

Abstract
Mutations in the parkin gene are linked to development of juvenile onset of Parkinson’s disease and recent studies have reported that parkin can protect against increased oxidative stress and mitochondrial dysfunction caused by a variety of oxidative and toxic insults. Overexpression of parkin has also been reported to selectively protect dopaminergic neurons from Mn toxicity. Accordingly, in this paper we compare the effect that mutations in parkin have on Mn toxicity and associated apoptotic signals in normal and human B lymphocyte cell lines containing a homozygous mutation in the gene. Results of these studies reveal that Mn toxicity was similar in both control and mutant parkin lymphocyte cells indicating that cell death caused by Mn was not altered in cells devoid of parkin activity. In contrast, Mn did inhibit mitochondrial function to a greater extent in cells devoid of active parkin as indicated by a decrease in ATP production although mitochondrial membrane potential was essentially unaffected. Consistent with inactive parkin influencing the Mn response is the observation of increased activity in the down-stream apoptotic signal, caspase 3. In summary, results reported in this paper demonstrate that mutations in parkin can lead to functional changes in potential signaling processes known to provoke Mn toxicity. The selectivity and magnitude of this response, however, does not necessarily lead to cell death in lymphocytes which are devoid of dopamine.
Keywords: manganese, parkin, Parkinson’s disease, manganism, lymphocytes, oxidative stress, apoptosis
1. Introduction
The syndrome of Mn toxicity, known as manganism, can express itself in a diverse set of symptoms that, in the more chronic stages, resemble the dystonic movements associated with Parkinson’s disease (Roth, 2006, 2009). This coupled with the observation that Mn is one of the most influential metals correlating with increased susceptibility to develop Parkinson’s disease implies there may be common biochemical defects between the two disorders (Gorell et al., 1999; Hudnell, 1999; Kim et al., 2002). In the initial stages of manganism, the preferential target of Mn is the globus pallidus as opposed to the dopaminergic neurons in the substantia nigra pars compacta seen in Parkinson’s disease. Although these neurons are preserved in manganism, there is functional overlap between the two disorders as Mn overexposure can result in disruption of dopaminergic function in the striatum (Guilarte et al., 2008; Verina et al., 2010).
Within the past two decades considerable progress has been made in regard to understanding the biochemical mechanism responsible for Mn toxicity although there is no explanation as to why there are wide deviations in both susceptibility and characteristics of the symptoms observed. It is reasonable to speculate that some of the differences observed may be due to underlying genetic variability either in signaling processes directly accountable for loss of pallidal neurons or possibly as a consequence to specific neurodegenerative processes responsible or relate to acquisition of Parkinson’s disease. Mutations in several genes have previously been identified as correlating with onset of Parkinson’s disease which include parkin (PARK2), α-synuclein, DJ-1, PINK1, ATP13A2 and LRRK2 (Lesage and Brice, 2009; Wider and Wszolek, 2007; Yang et al., 2009). Because of the association between the two disorders, the question is raised as to whether mutations in these genes can also lead to cellular changes which potentially can elicit development of Mn toxicity. In fact, two of these genes, parkin (Roth et al., 2010) and ATP13A2 (Gitler et al., 2009), have already been reported to protect cells from the neurotoxic actions of Mn. Thus, one-third of the known polymorphisms in genes associated with juvenile and/or late onset Parkinsonism have now been acknowledged to potentially correlate with development of manganism.
Mutations in parkin were the first genes to be recognized as being associated with pathogenesis of autosomal recessive-juvenile Parkinson’s disease (Kitada et al., 1998). Of the genes linked to early onset of the disorder, those involving parkin are the most prevalent encompassing approximately 50% of all recessive Parkinson’s disease cases (Mizuno et al., 2001; West et al., 2002). Parkin is one of over 600 E3 ligases responsible for the conjugation of ubiquitin to a variety of proteins which in the case of parkin include CDCrel-1, Pael receptor, O-glycosylated α-synculein, synphilin, cyclin E, α/β tubulin, p38/JTV-1, and synaptotagmin (Dev et al., 2003; Hilker et al., 2001; Huynh et al., 2003; Ikeuchi et al., 2009; Ko et al., 2005; Lim et al., 2005; Ren et al., 2003; Szargel et al., 2008). Although considered to be a recessive linked gene, there is evidence in the literature suggesting that mutations in a single allele may exert sufficient imbalance in dopaminergic activity within the substantia nigra to cause subclinical features of Parkinsonism (Hilker et al., 2001). Consistent with this is the fact that heterozygous mutations in parkin have also been reported to play a significant role in sporadic or late-onset of Parkinson’s disease (Oliveira et al., 2003; Satoh and Kuroda, 1999; West et al., 2002). Based on the correlation between parkin and Mn toxicity, it can also be anticipated that individuals that are heterozygous for parkin mutations may similarly be at greater risk to develop manganism.
