-HT2B ligands were used for virtual screening of ca. 59,000 molecules within the WDI chemical library; the selection of hits was limited by the applicability domains of each models.88 122 compounds were identified as VS hits (by consensus agreement between all accepted models, see Table S1 of Supporting Information for details) and 10 structurally diverse and commercially available hits were purchased from different suppliers and tested at PDSP in 5-HT2B radioligand binding assays.
Results and Discussion
Combinatorial QSAR Modeling of 5-HT2B Actives vs. Inactives
Balancing the Dataset
The original dataset of 146 actives and 608 inactives was first balanced by downsizing the class of inactives. Similarity searching between active and inactive compounds using Tc cutoff of 0.7 resulted in 195 inactives (that were similar to at least one active compound with Tc above 0.7), which were combined with the 146 actives to form the modeling set of 342 compounds. Dragon and MZ descriptors were generated for this 342-compound modeling set to be used separately with kNN. However, similarity searching using Dragon and MZ descriptors and applying Euclidean distance-based threshold resulted in a 304- (146 actives and 158 inactives) and 325-compound (146 actives and 179 inactives) modeling sets respectively. Thus, slightly different modeling sets were used depending on the type of descriptors.
k NN Classification
kNN method was used with each of the following descriptor types: DRAGON, MZ, MOE, and SG descriptors. Models were built for the three datasets resulting from the down-sampling of the original dataset. First, a validation set (15-20% of the dataset) was excluded from each of the resulting datasets randomly. The compounds in the remaining modeling set (85-80% of the original dataset) were divided into multiple training and test sets (28-40 divisions). Multiple QSAR models were generated independently for all training sets and applied to the test sets. Generally, we accepted models with CCR values for both the training and test set greater than 0.70. kNN combined with subgraphs and Dragon descriptors were the two best performing methods based on validation set statistics (Table 2). kNN-subgraphs (kNN-SG) had a CCRevs = 0.80, while kNN-Dragon gave a CCRevs = 0.72.
Table 2.
Performance of kNN classification methods to classify actives vs. inactives based on external validation set statistics.
| Model | Num. Modelsa | Confusion Matrix |
Statistics for the Models |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N(1)b | N(0)c | TP | TN | FP | FN | SE | SP | En(1) | En(0) | CCRevsd | CCRrande | ||
| Af | 908 | 26 | 34 | 20 | 23 | 11 | 6 | 0.77 | 0.68 | 1.41 | 1.49 | 0.72 | 0.49 |
| Bg | 235 | 38 | 36 | 22 | 20 | 16 | 16 | 0.58 | 0.56 | 1.13 | 1.14 | 0.57 | 0.50 |
| Ch | 619 | 32 | 38 | 17 | 29 | 9 | 15 | 0.53 | 0.76 | 1.38 | 1.24 | 0.65 | 0.53 |
| Di | 387 | 30 | 40 | 16 | 29 | 11 | 14 | 0.04 | 0.73 | 0.26 | 1.90 | 0.63 | 0.50 |
| Ej | 123 | 30 | 40 | 20 | 26 | 14 | 10 | 0.67 | 0.65 | 1.31 | 1.32 | 0.66 | 0.46 |
| Fk | 93 | 30 | 40 | 23 | 33 | 7 | 7 | 0.77 | 0.83 | 1.63 | 1.56 | 0.80 | 0.54 |
Num. Models, number of models with CCRtrain and CCRtest ≥ 0.70
N(1), number of actives
N(0), number of inactives
CCRevs, correct classification rate of the consensus models using the external validation set
CCRrand, correct classification rate of the random models using the external validation set
A, kNN-Dragon
B, kNN-MZ
C, kNN-Dragon-FP
D, kNN-MZ-FP
E, kNN-MOE-FP
F, kNN-SG.
Results of the Y-randomization test (Table 2) confirmed that kNN classification models with CCRtrain and CCRtest values ≥ 0.70 were robust. None of the models with randomized class labels of the training set compounds had CCRrand > 0.54 for any dataset.
Classification Based on Association (CBA)
The CBA method was applied to classify the dataset using SG descriptors. A dataset of 342 compounds (146 actives and 196 inactives), resulting from the downsizing process with FP and Tanimoto distances, was used. The dataset was split randomly into training (267 compounds) and validation sets (75 compounds). A total of 1371 closed frequent subgraphs were generated with FFSM (see Methods) from the training set using a support value of 12% and a maximum size limit of the fragments of 7. The training set consisting of 267 compounds (111 actives and 156 inactives) was then used to build the classifier in CBA. The classifier gave a CCRtrain of 0.79. Then the validation set consisting of 75 compounds (35 actives and 40 inactives) was used to assess the robustness of the classifier. The CCRevs was 0.65 which is not as high as the CCR value for the training set.
DWD Modeling
The DWD method was applied to classify the dataset using Dragon descriptors. A dataset of 304 compounds (146 actives and 158 inactives), resulting from the downsizing process with Dragon descriptors and Euclidean distances, was used. The dataset was split randomly into training (244 compounds) and validation sets (60 compounds). A total of 387 Dragon descriptors were generated for the training set. The training set consisting of 244 compounds (120 actives and 124 inactives) was then used to build the DWD model. The DWD model was able to group compounds in this dataset based on their biological classes with a CCRevs = 0.70 (TP=18, TN=24, FP=10, FN=8), setting the cutoff at “0.15”. DWD was further used to rank Dragon descriptors according to their importance for discriminating the two classes of compounds (actives vs. inactives). DWD uses class label information where positive (for actives) and negative (for inactives) signs are assigned to each descriptor value to indicate its importance to the corresponding class. The top 20 highly weighted descriptors (based only on weights’ values and ignoring the signs) are presented in Table S2 of Supporting Information.
Comparison of Binary QSAR Approaches for Classifying 5-HT2B Actives vs. Inactives
The performance of different binary QSAR approaches employed as part of combinatorial QSAR strategy for 5-HT2B, and based on validation set statistics, is summarized in Figure 2. kNNSG, and kNN-Dragon were the best performing methods for classifying 5-HT2B actives vs. inactives based on validation set statistics (Table 2), yielding the highest CCRevs of 0.80 in case of kNN-SG. On the contrary, kNN-MZ was the worst performing method with a CCRevs of 0.57 which was very close to random. It was also interesting to see that kNN-SG performed much better than CBA-SG with CCRevs = 0.80 in the former case and 0.65 in the latter. These results confirm the importance of employing the combinatorial QSAR approach to find the most predictive QSAR method/descriptor combination for each specific dataset.
Figure 2.

