

Abstract
Introduction
Biliary atresia (BA) is the leading indication for liver transplantation in the pediatric population. The murine model of BA supports a viral etiology as infection of neonatal mice with rhesus rotavirus (RRV) results in biliary obstruction. Viral infection targets the biliary epithelium and development of the model is viral strain dependent. No study has yet determined if human cholangiocytes are also susceptible to rotaviral infection. We established an in vitro human model utilizing an immortalized human cholangiocyte cell line and primary human cholangiocytes obtained from explanted livers to determine human cholangiocyte susceptibility to rotavirus infection.
Methods
Replication and binding assays were performed on immortalized mouse (mCL) and human (H69) cells using six different strains of rotavirus. Primary human cholangiocytes were isolated from cadaveric livers, characterized in culture, and infected with RRV which causes BA in mice and another simian strain, TUCH which does not cause BA in mice.
Results
Immortalized mouse and human cholangiocytes demonstrated similar patterns of infectivity and binding with different strains of rotavirus. Both cell lines produced a significantly higher viral yield with RRV infection than with the other strains tested. In primary human cholangiocytes, which maintained their epithelial characteristics as demonstrated by cytokeratin staining, RRV replicated to a yield 1000 fold higher than TUCH.
Conclusions
Both immortalized and primary human cholangiocytes are susceptible to RRV infection in a fashion similar to murine cholangiocytes. These novel findings suggest rotavirus infection could have a potential role in the pathogenesis of human BA.
Introduction
Biliary atresia (BA) is an obstructive cholangiopathy of neonates caused by inflammation and fibrosis of extrahepatic bile ducts. It affects approximately 1 in 5000–15000 live births worldwide.1,2 Early surgical treatment with Kasai portoenterostomy can re-establish bile flow, but the majority of infants progress to cirrhosis and end-stage liver disease. As a result, biliary atresia is the most common indication for liver transplantation in the pediatric population accounting for approximately 50% of the liver transplants performed.1
Despite numerous studies, the pathogenesis of biliary atresia is not understood. One proposed mechanism is perinatal viral infection triggering a host inflammatory response leading to obstruction of extrahepatic bile ducts.3 Evidence to support this virus-induced inflammatory cholangiopathy is twofold. First, in population studies, viruses including reovirus4–6, cytomegalovirus7, human papillomavirus8, Epstein-Barr virus9 and rotavirus10, have been found in the livers of BA patients. Second, a murine model of BA is well-defined that is induced by infection of neonatal mice with rhesus rotavirus (RRV) leading to liver inflammation, extrahepatic bile duct obstruction and histologic changes similar to BA afflicted human neonates.11
In the murine model of biliary atresia, RRV has unique tropism for the epithelium of the bile system.12,13 We previously established that induction of the murine model of BA was dependent on the specific rotavirus strain injected14. Certain viral strains (RRV, SA11-FM, and SA11-SM) can infect the hepatobiliary system in vivo and these infections lead to varying symptoms of cholangiopathy and liver disorder. RRV and SA11-FM specifically target the biliary epithelium and infected mice display obstructive symptoms of acholic stool and bilirubinuria. SA11-SM appears to target extra-portal hepatic cells.14 In contrast, other strains such as EDIM (murine strain) and Wa (human strain), do not infect the biliary system and do not cause clinical symptoms.14 Recently, we found a simian strain TUCH which can infect the biliary epithelium but does not induce biliary atresia. To identify which specific gene is responsible for the disease pathogenesis we utilized the rotavirus property of viral reassortment to generate a series of single gene substitutions between RRV and TUCH and found that RRV gene segment 4 governs specific tropism to the biliary epithelium and contributes to the development of murine BA.15 With identification of the biliary epithelium as the presumed target of the viral infection and inflammation, an in vitro model comprised of immortalized murine cholangiocyte cells was developed to better understand the pathogenic mechanism driving the in vivo model. Similar to what occurs in vivo, RRV binds to and replicates in vitro more efficiently in immortalized murine cholangiocytes as opposed to murine hepatocytes.13 As part of the characterization of the in vitro murine model, infection of cholangiocytes with different rotavirus strains was performed. In vitro infections with the six rotavirus strains correlated with their in vivo effect, further establishing the reliability of the in vitro model. This disease model has helped to build a foundation for the understanding of the pathogenesis of BA.
To date there have been no studies conducted that can directly correlate the viral-induced cholangiopathy found in the murine model with the development of BA in humans. Reports of rotavirus infection leading to viremia in human infants16 supports the possibility that perinatal rotavirus infection may result in targeting of and injury to biliary epithelium leading to the development of BA. The purpose of the present study was to develop an in vitro model based upon human cholangiocytes in which susceptibility to infection with various rotavirus strains were tested.