In regard to parkin, Higashi et al. (Higashi et al., 2004) have previously reported that the protective effect of parkin against Mn toxicity was preferentially occurred in dopaminergic neuronal cell lines including mouse CATH.a and human SH-SY5Y cells, but not in Neuro-2a cells devoid of dopamine. This implies the presence of the catecholamine as being a central component necessary for the selective Mn-induced neurotoxic actions of parkin. Recent studies in our laboratory (Roth et al., 2010) have demonstrated that parkin is responsible for the ubiquitination and subsequent degradation of the major Mn transport protein, divalent metal transporter 1 (DMT1) in human SH-SY5Y cells and human lymphocytes and therefore, is capable of regulating toxicity via uptake of Mn. Since mutations in parkin have also been reported to promote increased oxidative stress and mitochondrial dysfunction as a consequence to a variety of toxic insults (Hyun et al., 2005), it is equally possible that alterations in these signaling pathways can also contribute to parkin-induced Mn toxicity independent of its effect on transport. The availability of human lymphocytes which express either normal or a homozygous mutation in parkin makes it feasible to assess the effects of parkin deficiency on Mn viability as the cytotoxic mechanism of Mn has been well characterized in these cells (El Mchichi et al., 2007; Lima et al., 2008; Schrantz et al., 1999).
2. Materials and methods
2.1 Materials
The Epstein Barr-transformed human B lymphocytes ND01037 and ND00092 were provided by Dr. Jian Feng, Department of Physiology and Biophysics, University at Buffalo. ND01037 cells were derived from a PD patient (male Caucasian, 33 years of age at sampling) with homozygous deletion of exon 4 of parkin whereas ND00092 was obtained from an unrelated and unaffected individual (male, Caucasian, 37 years). Both cell lines are commercially available from Coriell Cell Repositories (Camden, NJ) and have been used as a model to examine the effects of parkin deletion on a number of cellular systems (Jiang et al., 2006; Ren et al., 2009; Roth et al., 2010). RPMI 1640 medium and L-glutamine were obtained from Invitrogen (BRL, Grand Island, NY).
2.2. Cell culture conditions and cell viability assay
The lymphocyte cell lines, ND01037 and ND00092 were maintained in RPMI 1640 medium containing 15% fetal bovine serum and 2 mM L-glutamine as described previously (Roth et al., 2010). Cells were grown as a suspension culture at 37° C in a humidified atmosphere containing 5% CO2 and subcultured every 3 – 4 days. The lymphocyte cell lines were treated with vehicle or MnCl2 at varying concentrations and cell viability was measured by trypan blue dye exclusion method over varying time points as indicated.
2.3 Caspase assay
To measure caspase 3 activity, cell lysates were incubated for 30 min. on ice in lysis buffer containing complete protease inhibitor and subsequently centrifuged to remove particulates. Protein concentration was determined using BCA assay. Aliquots of the lysates were transferred to a 96 well plate and assayed for caspase-3 activity using the Enzchek Caspase-3 assay kit (Molecular Probes, Grand Island, NY). Solutions of Z-DEVD-AMC substrate were added to all the wells and incubated for 30 min at room temperature. Fluorescence was measured using excitation wavelength at 350 nm and emission detection at 450 nm on a Fluorescence multi-well plate reader (Biotek, VT).
2.4 ATP assay
The CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega, Madison, WI) was used to assess ATP levels. The two lymphocyte cell lines were treated with 0.5 mM MnCl2 for 12 and 24 hrs. and subsequently transferred to a 96 well plate. A volume of CellTiter-Glo® Reagent equal to the volume of cell culture medium present was added to each well. The plates were incubated for 2 min. to induce cell lysis followed by a 10 min. incubation to stabilize the luminescent signal which was recorded using a Biotek Plate Reader (Winooski, VT).
2.5 Mitochondrial membrane potential
Mitochondrial membrane potential was assessed in the lymphocyte cell lines treated in the presence and absence of 0.5 mM MnCl2 for 12 hrs. The cells were harvested by centrifuging at 400 × g and resuspended in 1X PBS. The MitoProbe JC-1 assay kit (Molecular Probes, Inc., Eugene, OR) was used to measure the mitochondrial membrane potential. The JC-1 dye was added to the cell suspension at a final concentration of 2 μM and incubated for 15–30 min. As a positive control, carbonyl cyanide 3-chlorophenylhydrazone (CCCP) was added to a final concentration of 50 μM. The cells were washed once with 1X PBS and analyzed using a flow cytometry for production of red color indicative of healthy mitochondria.
2.6 Mn Uptake measurement
The amount of Mn taken up by the two lymphocyte cell lines was measured by Dr. Andy Ghio at the US EPA Human Studies Facility using Inductively Coupled Plasma Atomic Adsorption Spectroscopy. Cells were treated with 0.5 mM MnCl2 at varying time points as indicated. Cells were washed once with 1X PBS and resuspended in ultraclean 3 M HCl/10% TCA. Approximately equal number of cells was used for analyzing the Mn levels and all samples were blinded for this study.