Comparison of CCR values for the external validation set (CCRevs) for different QSAR models developed to classify actives vs. inactives. CCRevs values for models built with both real (blue) and randomized (red) activities of the training sets are shown (see text for abbreviations).
Our models also indicated that the nature of the descriptors used has a dramatic effect on the performance of the modeling methods. It was clear that MOE and MolConnZ descriptors did not perform very well in all tested cases irrespective of the applied modeling techniques. On the contrary, Dragon descriptors afforded most significant models with all methods and in all tests, for both validation and external sets.
Additional Model Validation
Model Validation by Predicting Drugs Known to be 5-HT2B Actives and Valulopathogens
Both fenfluramine and dexfenfluramine (known to be 5-HT2B actives and agonists, which were not included in our modeling sets) were predicted as 5-HT2B actives using consensus models to classify actives vs. inactives. The consensus scores using kNN-Dragon were 0.79 for both compounds. Our previous studies suggest that consensus prediction that is based on the results obtained by all validated predictive models always provides the most stable solution.87 A 5-HT2B active compound can have consensus scores in the interval [0.5-1.0]. The closer value to 1.0 the greater is the confidence in the prediction. Therefore, we can claim that both compounds were predicted as actives with statistically significant consensus scores.
These results highlight the predictive power of our validated models that could have predicted the possible dangerous side effects of these two drugs by suggesting that they may be 5-HT2B actives. This prediction would have suggested that these compounds should be tested experimentally in 5-HT2B functional assays and prevented from further development as potentially unsafe medicines. This example illustrates the potential use of models developed in this study as computational drug safety alerts.
Model Validation by Predicting an External Set
An additional 16-compound set was obtained from PDSP after we finished out modeling studies. This external set was used to further assess the robustness and the predictive power of our models. All 16 compounds were 5-HT2B actives including 4 agonists and 12 antagonists.
The 16 external compounds were predicted using all consensus models built to classify actives vs. inactives. kNN-Dragon was the best performing method on this external set with a CCRex of 0.81. Predictions were made by applying local model applicability domains with Z = 0.5 (see Applicability Domain of kNN QSAR Models). It was interesting to find that kNN-Dragon had CCR ≥ 0.72 with both the validation (CCRevs = 0.72) and the external (CCRex = 0.81) sets. However, kNN-SG (the best performing method on validation sets) was not as good with the external set (CCRex = 0.65) as it was with the validation set (CCRevs = 0.80). CBA-SG gave a CCRex = 0.65, which was consistent with its performance with the validation set (CCRevs = 0.65) but less than CCRtrain (0.79). The latter results using SG descriptors with kNN and CBA might be due to the limitation that frequent subgraphs are derived from the training set compounds; therefore, it is possible that fragments that are frequent in the external set are not represented in the frequent subgraphs used for prediction. Our current applicability domain filter, which is calculated using the fragments in the training set, does not account for this possibility. It is clear that a more stringent applicability domain filter could be applied in this case, which uses the distribution of subgraphs counts between the training and test set, but this has not been implemented yet within our current method.
The Importance of Variable Selection
Since kNN-Dragon was the best performing method to classify actives vs. inactives based on the results for all validation sets, we thought it would be interesting to check the performance of kNN using all 387 Dragon descriptors, generated for the actives vs. inactives modeling set, without variable selection. The results of this test are shown in Table 3. Comparison of modeling results for kNN-variable-selection (CCRevs = 0.72) vs. kNN-without-variable-selection (CCRevs = 0.52) clearly indicates that variable selection is a vital part of modeling. Furthermore, the top 20 most frequent descriptors (MFD) selected by kNN models (Table S3 of Supporting Information) and top 20 highly weighted descriptors by DWD based only on weights and ignoring the sign (Table S2 of Supporting Information) were used independently with the kNN method (with no variable selection) to predict actives vs. inactives (Table 3). Models built with either the top 20 DWD-selected Dragon descriptors or MFD from Dragon-kNN and using 1-5 nearest neighbors gave CCRevs ~ 0.5 (Table 3). These results illustrated again that SA-based variable selection procedures implemented in our kNN QSAR method69 lead to models with the highest external predictive power as compared to any other approach not relying on variable selection for model optimization.
Table 3.
Comparison between different kNN-Dragon QSAR models generated with or without variable selection.
| Model | Num. Modelsa | Confusion Matrix |
Statistics for the Models |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N(1)b | N(0)c | TP | TN | FP | FN | SE | SP | En(1) | En(0) | CCRevsd | Coveragee | ||
| Af | 908 | 26 | 34 | 20 | 23 | 11 | 6 | 0.77 | 0.68 | 1.41 | 1.49 | 0.72 | 100% |
| Bg | 1 | 26 | 34 | 10 | 22 | 10 | 8 | 0.38 | 0.65 | 1.13 | 1.36 | 0.52 | 83% |
| Ch | 1 | 26 | 34 | 14 | 15 | 19 | 9 | 0.54 | 0.44 | 0.98 | 1.12 | 0.49 | 95% |
| Di | 1 | 26 | 34 | 14 | 15 | 19 | 9 | 0.54 | 0.44 | 0.98 | 1.12 | 0.49 | 95% |
Num. models, number of models with CCRtrain and CCRtest ≥ 0.70
N(1), number of actives
N(0), number of inactives
CCRevs, correct classification rate of the consensus models using the external validation set
Coverage: percentage of predicted compounds, and coverage = % of the external set compounds predicted by the models
A, kNN-Dragon
B, kNN-Dragon-NVS where kNN model was generated using all 387 Dragon descriptors with no variable selection and 1 nearest neighbor (NN)
C, kNN-Dragon-MFD where the kNN model was generated with top 20 most frequent Dragon descriptors and 1NN
D, kNN-Dragon-DWD where the kNN model was generated with top 20 highly weighted Dragon descriptors by DWD and 1 NN.
Mechanistic interpretability is frequently regarded as very important feature of QSAR models. We generally argue that only models that have been extensively validated on external datasets and identified experimentally-confirmed hits should be subjected to interpretation. Furthermore, very few classes of models, specifically, those based on (multiple) linear regression and small number of descriptors can afford a relatively straightforward interpretation. The interpretation of multi-parametric statistical models developed with non-linear optimization algorithms (as in this study) should be attempted with great care because of strong and often poorly understood interplay between descriptors. Furthermore, although we could foresee that in some cases medicinal chemists may want to modify their candidate compounds to prevent 5HT2B binding, the tools developed in this study are predominantly intended for virtual screening of libraries of drug candidates to flag and possibly eliminate compounds that are likely to bind 5HT2B receptor, not to design new compounds; and any compound designed by chemists could be passed through our models. Therefore, we only restricted the discussion in this paper to the most frequent descriptors found by all acceptable kNN models and the most highly weighted descriptors selected by DWD to stress that the process of variable selection employed as part of model optimization has indeed converged on a small number of descriptors.
Virtual Screening of the World Drug Index Database to Identify Putative 5-HT2B Ligands
Since our models proved to be reasonably accurate based on two external validation sets, we used the best models to mine a large external database of approved and potential drugs for putative 5-HT2B actives. An important condition that assures reliable predictions by the model is the use of AD. Therefore, two types of AD were employed in the virtual screening of compound databases. The first is a global AD that acts as a filter and ensures some level of global similarity between the predicted compounds and the compounds in the modeling set. The second is a local AD which is defined for each of the individual classification models.
The WDI database of ca. 59,000 compounds (approved or investigational drugs) was used for virtual screening (Fig. 3). This original collection had many duplicates (i.e., many salt forms for the same chemical entity). The duplicates were removed using MOE: keeping unique structures and deleting duplicates. We also removed all compounds included in our modeling and external validation sets. Dragon descriptors were generated for the remaining 46,859 unique compounds in the database; 926 compounds were excluded because Dragon was unable to calculate at least one of the descriptors generated for the modeling set. The remaining 45,933 compounds were then subjected to a global AD filter for the actives vs. inactives modeling set using a strict Z cutoff of 0.5 (which formally places the allowed pairwise distance threshold at the mean of all pairwise distance distribution for the training set plus one-half of the standard deviation). Obviously, increasing the AD would increase the number of computational hits identified by virtual screening. However, our experience suggests that such increase is typically accompanied by the decrease in prediction accuracy. Additionally, we required that the nearest neighbor in the modeling set of a compound from the virtual library be an active. The resulting 7,286 compounds were then classified into actives vs. inactives using DWD-Dragon classifier resulting in 891 actives. Next, all kNN-Dragon models with CCRtrain and CCRtest ≥ 0.70 were employed in consensus fashion to predict these 891 compounds resulting in a selection of the 500 active hits. At this point, SG descriptors were generated for these 500 molecules. CBA-SG classifier followed by kNN-SG consensus models were used as final filters for the determination of 122 compounds regarded as putative 5-HT2B actives.
Figure 3.