2.7 Statistical considerations
All data presented are based on results from a minimum of three independent experiments performed in duplicate. Statistical significance was based on a two-tailed or paired t-test (ATP) with P values equal to or less than 0.05 as being considered significant. Analysis of variance was performed for the trypan blue dye exclusion and MnCl2 uptake experiments with P values equal to or less than 0.05 being considered as significant.
3. Results
Based on prior studies demonstrating that overexpression of parkin can protect against Mn toxicity, studies were performed to examine the effect of Mn in cells devoid of parkin. For these studies we utilized two human lymphocyte cell lines, one with normal parkin ND00092, and the other containing an inactive mutant form of the ligase ND01037. Since protection from Mn-induced toxicity was only observed in cells containing dopamine (Higashi et al., 2004), application of the lymphocytes allowed us to assess only the direct interactions of mutant parkin on cellular processes independent of the presence of dopamine. Initial studies were conducted to determine the effect of Mn on cell viability as both a function of time and dose. As illustrated in Fig. 1, there was no significant difference in percentage of cell survival between the ND00092 and ND01037 cells when treated with Mn for 24 hrs. at all concentrations tested. At 0.5 mM Mn, there was approximately 25% inhibition of cell viability and at 1.0 mM this value was slightly greater than 40%. As illustrated in Fig. 2, cell toxicity at 0.5 mM Mn was essentially linear over the range of times tested and, again, there was no significant difference between the two cell lines. These results were somewhat surprising as it had previously been shown that the parkin mutant cell line used in this experiment possessed higher levels of the Mn transporter, DMT1 (Roth et al., 2010).
Fig. 1.

The effect of Mn toxicity in the lymphocyte cell lines, ND00092 and ND01037. The cells were treated for 24 hours with varying concentration. Percent survival was measured by trypan blue dye exclusion assay. Data is the mean ± S.E of three independent experiments.
Fig. 2.

The effect time on Mn toxicity in the lymphocyte cell lines, ND00092 and ND01037. Cells were treated with 0.5 mM Mn for various lengths of time as indicated. Percent cell death was measured by Trypan blue dye exclusion assay. Data is the mean ± S.E of three independent experiments.
Because of the differences in DMT1 expression, it was of interest to determine whether the parkin negative mutant cells would, in fact accumulate more Mn as compared to the wild-type lymphocyte cell line. Despite difference in DMT1 expression, results of these studies, shown in Fig. 3, indicate there was no significant increase in the mean uptake of Mn over the four time points examined between the parkin mutant cells and the control cultures suggesting that DMT1 may not be the only transporter for Mn in these cells.
Fig. 3.

Mn accumulation (ppm/mg protein) in lymphocyte cell lines, ND00092 and ND01037, treated in the absence and presence of 0.5 mM manganese (Mn) for varying lengths of time as indicated. Mn levels were determined using Inductively Coupled Plasma Atomic Adsorption Spectroscopy. Approximately an equal number of cells were used for analyzing Mn levels. Data is the mean ± S.E of three independent experiments.
Besides the involvement of parkin in the ubiquitination of proteins, it has also been shown to be implicated in maintenance of mitochondrial membrane potential (Kuroda et al., 2011). Based on these findings and the fact that Mn has previously been reported to lead to opening of the permeability transition pore (Gunter et al., 2009), studies were performed to compare the effect Mn on mitochondrial membrane potential in the two cell lines. To measure Mn-induced changes in mitochondrial membrane potential, the fluorescent dye, JC-1, was employed. For these experiments loss of mitochondrial membrane potential is indicated by the decrease in red fluorescence and increase in green fluorescent intensity of the dye. The histograms for both the cell lines are shown in Fig. 4A and quantification of this data from three separate experiments is reported in Fig. 4B. Results of this study reveal that Mn produces a small but non-significant increase in membrane potential in both cell lines. Consistent with the findings noted above, there is little difference in in the response to Mn between the two cell lines under the conditions employed.
Fig. 4.


Effect of Mn on relative mitochondrial membrane potential (ψ) in lymphocyte cell lines, ND00092 and ND01037. Cells were treated with 0.5 mM Mn for 12 hrs. and then incubated for 15 min. with the JC-1 dye. The cells were then washed once with 1X PBS and subsequently analyzed by flow cytometry. (A) The histograms for both the cell lines obtained from the scatter plots. using the FL2 channel as a measure of red fluorescence. The solid lines indicate the untreated cells; dash lines represent the cells treated with Mn and dotted lines served as the positive control (cyanide 3-chlorophenylhydrazone; CCCP)). The marker M1 represents percentage of gated cells with healthy mitochondria, while the marker M2 represents percentage of gated cells with depolarized mitochondria. (B) The average relative ψ indicates there is no significant difference between the 2 cell lines. The data is the mean ± S.E of three independent experiments.
Since mutations in parkin have also been reported to cause a decrease in ATP production after oxidative insult, it was of interest to determine whether Mn would differentially alter ATP production in the cell line devoid of parkin activity. This is especially relevant as Mn has been shown to have a direct effect on oxidative phosphorylation by inhibiting both mitochondrial F1-ATPase (Gavin et al., 1992, 1999) and complex I (Galvani et al., 1995), causing depletion of ATP (Brouillet et al., 1993; Chen and Liao, 2002; Roth et al., 2000; Zhang et al., 2005). Studies were thus, performed to examine the effect of Mn in both the control and parkin-deficient lymphocytes. In contrast to results obtained with the mitochondrial membrane potential, the data presented in Fig. 5 reveal there is a significant decrease in ATP production in the two cell lines at both the 12 and 24 hrs. time points after initiating treatment with Mn. At 12 hrs., there was almost a 40% decrease in ATP formation in the control cultures and a 50% decrease in the parkin deficient cells. At 24 hrs. there was even a greater loss of ATP content with control cultures losing almost 60% compared to untreated cells and the mutant parkin cells almost 80%. The difference in ATP levels between the control and mutant cell lines was also statistically significant at both the 12 hrs. (p < 0.05) and at 24 hrs. (p < 0.04) time points.
Fig. 5.

The effect Mn on ATP production in lymphocyte cell lines, ND00092 and ND01037. Cells were treated with 0.5 mM Mn for 12 (black bars) and 24 hrs (white bars). The relative ATP formed per mg protein in treated cultures with respect to the untreated samples was calculated for each time point. The decrease in ATP levels produced by Mn was significantly different for ND00092 cell (12 hrs. is ap < 0.02 and at 24 hrs. bp < 0.001) and for the ND01037 cells (12 hours is cp < 0.001and at 24 hrs. dp < 0.005). Statistically significant difference for ATP levels were observed between the ND00092 and ND01037 cell lines at 12 hrs. (p < 0.05) and at 24 hrs. (p < 0.04). The data is the mean ± S.E of three independent experiments.
The decrease in ATP implies that Mn can disrupt mitochondrial function even in the absence of changes in permeability transition pore. Disruption of mitochondria can cause release of cytochrome c which subsequently activates a caspase cascade leading to activation of caspase 3 and apoptosis. Several studies have reported that Mn can promote caspase 3 activity (Chun et al., 2001; Hirata, 2002; Roth et al., 2000; Schrantz et al., 1999). Accordingly, studies were performed to compare the effect of Mn on caspase-3 activity in the two lymphocyte cell lines. Results of these studies, shown in Fig 6, demonstrate a significant difference in the activation of caspase 3 by Mn in both cell lines, consistent with its effects on ATP formation. The increase in caspase activity was approximately 4-fold greater in the normal cells and almost 7-fold in the cell line devoid of parkin. The difference in ATP levels between the two cell lines was significant to p < 0.025.
Fig. 6.
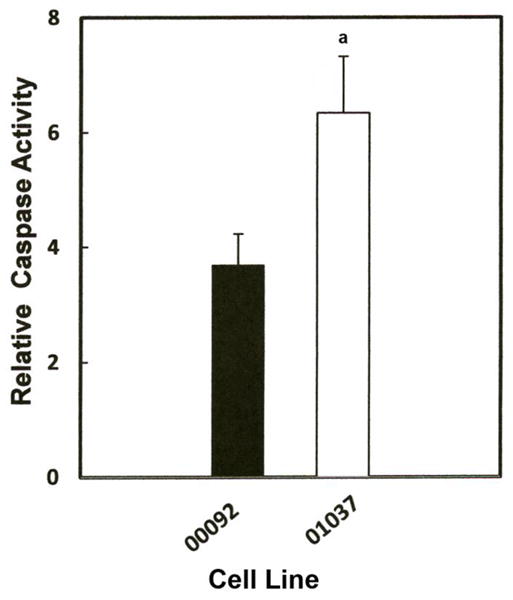
Effect of Mn on caspase-3 activity in lymphocyte cell lines, ND00092 and ND01037. Cells were treated with the 0.5 mM Mn for 12 hrs. Fifty μl of the cell lysate from each sample was added to the 96 well plates in triplicate and incubated with Z-DEVD-AMC for 30 minutes at room temperature. All activities measured were normalized to protein concentration and fluorescence was measured using the CytofluorII Multiplate Reader with excitation set at 380 nm and emission set at 460 nm. Data is the mean ± S.E of three independent experiments. Results indicate there was significant increase in the relative caspase activity in ND01037 when compared to ND00092, ap < 0.025.