Steps of the virtual screening of the WDI database to identify putative 5-HT2B ligands (see text for the abbreviations).
Experimental Validation
Ten structurally diverse hits (1-10, see Table 4) were selected from the final consensus virtual screening hits for further experimental validation taking into account both their commercial availability and cost (see Table 4). To our satisfaction, nine compounds were confirmed to inhibit 5-HT2B radioligand binding, which implies a hit rate of 90 %. Ki values were in the range 0.8 – 3,127 nM, with 4 compounds having Ki values < 100 nM. The four highest affinity compounds were: 4 (Ki=33 nM, see Fig. 4 (A)), 7 (Ki=0.8 nM, see Setola et al, 20039), 9 (Ki=70 nM, see Fig. 4 (B)), and 10 (Ki=69 nM, see Fig. 4 (C)).. It should be noted that methylergometrine, though not included initially in our dataset, was known to be a valvulopathic compound and had been tested against 5-HT2B receptors in both binding (K =0.8 nM)9 and functional assays (pEC50 for 5-HT2B-Mediated calcium flux = 7.67)23. In order to determine the activity of the remaining eight 5-HT2B ligands, all compounds were tested at the PDSP in 5-HT2B functional assays. Results indicated that methylergometrine was the only compound among the 9 5-HT2B ligands that possessed strong agonist activity.
Table 4.
Experimental validation results for the 10 computational hits predicted as 5-HT2B ligands as a result of QSAR-based mining of the WDI chemical screening library.
| Compound ID | Chemical Stucture/Name | PubChem CID | PDSP ID | Predicted 5-HT2B Activity | Experimental Ki (nM) |
|---|---|---|---|---|---|
| 1 |

|
43922 | 14809 | Active | 2,495 |
| 2 |
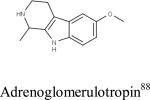
|
71028 | 14807 | Active | 491 |
| 3 |

|
114709 | 14806 | Active | >10,000 |
| 4 |

|
3038495 | 14814 | Active | 33.1 |
| 5 |

|
3336 | 14821 | Active | 3,217 |
| 6 |

|
1715104 | 14815 | Active | 151.4 |
| 7 |

|
4140 | 27769 | Active | 0.8 |
| 8 |

|
195658 | 14805 | Active | 1,617 |
| 9 |

|
9909648 | 13513 | Active | 69.6 |
| 10 |

|
15940170 | 13505 | Active | 69 |
Figure 4.

Competition binding at 5-HT2B receptors for (A) 4 (triangle) and SB206553 (square), (B) 9 (triangle) and SB206553 (square), and (C) 10 (triangle) and chlorpromazine (square), versus [3H]LSD.
This low hit rate of 11.1% for identifying validated agonists is in fact not surprising in light of Huang et al23 major finding that potent 5-HT2B receptor agonism is a relatively rare occurrence among drugs and drug-like compounds. However, to arrive at such conclusions, Huang et al screened a composite library containing three publicly available collections of FDA-approved and investigational medications and one internally compiled library. Of the approximately 2200 compounds screened, 27 5-HT2B receptor agonists were identified; thus, the validated hit rate was 1.2%.
These results illustrate that the validated QSAR workflow, as employed in this paper, could be used as a general tool for identifying 5-HT2B ligands by the means of virtual screening of chemical libraries using rigorously built QSAR models. As we demonstrated in this study, our models identify a relatively small number of VS hits making it feasible to employ experimental tools to validate predictions in 5-HT2B binding and functional assays. Ten compounds selected from a large external library have been tested experimentally in this proof-of-concept study resulting in very high experimentally confirmed hit rate. The list of all compounds predicted to be 5-HT2B actives is available in the Supporting Information (Table S1).
To verify the diversity of the experimentally validated hits, we have compared the results of QSAR-based virtual screening with simple similarity searches. Similarity calculations were done using two different descriptor-metric combinations: (1) MACCS structural keys and Tanimoto coefficients (as a standard similarity searching approach, see Table S9 and Figure S1 in Supporting Information) and (2) Dragon descriptors and Euclidean distances (to compare directly with our best performing QSAR models of kNN-Dragon, see Table S10 and Figure S2 in Supporting Information). The nearest neighbor compounds (based on Tanimoto similarities and MACCS keys) from the active compounds in the dataset and the 10 experimentally validated VS hits are reported in Table 5. Results of similarity analyses indicated that neither technique would be able to efficiently identify the diverse hits obtained with our methods (see Supporting Information for details). Hence, our studies illustrated the power of combi-QSAR-based VS in prioritizing compounds (which are not just close analogs of the modeling set compounds) from screening libraries to achieve high success rates when experimentally validated.
Table 5.
Nearest neighbor compounds from the active compounds in the dataset and the 10 experimentally validated VS hits.
| Virtual screening hits | Nearest neighbor from the modeling set compounds based on MACCS structural keys and Tanimoto distances |
|---|---|