4. Discussion
Although the initial lesion observed in the basal ganglia in manganism and Parkinson’s disease differ, the two disorders are not mutually exclusive based on both biochemical and epidemiological evidence suggesting reciprocal dependent mechanisms regulating both their onset and symptoms. Two genes associated with early onset of Parkinsonism, parkin and ATP13A2, have already be shown to be linked to manganese toxicity (Higashi et al., 2004; Roth et al., 2010) and a number of epidemiological studies in a corresponding fashion suggest that exposure to excess Mn may correspondingly provoke premature onset of Parkinson’s disease (Gorell et al., 1999; Hudnell, 1999; Kim et al., 2002). Overlap between the two disorders is not totally unexpected as excess Mn can reduce dopamine transporter levels in the striatum as well as inhibit amphetamine-induced dopamine release both of which will lead to symptoms associated with dopaminergic neuronal cell loss observed in substantia nigra pars compacta in Parkinson’s disease (Guilarte et al., 2008). Thus it is not unreasonable to presume that genes linked with early or late onset of Parkinsonism which accompany dopaminergic cell loss may equally foster Mn-induced cell toxicity.
Recent findings in my laboratory (Roth et al., 2010) and that of Higashi et al. (2004) demonstrate that the gene linked with early onset of Parkinson’s disease, parkin, has the potential to prevent Mn-induced cell death. The studies of Higashi et al. (2004) suggested that parkin preferentially preserves cell viability only in dopaminergic neurons as protection was not observed in non-dopaminergic containing neuronal cultures although the mechanism for this selectivity was not described. These findings imply that parkin may selectively suppress the stimulatory effects of dopamine on the neurotoxic actions produced by Mn. Mechanistically, Mn predominantly promotes apoptosis via its interaction with mitochondria (Roth, 2006, 2009). Disruption of mitochondrial activity invariably leads to decreased production of ATP production in addition to formation of highly reactive oxygen species provoking either apoptosis and/or necrosis which would only be enhanced in the presence of dopamine. Thus, for parkin to protect against Mn toxicity, it would have to attenuate biological signals which lead to production of these cytotoxic events. Thus, it is of interest to determine the effect that parkin may have on the primary stress related processes independent of the presence of dopamine. This is significant since GABAminergic neurons within the globus pallidus which are the preferential targets of Mn are devoid of dopamine.
Prior studies (Higashi et al., 2004; Roth et al., 2010) examining the effects of parkin on Mn toxicity only examined the protective actions in cells overexpressing parkin and did not investigate the effects in cells devoid of parkin activity. Since mutated parkin has been reported to augment cellular responses to a variety of toxic insults (Hyun et al., 2005), we wanted to analyze the consequence that mutations in parkin may have on Mn toxicity. To accomplish this, we utilized transformed human B lymphocyte cell lines possessing either normal parkin or an inactive homozygous mutant form of the gene. Since these cells lack dopamine, the effects of Mn on signaling mechanisms in the two cell lines will be autonomous of its effect on dopamine stability. Parkin deficient lymphocytes have previously been used to study the effects of mutated parkin on a number of biological systems (Jiang et al., 2006; Jimenez Del Rio et al., 2004; Ren et al., 2003; Roth et al., 2010). In this regard, these cells have been reported to lose their ability to suppress monoamine oxidase activity (Jiang et al., 2006) as well as alter MAP kinase activation and microtubule depolymerization (Ren et al., 2003). Homozygote parkin mutation in AR-JP patients was also reported to render lymphocytes sensitive to apoptotic damage caused by dopamine, iron and hydrogen peroxide (Jimenez Del Rio et al., 2004). In addition, the mutant lymphocyte cell line devoid of parkin has also been shown to overexpress DMT1, the major protein responsible for transport of Mn (Roth et al., 2010) which may account for the increased sensitivity of the AR-JP patients to iron toxicity. Mn has been shown to affect lymphocyte cell cycle and promote clastogenicity and DNA damage (Lima et al., 2008).
Similar to the results of Higashi et al. (2004), the data reported in this paper demonstrate that the presence or absence of parkin does not alter Mn toxicity in cells devoid of dopamine. We did observe a Mn-induced decrease in ATP production in both cell lines implying there was an effect on mitochondrial function despite the fact there no difference in mitochondrial membrane potential between the two cell lines. The effect on ATP production is consistent with Mn inhibiting either F1-ATPase (Gavin et al., 1999) and/or complex I (Galvani et al., 1995) within the mitochondria. Why changes in mitochondrial membrane potential were not observed is not known but suggests that inhibition of ATP formation in these cells is a more sensitive and direct consequence of Mn and thus, may be a preferential inhibitory response in the mitochondria. Also, for Mn to influence mitochondrial membrane potential may require greater increases in calcium accumulation in these cells to observe an effect (Gunter et al., 2009) under the conditions employed. More importantly, when comparing the response in mitochondria to Mn, the data demonstrates there was a greater decrease in ATP generation in the cells devoid of parkin activity relative to control lymphocytes. The response of mitochondria to Mn in these mutant cells is likely not caused by changes in internal Mn concentration but as noted by Pacelli et al (Pacelli et al., 2011) may be associated with impaired mitochondrial homeostasis, resulting from mutations in the parkin gene. This may explain the mechanism of Mn induced toxicity in the parkin mutant lymphocytes used in this paper. Changes in ATP production are also accompanied by an increase in Mn-induced apoptotic signaling as revealed by the subsequent increase in activity of the down-stream apoptotic marker caspase 3. Although these measures of apoptotic cell toxicity were clearly elevated in the parkin deficient cells, they failed to significantly alter lymphocyte viability. This conflict may be due to the fact that activation of caspase 3 and loss of ATP may be consequences that are indicative of early stage apoptosis prior to loss of membrane integrity required for trypan blue uptake. Nevertheless, these results imply that the interaction of Mn with dopamine must play an important role in the neurotoxic actions in cells devoid of active parkin as previously suggested by Higashi et al. (2004).