|

|
|

|

|
|

|

|

|

|

|

|

|

|

|

|

|
|

|

|
We also think that agonist vs. antagonist models will be highly useful as more data about agonist compounds become available. The small number of known 5-HT2B agonists made it impossible at this stage to develop statistically significant models that could distinguish agonists from antagonists. Thus, the current study was limited to building binder vs. non-binder models. We will continue with our efforts to develop quantitative 5-HT2B agonist predictors as we accumulate more experimental data.
Conclusions
QSAR models are becoming increasingly attractive as robust computational tools for virtual screening due to both their computational efficiency and success rates [reviewed in26 as well as in a recent monograph21]. In this study, we have applied a combinatorial QSAR approach to a dataset of 800 compounds experimentally annotated as 5-HT2B receptor agonists, antagonists and inactives resulting in statistically validated and externally predictive models. Specifically, we have applied a combi-QSAR approach utilizing three different classification methods (kNN, CBA and DWD) and four different descriptor types (Dragon, MZ, MOE and SGs) to generate classification QSAR models to discriminate between 5-HT2B actives (agonists and antagonists) from inactives. Predictive models with classification accuracies as high as 0.80 for actives vs. inactives, as estimated on external validation sets, were obtained.
Classification models for actives vs. inactives were further validated by predicting an external validation set obtained after we completed the modeling studies. The high accuracy of prediction for the second external validation set proved that our models were indeed rigorous. Therefore, we posited that our studies afforded a robust computational tool to predict potential 5-HT2B activity and consequently prioritize hits for testing in functional 5-HT2B assays to predict valvulopathic side effects of drugs and drug candidates that act as 5-HT2B agonists. We suggested that this computational predictor could be used to eliminate high risk compounds at the early stages of the drug development process. To illustrate this point, we have used this predictor retrospectively to evaluate the valvulopathic potential of two drugs withdrawn from the U.S. market for this reason, i.e., fenfluramine and dextrofenfluramine. Both drugs were not included in our modeling set and both were indeed predicted with high confidence as actives for binding to 5-HT2B receptors.
Encouraged by our model validation results, we have applied these models for virtual screening of the 59,000 compounds in WDI database. Our classification strategies identified 122 potential 5-HT2B ligands. Ten structurally diverse VS hits were experimentally tested at PDSP. Nine compounds were experimentally confirmed as 5-HT2B ligands thereby demonstrating a very high success rate of 90%.
The predictor developed in this report is similar in its potential use to other predictors of drug liability such as carcinogenicity and mutagenicity that are widely used in pharmaceutical industry. For instance, the TOPKAT program available in the Discovery Studio,89 is a QSAR-based system that generates and validates accurate, rapid assessments of various types of chemical toxicity solely from a chemical's molecular structure. In contrast, our predictor is a unique specialized tool for the prediction of 5-HT2B activity and therefore prioritizing compounds for functional testing against 5-HT2B receptors to assess their valvulopathic potential. Therefore, this predictor can be used, along with other computational chemical health risk assessment tools, to evaluate compounds’ safety at early stages of the drug development. It can be used as well to verify that all drugs available on the market are free from possibly fatal valvulopathic risk. This predictor will be made publicly available at the ChemBench server established in the Laboratory for Molecular Modeling (chembench.mml.unc.edu). We will also gladly apply this predictor to any compound library that may be of interest to any researcher.
Experimental Section
Radioligand Binding Assays
This screen was performed by the National Institute of Mental Health Psychoactive Drug Screening Program (PDSP). Radioligands were purchased by PDSP from Perkin-Elmer or GE Healthcare. Competition binding assays were performed using transfected or stably expressing cell membrane preparations as previously described (Shapiro et al. 2003;90 Roth et al. 200291) and are available online (http://pdsp.med.unc.edu). All experimental details are available online (http://pdsp.med.unc.edu/UNC-CH%20Protocol%20Book.pdf).
Chemistry
Chemical compounds predicted as hits from the virtual screening were obtained from commercial suppliers according to their availability. All compounds were ordered to have ≥ 95% purity. Additionally, all compounds were subjected to purity assessment using LC/MS by the Center for Integrative Chemical Biology and Drug Discovery at UNC-Chapel Hill. LC/MS spectra of all compounds were acquired from an Agilent 6110 Series system with UV detector set to 220 nm. Samples were injected (5 uL) onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 uM, C18 column at room temperature. A linear gradient from 10% to 100% B (MeOH + 0.1% Acetic Acid) in 5.0 min was followed by pumping 100% B for another 2 minutes with A being H2O + 0.1% acetic acid. The flow rate was 1.0 mL/min.
Supplementary Material
Acknowledgements
We are grateful to Dr. Steve Marron for providing us with the DWD program and to Drs. Weifan Zheng and Raed Khashan for developing the SG descriptors and helpful discussions. We thank Tripos, Chemical Computing Group, and eduSoft for software grants. We also thank Ms. Xin Chin from the Center for Integrative Chemical Biology and Drug Discovery at UNC-Chapel Hill for performing the purity control tests. Finally, we acknowledge the access to the computing facilities at the ITS Research Computing Division of the University of North Carolina at Chapel Hill. The studies reported in this paper were supported in part by the NIH research grant GM066940 and the planning grant HG003898 (awarded to AT); RO1MH61887 and an NIH contract U19MH82441, supporting the NIMH Psychoactive Drug Screening Program (awarded to BR); and by the University of Jordan scholarship (awarded to RH).