As noted previously, the mutant lymphocytes possess more DMT1 than the wild-type cells which presumably should cause a greater accumulation of Mn in these cells (Roth et al., 2010). Although this was not observed, we cannot totally rule out the possibility that the small increase in Mn uptake which was observed may have been due to elevated DMT1 levels. It is also feasible that the lack of difference in the accumulation of Mn between the two cell lines may also have been caused by the fact that these cells contain two other Mn transporters, ZIP8 and ZIP 14 (Aydemir et al., 2009). Both ZIP proteins have relatively high affinity for Mn similar to that for DMT1. Thus, any difference in the accumulation of Mn caused by elevated DMT1 levels may have been obscured by uptake by these other transporters. Thus, the results observed in this paper suggest that the differences observed in both ATP generation and caspase 3 activity in the mutant cells were most likely not caused by difference in Mn levels in the cell but, instead, by the direct consequence of mutant parkin altering the neurotoxic signaling responsible for Mn-induced cell toxicity. Results of these studies confirm that mutations in the parkin gene has the potential to contribute to the toxic actions of Mn in vivo which may, at least in part, be independent of its effect on DMT1 expression and Mn uptake.
Highlights.
Mn-induced toxicity was not different between control and a lymphocyte cell line lacking active parkin, however, Mn-induced apoptotic markers were increased and ATP production was decreased in the lymphocyte cell line lacking active parkin.
Acknowledgments
This research supported in part by grants from the NIH, R21 ES015762 and RC1 ES0810301. We acknowledge the assistance of the Confocal Microscope and Flow Cytometry Facility in the School of Medicine and Biomedical Sciences, University at Buffalo.
Footnotes
There is no conflict of interest on the part of any of the authors of this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aydemir TB, Liuzzi JP, McClellan S, Cousins RJ. Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-gamma expression in activated human T cells. J Leukoc Biol. 2009;86:337–348. doi: 10.1189/jlb.1208759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet EP, Shinobu L, McGarvey U, Hochberg F, Beal MF. Manganese injection into the rat striatum produces excitotoxic lesions by impairing energy metabolism. Exp Neurol. 1993;120:89–94. doi: 10.1006/exnr.1993.1042. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Liao SL. Oxidative stress involves in astrocytic alterations induced by manganese. Exp Neurol. 2002;175:216–225. doi: 10.1006/exnr.2002.7894. [DOI] [PubMed] [Google Scholar]
- Chun HS, Lee H, Son JH. Manganese induces endoplasmic reticulum (ER) stress and activates multiple caspases in nigral dopaminergic neuronal cells, SN4741. Neurosci Lett. 2001;316:5–8. doi: 10.1016/s0304-3940(01)02341-2. [DOI] [PubMed] [Google Scholar]
- Dev KK, van der Putten H, Sommer B, Rovelli G. Part I: parkin-associated proteins and Parkinson’s disease. Neuropharmacology. 2003;45:1–13. doi: 10.1016/s0028-3908(02)00337-4. [DOI] [PubMed] [Google Scholar]
- El Mchichi B, Hadji A, Vazquez A, Leca G. p38 MAPK and MSK1 mediate caspase-8 activation in manganese-induced mitochondria-dependent cell death. Cell Death Differ. 2007;14:1826–1836. doi: 10.1038/sj.cdd.4402187. [DOI] [PubMed] [Google Scholar]
- Galvani P, Fumagalli P, Santagostino A. Vulnerability of mitochondrial complex I in PC12 cells exposed to manganese. Eur J Pharmacol. 1995;293:377–383. doi: 10.1016/0926-6917(95)90058-6. [DOI] [PubMed] [Google Scholar]
- Gavin CE, Gunter KK, Gunter TE. Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol Appl Pharmacol. 1992;115:1–5. doi: 10.1016/0041-008x(92)90360-5. [DOI] [PubMed] [Google Scholar]
- Gavin CE, Gunter KK, Gunter TE. Manganese and calcium transport in mitochondria: implications for manganese toxicity. Neurotoxicology. 1999;20:445–453. [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, Lindquist S. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology. 1999;20:239–247. [PubMed] [Google Scholar]
- Guilarte TR, Burton NC, McGlothan JL, Verina T, Zhou Y, Alexander M, Pham L, Griswold M, Wong DF, Syversen T, Schneider JS. Impairment of nigrostriatal dopamine neurotransmission by manganese is mediated by pre-synaptic mechanism(s): implications to manganese-induced parkinsonism. J Neurochem. 2008;107:1236–1247. doi: 10.1111/j.1471-4159.2008.05695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Gavin CE, Gunter KK. The case for manganese interaction with mitochondria. Neurotoxicology. 2009;30:727–729. doi: 10.1016/j.neuro.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi Y, Asanuma M, Miyazaki I, Hattori N, Mizuno Y, Ogawa N. Parkin attenuates manganese-induced dopaminergic cell death. J Neurochem. 2004;89:1490–1497. doi: 10.1111/j.1471-4159.2004.02445.x. [DOI] [PubMed] [Google Scholar]
- Hilker R, Klein C, Ghaemi M, Kis B, Strotmann T, Ozelius LJ, Lenz O, Vieregge P, Herholz K, Heiss WD, Pramstaller PP. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. 2001;49:367–376. [PubMed] [Google Scholar]
- Hirata Y. Manganese-induced apoptosis in PC12 cells. Neurotoxicol Teratol. 2002;24:639–653. doi: 10.1016/s0892-0362(02)00215-5. [DOI] [PubMed] [Google Scholar]
- Hudnell HK. Effects from environmental Mn exposures: a review of the evidence from non-occupational exposure studies. Neurotoxicology. 1999;20:379–397. [PubMed] [Google Scholar]
- Huynh DP, Scoles DR, Nguyen D, Pulst SM. The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum Mol Genet. 2003;12:2587–2597. doi: 10.1093/hmg/ddg269. [DOI] [PubMed] [Google Scholar]
- Hyun DH, Lee M, Halliwell B, Jenner P. Effect of overexpression of wild-type or mutant parkin on the cellular response induced by toxic insults. J Neurosci Res. 2005;82:232–244. doi: 10.1002/jnr.20638. [DOI] [PubMed] [Google Scholar]
- Ikeuchi K, Marusawa H, Fujiwara M, Matsumoto Y, Endo Y, Watanabe T, Iwai A, Sakai Y, Takahashi R, Chiba T. Attenuation of proteolysis-mediated cyclin E regulation by alternatively spliced Parkin in human colorectal cancers. Int J Cancer. 2009;125:2029–2035. doi: 10.1002/ijc.24565. [DOI] [PubMed] [Google Scholar]
- Jiang H, Jiang Q, Liu W, Feng J. Parkin suppresses the expression of monoamine oxidases. J Biol Chem. 2006;281:8591–8599. doi: 10.1074/jbc.M510926200. [DOI] [PubMed] [Google Scholar]
- Jimenez Del Rio M, Moreno S, Garcia-Ospina G, Buritica O, Uribe CS, Lopera F, Velez-Pardo C. Autosomal recessive juvenile parkinsonism Cys212Tyr mutation in parkin renders lymphocytes susceptible to dopamine- and iron-mediated apoptosis. Mov Disord. 2004;19:324–330. doi: 10.1002/mds.10670. [DOI] [PubMed] [Google Scholar]
- Kim Y, Kim JM, Kim JW, Yoo CI, Lee CR, Lee JH, Kim HK, Yang SO, Chung HK, Lee DS, Jeon B. Dopamine transporter density is decreased in parkinsonian patients with a history of manganese exposure: what does it mean? Mov Disord. 2002;17:568–575. doi: 10.1002/mds.10089. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Ko HS, von Coelln R, Sriram SR, Kim SW, Chung KK, Pletnikova O, Troncoso J, Johnson B, Saffary R, Goh EL, Song H, Park BJ, Kim MJ, Kim S, Dawson VL, Dawson TM. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci. 2005;25:7968–7978. doi: 10.1523/JNEUROSCI.2172-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda Y, Sako W, Goto S, Sawada T, Uchida D, Izumi Y, Takahashi T, Kagawa N, Matsumoto M, Takahashi R, Kaji R, Mitsui T. Parkin interacts with Klokin1 for mitochondrial import and maintenance of membrane potential. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr530. [DOI] [PubMed] [Google Scholar]
- Lesage S, Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci. 2005;25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima PD, Vasconcellos MC, Bahia MO, Montenegro RC, Pessoa CO, Costa-Lotufo LV, Moraes MO, Burbano RR. Genotoxic and cytotoxic effects of manganese chloride in cultured human lymphocytes treated in different phases of cell cycle. Toxicol In Vitro. 2008;22:1032–1037. doi: 10.1016/j.tiv.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Hattori N, Mori H, Suzuki T, Tanaka K. Parkin and Parkinson’s disease. Curr Opin Neurol. 2001;14:477–482. doi: 10.1097/00019052-200108000-00008. [DOI] [PubMed] [Google Scholar]