Abbreviations
- 5-HT2B
5-Hydroxy Tryptamine subtype 2B receptors
- AD
Applicability Domain
- CARs
Class Association Rules
- CBA
Classification Based on Association
- CCR
Correct Classification Rate
- CCRtrain
Correct Classification Rate for training set
- CCRtest
Correct Classification Rate for test set
- CCRevs
Correct Classification Rate for external validation set
- CCRex
Correct Classification Rate for external set
- CCRrand
Correct Classification Rate of the random models using the external validation set
- CV
Cross Validation
- DWD
Distance Weighted Discrimination
- E
Enrichment
- En
Normalized Enrichment
- FN
False Negative
- FP
False Positive
- HTS
High Throughput Screen
- kNN
K Nearest Neighbor
- LOO-CV
Leave-One-Out Cross Validation
- MFD
Most Frequent Descriptors
- MOE
Molecular Operating Environment
- MZ
MolConnZ descriptors
- PDSP
NIMH Psychoactive Drug Screening Program
- QSAR
Quantitative Structure Activity Relationships
- SA
Simulated Annealing
- SE
Sensitivity
- SG
Subgraph
- SP
Specificity
- TP
True Positive
- TN
True Negative
- VHD
Valvular Heart Disease
- VS
Virtual Screening
- WDI
World Drug Index
Footnotes
Supporting Information Available: 5-HT2B datasets and external validation sets, virtual screening hits, and further computational details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Connolly HM, Crary JL, Mcgoon MD, Hensrud DD, Edwards BS, Edwards WD, Schaff HV. Valvular heart disease associated with fenfluramine-phentermine. New England Journal of Medicine. 1997;337:581–588. doi: 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- 2.Pritchett AM, Morrison JF, Edwards WD, Schaff HV, Connolly HM, Espinosa RE. VaIvular heart disease in patients taking pergolide. Mayo Clinic Proceedings. 2002;77:1280–1286. doi: 10.4065/77.12.1280. [DOI] [PubMed] [Google Scholar]
- 3.Peralta C, Wolf E, Alber H, Seppi K, Muller S, Bosch S, Wenning GK, Pachinger O, Poewe W. Valvular heart disease in Parkinson's disease vs. controls: An echocardiographic study. Movement Disorders. 2006;21:1109–1113. doi: 10.1002/mds.20887. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto M, Uesugi T, Nakayama T. Dopamine agonists and cardiac valvulopathy in Parkinson disease - A case-control study. Neurology. 2006;67:1225–1229. doi: 10.1212/01.wnl.0000238508.68593.1d. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto M, Uesugi T. Dopamine agonists and valvular heart disease in patients with Parkinson's disease: evidence and mystery. Journal of Neurology. 2007;254:74–78. [Google Scholar]
- 6.Schade R, Andersohn F, Suissa S, Haverkamp W, Garbe E. Dopamine agonists and the risk of cardiac-valve regurgitation. New England Journal of Medicine. 2007;356:29–38. doi: 10.1056/NEJMoa062222. [DOI] [PubMed] [Google Scholar]
- 7.Zanettini R, Antonini A, Gatto G, Gentile R, Tesei S, Pezzoli G. Valvular heart disease and the use of dopamine agonists for Parkinson's disease. New England Journal of Medicine. 2007;356:39–46. doi: 10.1056/NEJMoa054830. [DOI] [PubMed] [Google Scholar]
- 8.Droogmans S, Cosyns B, D'haenen H, Creeten E, Weytjens C, Franken PR, Scott B, Schoors D, Kemdem A, Close L, Vandenbossche JL, Bechet S, Van Camp G. Possible association between 3,4-methylenedioxymethamphetamine abuse and valvular heart disease. Am. J. Cardiol. 2007;100:1442–1445. doi: 10.1016/j.amjcard.2007.06.045. [DOI] [PubMed] [Google Scholar]
- 9.Setola V, Hufeisen SJ, Grande-Allen KJ, Vesely I, Glennon RA, Blough B, Rothman RB, Roth BL. 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) induces fenfluramine-like proliferative actions on human cardiac valvular interstitial cells in vitro. Mol. Pharmacol. 2003;63:1223–1229. doi: 10.1124/mol.63.6.1223. [DOI] [PubMed] [Google Scholar]
- 10.Setola V, Roth BL. The emergence of 5-HT2B receptors as targets to avoid in designing and refining pharmaceuticals. In: Roth BL, editor. The Serotonin Receptors: From Pharmacology to Human Therapeutics. Human Press; Totowa, NJ: 2008. p. 419. [Google Scholar]
- 11.Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ, Roth BL. Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation. 2000;102:2836–2841. doi: 10.1161/01.cir.102.23.2836. [DOI] [PubMed] [Google Scholar]
- 12.Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu. Rev. Med. 2009;60:355–366. doi: 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roth BL. Drugs and valvular heart disease. N. Engl. J. Med. 2007;356:6–9. doi: 10.1056/NEJMp068265. [DOI] [PubMed] [Google Scholar]
- 14.Setola V, Roth BL. Screening the receptorome reveals molecular targets responsible for drug-induced side effects: focus on ‘fen-phen’. Expert Opin. Drug Metab Toxicol. 2005;1:377–387. doi: 10.1517/17425255.1.3.377. [DOI] [PubMed] [Google Scholar]
- 15.Newman-Tancredi A, Cussac D, Quentric Y, Touzard M, Verriele L, Carpentier N, Millan MJ. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. III. Agonist and antagonist properties at serotonin, 5-HT(1) and 5-HT(2), receptor subtypes. J. Pharmacol. Exp. Ther. 2002;303:815–822. doi: 10.1124/jpet.102.039883. [DOI] [PubMed] [Google Scholar]
- 16.Nebigil CG, Choi DS, Dierich A, Hickel P, Le Meur M, Messaddeq N, Launay JM, Maroteaux L. Serotonin 2B receptor is required for heart development. Proc. Natl. Acad. Sci. U. S A. 2000;97:9508–9513. doi: 10.1073/pnas.97.17.9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nebigil CG, Launay JM, Hickel P, Tournois C, Maroteaux L. 5-hydroxytryptamine 2B receptor regulates cell-cycle progression: cross-talk with tyrosine kinase pathways. Proc. Natl. Acad. Sci. U. S A. 2000;97:2591–2596. doi: 10.1073/pnas.050282397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL, Hartig PR, Hollis GF, Meunier PC, Robichaud AJ, Robertson DW. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol. Pharmacol. 2000;57:75–81. [PubMed] [Google Scholar]
- 19.Levy RJ. Serotonin transporter mechanisms and cardiac disease. Circulation. 2006;113:2–4. doi: 10.1161/CIRCULATIONAHA.105.593459. [DOI] [PubMed] [Google Scholar]
- 20.Stahura FL, Bajorath J. Virtual screening methods that complement HTS. Combinatorial Chemistry & High Throughput Screening. 2004;7:259–269. doi: 10.2174/1386207043328706. [DOI] [PubMed] [Google Scholar]
- 21.Chemoinformatics Approaches to Virtual Screening. RSCPublishing; Cambridge, UK: 2008. [Google Scholar]
- 22.Chekmarev DS, Kholodovych V, Balakin KV, Ivanenkov Y, Ekins S, Welsh WJ. Shape signatures: New descriptors for predicting cardiotoxicity in silico. Chemical Research in Toxicology. 2008;21:1304–1314. doi: 10.1021/tx800063r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang XP, Setola V, Yadav PN, Allen JA, Rogan SC, Hanson BJ, Revankar C, Robers M, Doucette C, Roth BL. Parallel functional activity profiling reveals valvulopathogens are potent 5-hydroxytryptamine(2B) receptor agonists: implications for drug safety assessment. Molecular Pharmacology. 2009;76:710–722. doi: 10.1124/mol.109.058057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Cerqueira LP, Golbraikh A, Oloff S, Xiao Y, Tropsha A. Combinatorial QSAR modeling of P-glycoprotein substrates. J. Chem. Inf. Model. 2006;46:1245–1254. doi: 10.1021/ci0504317. [DOI] [PubMed] [Google Scholar]
- 25.Kovatcheva A, Golbraikh A, Oloff S, Xiao YD, Zheng W, Wolschann P, Buchbauer G, Tropsha A. Combinatorial QSAR of ambergris fragrance compounds. J. Chem. Inf. Comput. Sci. 2004;44:582–595. doi: 10.1021/ci034203t. [DOI] [PubMed] [Google Scholar]
- 26.Tropsha A, Golbraikh A. Predictive QSAR Modeling Workflow, Model Applicability Domains, and Virtual Screening. Curr. Pharm. Des. 2007;13:3494–3504. doi: 10.2174/138161207782794257. [DOI] [PubMed] [Google Scholar]
- 27.Mohan CG, Gandhi T, Garg D, Shinde R. Computer-assisted methods in chemical toxicity prediction. Mini-Reviews in Medicinal Chemistry. 2007;7:499–507. doi: 10.2174/138955707780619554. [DOI] [PubMed] [Google Scholar]
- 28.Simon-Hettich B, Rothfuss A, Steger-Hartmann T. Use of computer-assisted prediction of toxic effects of chemical substances. Toxicology. 2006;224:156–162. doi: 10.1016/j.tox.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 29.Benfenati E, Benigni R, DeMarini DM, Helma C, Kirkland D, Martin TM, Mazzatorta P, Ouedraogo-Arras G, Richard AM, Schilter B, Schoonen WGEJ, Snyder R, Yang C. Predictive Models for Carcinogenicity and Mutagenicity: Frameworks, State-of-the-Art, and Perspectives. Journal of Environmental Science and Health Part C-Environmental Carcinogenesis & Ecotoxicology Reviews. 2009;27:57–90. doi: 10.1080/10590500902885593. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz P, Faroon O, Moudgal CJ, Hansen H, De Rosa CT, Mumtaz M. Prediction of the health effects of polychlorinated biphenyls (PCBs) and their metabolites using quantitative structure-activity relationship (QSAR). Toxicology Letters. 2008;181:51–63. doi: 10.1016/j.toxlet.2008.06.870. [DOI] [PubMed] [Google Scholar]
- 31.Venkatapathy R, Wang CY, Bruce RM, Moudgal C. Development of quantitative structure-activity relationship (QSAR) models to predict the carcinogenic potency of chemicals I. Alternative toxicity measures as an estimator of carcinogenic potency. Toxicology and Applied Pharmacology. 2009;234:209–221. doi: 10.1016/j.taap.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 32.Papa E, Pilutti P, Gramatica P. Prediction of PAH mutagenicity in human cells by QSAR classification. Sar and Qsar in Environmental Research. 2008;19:115–127. doi: 10.1080/10629360701843482. [DOI] [PubMed] [Google Scholar]
- 33.Zhang ZY, Niu JF, Zhi X. A QSAR model for predicting mutagenicity of nitronaphthalenes and methylnitronaphthalenes. Bulletin of Environmental Contamination and Toxicology. 2008;81:498–502. doi: 10.1007/s00128-008-9540-4. [DOI] [PubMed] [Google Scholar]
- 34.Ekins S, Crumb WJ, Sarazan RD, Wikel JH, Wrighton SA. Three-dimensional quantitative structure-activity relationship for inhibition of human ether-a-go-go-related gene potassium channel. Journal of Pharmacology and Experimental Therapeutics. 2002;301:427–434. doi: 10.1124/jpet.301.2.427. [DOI] [PubMed] [Google Scholar]
- 35.Garg D, Gandhi T, Mohan CG. Exploring QSTR and toxicophore of hERG K+ channel blockers using GFA and HypoGen techniques. Journal of Molecular Graphics & Modelling. 2008;26:966–976. doi: 10.1016/j.jmgm.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Seierstad M, Agrafiotis DK. A QSAR model of hERG binding using a large, diverse, and internally consistent training set. Chemical Biology & Drug Design. 2006;67:284–296. doi: 10.1111/j.1747-0285.2006.00379.x. [DOI] [PubMed] [Google Scholar]
- 37.Yoshida K, Niwa T. Quantitative structure-activity relationship studies on inhibition of HERG potassium channels. Journal of Chemical Information and Modeling. 2006;46:1371–1378. doi: 10.1021/ci050450g. [DOI] [PubMed] [Google Scholar]
- 38.PubChem NIH's Molecular Libraries Roadmap Initiative . 2009.
- 39.MOE Chemical Computing Group [2007.09 ] 2008.
- 40.ChemAxon JChem. 2010.
- 41.Golbraikh A, Tropsha A. Beware of q2! J. Mol. Graph. Model. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- 42.Golbraikh A, Tropsha A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Aided Mol. Des. 2002;16:357–369. doi: 10.1023/a:1020869118689. [DOI] [PubMed] [Google Scholar]
- 43.Talete s.r.l. Dragon. [5.4.2006] Milan; Italy: 2007. [Google Scholar]
- 44.MolconnZ 2006 http://www.edusoft-lc.com/molconn/
- 45.Khashan R, Zheng WF, Huan J, Wang W, Tropsha A. Development of fragment-based chemical descriptors using novel frequent common subgraph mining approach and their application in QSAR modeling. Abstracts of Papers of the American Chemical Society. 2005;230:U1335–U1336. [Google Scholar]
- 46.Todeschini R, Consonni V. Handbook of molecular descriptors. Wiley; Weinheim (Germany): 2000. [Google Scholar]
- 47.Kier LB, Hall LH. Molecular connectivity in structure–activity analysis. Wiley; New York: 1986. [Google Scholar]
- 48.Kier LB, Hall LH. Molecular connectivity in chemistry and drug research. Academic Press; New York: 1976. [Google Scholar]
- 49.Randic M. Characterization of Molecular Branching. Journal of the American Chemical Society. 1975;97:6609–6615. [Google Scholar]
- 50.Kier LB. Inclusion of Symmetry As A Shape Attribute in Kappa-Index Analysis. Quantitative Structure-Activity Relationships. 1987;6:8–12. [Google Scholar]
- 51.Kier LB. A Shape Index from Molecular Graphs. Quantitative Structure-Activity Relationships. 1985;4:109–116. [Google Scholar]