- Oliveira SA, Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, Lyons KE, Pahwa R, Stern MB, Hiner BC, Jankovic J, Ondo WG, Allen FH, Jr, Scott BL, Goetz CG, Small GW, Mastaglia FL, Stajich JM, Zhang F, Booze MW, Reaves JA, Middleton LT, Haines JL, Pericak-Vance MA, Vance JM, Martin ER. Association study of Parkin gene polymorphisms with idiopathic Parkinson disease. Arch Neurol. 2003;60:975–980. doi: 10.1001/archneur.60.7.975. [DOI] [PubMed] [Google Scholar]
- Pacelli C, De Rasmo D, Signorile A, Grattagliano I, di Tullio G, D’Orazio A, Nico B, Comi GP, Ronchi D, Ferranini E, Pirolo D, Seibel P, Schubert S, Gaballo A, Villani G, Cocco T. Mitochondrial defect and PGC-1alpha dysfunction in parkin-associated familial Parkinson’s disease. Biochim Biophys Acta. 2011;1812:1041–1053. doi: 10.1016/j.bbadis.2010.12.022. [DOI] [PubMed] [Google Scholar]
- Ren Y, Jiang H, Yang F, Nakaso K, Feng J. Parkin protects dopaminergic neurons against microtubule-depolymerizing toxins by attenuating microtubule-associated protein kinase activation. J Biol Chem. 2009;284:4009–4017. doi: 10.1074/jbc.M806245200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Zhao J, Feng J. Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J Neurosci. 2003;23:3316–3324. doi: 10.1523/JNEUROSCI.23-08-03316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JA. Homeostatic and toxic mechanisms regulating manganese uptake, retention, and elimination. Biol Res. 2006;39:45–57. doi: 10.4067/s0716-97602006000100006. [DOI] [PubMed] [Google Scholar]
- Roth JA. Are There Common Biochemical and Molecular Mechanisms Controlling Manganism and Parkisonism. Neuromolecular Med. 2009 doi: 10.1007/s12017-009-8088-8. [DOI] [PubMed] [Google Scholar]
- Roth JA, Feng L, Walowitz J, Browne RW. Manganese-induced rat pheochromocytoma (PC12) cell death is independent of caspase activation. J Neurosci Res. 2000;61:162–171. doi: 10.1002/1097-4547(20000715)61:2<162::AID-JNR7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Roth JA, Singleton S, Feng J, Garrick M, Paradkar PN. Parkin regulates metal transport via proteasomal degradation of the 1B isoforms of divalent metal transporter 1. J Neurochem. 2010;113:454–464. doi: 10.1111/j.1471-4159.2010.06607.x. [DOI] [PubMed] [Google Scholar]
- Satoh J, Kuroda Y. Association of codon 167 Ser/Asn heterozygosity in the parkin gene with sporadic Parkinson’s disease. Neuroreport. 1999;10:2735–2739. doi: 10.1097/00001756-199909090-00008. [DOI] [PubMed] [Google Scholar]
- Schrantz N, Blanchard DA, Mitenne F, Auffredou MT, Vazquez A, Leca G. Manganese induces apoptosis of human B cells: caspase-dependent cell death blocked by bcl-2. Cell Death Differ. 1999;6:445–453. doi: 10.1038/sj.cdd.4400508. [DOI] [PubMed] [Google Scholar]
- Szargel R, Rott R, Engelender S. Synphilin-1 isoforms in Parkinson’s disease: regulation by phosphorylation and ubiquitylation. Cell Mol Life Sci. 2008;65:80–88. doi: 10.1007/s00018-007-7343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verina T, Kiihl SF, Schneider JS, Guilarte TR. Manganese exposure induces microglia activation and dystrophy in the substantia nigra of non-human primates. Neurotoxicology. 2010 doi: 10.1016/j.neuro.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Maraganore D, Crook J, Lesnick T, Lockhart PJ, Wilkes KM, Kapatos G, Hardy JA, Farrer MJ. Functional association of the parkin gene promoter with idiopathic Parkinson’s disease. Hum Mol Genet. 2002;11:2787–2792. doi: 10.1093/hmg/11.22.2787. [DOI] [PubMed] [Google Scholar]
- Wider C, Wszolek ZK. Clinical genetics of Parkinson’s disease and related disorders. Parkinsonism Relat Disord. 2007;13(Suppl 3):S229–232. doi: 10.1016/S1353-8020(08)70007-5. [DOI] [PubMed] [Google Scholar]
- Yang YX, Wood NW, Latchman DS. Molecular basis of Parkinson’s disease. Neuroreport. 2009;20:150–156. doi: 10.1097/WNR.0b013e32831c50df. [DOI] [PubMed] [Google Scholar]
- Zhang S, Fu J, Zhou Z. Changes in the brain mitochondrial proteome of male Sprague-Dawley rats treated with manganese chloride. Toxicol Appl Pharmacol. 2005;202:13–17. doi: 10.1016/j.taap.2004.06.001. [DOI] [PubMed] [Google Scholar]