- 52.Hall LH, Kier LB. Determination of Topological Equivalence in Molecular Graphs from the Topological State. Quantitative Structure-Activity Relationships. 1990;9:115–131. [Google Scholar]
- 53.Kier LB, Hall LH. Molecular structure description: The electrotopological state. Academic Press; New York: 1999. [Google Scholar]
- 54.Kellogg GE, Kier LB, Gaillard P, Hall LH. E-state fields: Applications to 3D QSAR. Journal of Computer-Aided Molecular Design. 1996;10:513–520. doi: 10.1007/BF00134175. [DOI] [PubMed] [Google Scholar]
- 55.Hall LH, Mohney B, Kier LB. The Electrotopological State - Structure Information at the Atomic Level for Molecular Graphs. Journal of Chemical Information and Computer Sciences. 1991;31:76–82. [Google Scholar]
- 56.Hall LH, Mohney B, Kier LB. The Electrotopological State - An Atom Index for Qsar. Quantitative Structure-Activity Relationships. 1991;10:43–51. [Google Scholar]
- 57.Kier LB, Hall LH. A Differential Molecular Connectivity Index. Quantitative Structure-Activity Relationships. 1991;10:134–140. [Google Scholar]
- 58.Petitjean M. Applications of the Radius Diameter Diagram to the Classification of Topological and Geometrical Shapes of Chemical-Compounds. Journal of Chemical Information and Computer Sciences. 1992;32:331–337. [Google Scholar]
- 59.Wiener HJ. Structural determination of paraffin boiling points. J. Am. Chem. Soc. 1947;69:17–20. doi: 10.1021/ja01193a005. [DOI] [PubMed] [Google Scholar]
- 60.Platt JR. Influence of neighbor bonds on additive bond properties in paraffins. J. Chem. Phys. 1947;15:419–420. [Google Scholar]
- 61.Shannon C, Weaver W. In mathematical theory of communication. University of Illinois; p. 1949. [Google Scholar]
- 62.Bonchev D, Mekenyan O, Trinajstic N. Isomer Discrimination by Topological Information Approach. Journal of Computational Chemistry. 1981;2:127–148. [Google Scholar]
- 63.Balaban AT. Highly Discriminating Distance-Based Topological Index. Chemical Physics Letters. 1982;89:399–404. [Google Scholar]
- 64.Balaban AT. Five new topological indices for the branching of tree-like graphs. Theor. Chim. Acta. 1979:355–375. [Google Scholar]
- 65.Huan J, Prins J, Wang W. Efficient Mining of Frequent Subgraph in the Presence of Isomorphism. 2003. pp. 549–552.
- 66.Sheridan RP, Kearsley SK. Why do we need so many chemical similarity search methods? Drug Discov. Today. 2002;7:903–911. doi: 10.1016/s1359-6446(02)02411-x. [DOI] [PubMed] [Google Scholar]
- 67.Holliday JD, Hu CY, Willett P. Grouping of coefficients for the calculation of inter-molecular similarity and dissimilarity using 2D fragment bit-strings. Comb. Chem. High Throughput Screen. 2002;5:155–166. doi: 10.2174/1386207024607338. [DOI] [PubMed] [Google Scholar]
- 68.MDL Ltd. MACCS. MDL Ltd.; San Leandro, CA: 1992. [Google Scholar]
- 69.Zheng W, Tropsha A. Novel variable selection quantitative structure--property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci. 2000;40:185–194. doi: 10.1021/ci980033m. [DOI] [PubMed] [Google Scholar]
- 70.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953;21:1087–1092. [Google Scholar]
- 71.Kirkpatrick S, Gelatt CD, Vecchi MP. Optimization by Simulated Annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 72.Golbraikh A, Shen M, Tropsha A. Enrichment: A new estimator of classification accuracy of QSAR models. Abstracts of Papers of the American Chemical Society. 2002;223:U494–U495. [Google Scholar]
- 73.Breiman L, Friedman J, Olshen R, Stone C. Classification and regression trees. Wadsworth; Belmont: 1984. [Google Scholar]
- 74.Quinlan JR. C4.5: program for machine learning. Morgan Kaufmann; 1992. [Google Scholar]
- 75.Agrawal R, Srikant R. Fast algorithms for mining association rules. VLDB-94; 1994. [Google Scholar]
- 76.Liu B, Hsu W, Ma Y. Integrating classification and association rule mining; Fourth International conference on Knowledge Discovery and Data Mining (KDD-98, Plenary Presentation); New York: 1998. pp. 80–68. [Google Scholar]
- 77.Liu B, Hsu W, Ma Y. Pruning and summarizing the discovered associations.. Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery & Data Mining (KDD-99, full paper); San Diego, CA, USA.. 8-15-1999.1999. [Google Scholar]
- 78.Liu B, Hsu W, Ma Y. Classification Based on Association (CBA). [v2.1] School of Computing, National University of Singapore; 2001. [Google Scholar]
- 79.Marron JS, Todd MJ, Ahn J. Distance-weighted discrimination. Journal of the American Statistical Association. 2007;102:1267–1271. doi: 10.1198/jasa.2010.tm08487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cristiainini N, Shawe-Taylor J. An introduction to support vector machines. Cambridge University Press; Cambridge, United Kingdom: 2000. [Google Scholar]
- 81.Vapnik VN. In the nature of statistical learning theory. Springer Verlag; New York: 1995. [Google Scholar]
- 82.Alizadeh F, Goldfarb D. Second-order cone programming. Mathematical Programming. 2003;95:3–51. [Google Scholar]
- 83.Marron JS. MATLAB software for smoothing, functional data analysis and distance weighted discrimination. 2002.
- 84.Matlab 2010.
- 85.Wold S.a.E.L. Statistical Validation of QSAR Results. In: H., v. d. W., editor. Chemometrics Methods in Molecular Design. VCH; Weinheim: 1995. pp. 309–318. [Google Scholar]
- 86.Shen M, Beguin C, Golbraikh A, Stables JP, Kohn H, Tropsha A. Application of predictive QSAR models to database mining: identification and experimental validation of novel anticonvulsant compounds. J. Med. Chem. 2004;47:2356–2364. doi: 10.1021/jm030584q. [DOI] [PubMed] [Google Scholar]
- 87.Zhu H, Tropsha A, Fourches D, Varnek A, Papa E, Gramatica P, Oberg T, Dao P, Cherkasov A, Tetko IV. Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis. Journal of Chemical Information and Modeling. 2008;48:766–784. doi: 10.1021/ci700443v. [DOI] [PubMed] [Google Scholar]
- 88.Daylight, World Drug Index (WDI) 2004.
- 89.Discovery Studio . Accelrys [2.1] Accelrys; 2008. [Google Scholar]
- 90.Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, Roth BL, Mailman R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology. 2003;28:1400–1411. doi: 10.1038/sj.npp.1300203. [DOI] [PubMed] [Google Scholar]
- 91.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: A potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.