Materials and Methods
Rotavirus strains
We utilized six strains of rotavirus for these studies including: simian strains RRV (provided by H. Greenberg, Stanford University, Palo Alto, CA), SA11-fast moving (SA11-FM, as previously described by Gorziglia et al.), SA11-slow moving (SA11-SM, provided by Y. Hoshino, National Institutes of Health, Bethesda, MD), and TUCH (Tulane National Primate Research Center and Cincinnati Children’s Hospital), mouse strain epizootic diarrhea of infant mice (EDIM provided by M. Collins, Microbiological Associates, Bethesda, MD) and the human strain Wa (provided by R. Wyatt, National Institutes of Health, Bethesda, MD).
Immortalized cell lines
Two immortalized cholangiocyte cell lines were utilized. The mouse cholangiocyte cell line (mCL) which was derived from primary BALB/c mice and immortalized with SV40 large T-cell antigen-kindly provided by the laboratory of James Boyer (Yale Liver Care Center, Hartford, CT) and cultured as previously described.13 The human cholangiocytes (H69) were provided by Douglas Jefferson (New England Medical Center, Tufts University, Boston, Ma) and cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco/BRL, Gaithersburg, MD), 0.01% penicillin-streptomycin (Gibco/BRL), Adenosine 5-Triphosphate (2.43 μg/ml)(New England Biolabs), Insulin-Transferrin-Selenium (0.124 μg/ml) (Invitrogen), epithelial growth factor (10 ng/ml), hydrocortisone (2 μg/ml), and tri-iodo-thyronine (2 nM) (all from Sigma). MA104 cells, (Bio Whittaker, Walkersville, MD) and an immortalized human hepatocyte-like hepatoblastoma cell line (Hep G2)(HB8065)( ATCC, Manassas, VA) were grown in DMEM (Cellgro) supplemented with 10% FBS, 0.01% penicillin-streptomycin, 0.01% L-glutamine (Gibco/BRL), and 0.005% amphotericin B (Cellgro).
Isolation of primary human cholangiocytes
Primary human cholangiocytes were isolated from deceased donors in which donation for research purposes had been obtained. Liver specimens of about 30 grams were finely diced and incubated with collagenase type 1A (Sigma, St. Louis, MO). The digest was layered onto a 33% and 77% iso-osmotic Percoll gradient and centrifuged at 2000 rpm for 30 min. The interface layer was collected, washed three times in PBS, and incubated with cholangiocyte-specific mouse monoclonal antibody (HEA-125) (PROGEN Heidelberg, Germany). Cholangiocytes were positively selected by magnetic separation after incubating with anti-mouse IgG1-coated Dynabeads (Dynal). The positively selected cells were cultured in plating media: Hams F12, DMEM, FBS (10% v/v), penicillin-streptomycin (100 μg/ml), glutamine (2 mM), epidermal growth factor (10 ng/ml), hydrocortisone (2 μg/ml), choleratoxin (10 ng/ml), tri-iodothyronine (2 nM)(all from Sigma), and insulin (0.124 μg/ml) (Invitrogen). After 1–2 days in culture, the plating medium was exchanged with growth media containing 5% fetal calf serum and 10 ng/ml hepatocyte growth factor (Sigma). Cells were expanded in 25 ml tissue culture flasks with regular medium exchanges until they became a confluent monolayer. For passage and harvesting, cells were subjected to gentle trypsinization (5 min in 0.25% bovine pancreatic trypsin in basal media (Sigma), followed by neutralization with medium containing 10% fetal calf serum). In all subsequent experiments, cells were used between passage 2 and 5 depending on the initial yield of the primary isolate.
Immunohistochemistry for detection of cytokeratin or RRV in human cholangiocytes
The H69 cells and primary human cholangiocytes were grown in collagen coated plates and fixed with 75% cold acetone for 15 min at room temperature. After blocking in phosphate buffered saline (PBS) containing blocking reagent (Vector Laboratories US), the cells were washed with PBS and incubated for 12 hrs at 4°C using an antibody to cytokeratin-19 (CK-19, called TROMA III) (Developmental Studies Hybridoma Bank, University of Iowa) or anti-cytokeratin (pan-CK) antibody (Dako, Denmark) a mixture of different keratins. Following washing, the cells were incubated with DyLight 488 conjugated donkey anti-mouse IgG or anti-rabbit IgG (1:100, Jackson Immunoresearch Laboratories, West Grove, PA) for 1 hr at room temperature. After a final wash, the nuclei were counterstained with DAPI (Vector Laboratories US) and observed under a Nikon Eclipse E600 microscope and photographed with a Nikon Digital Camera DXM1200F.
H69 cells also underwent immunohistochemistry following infection with RRV to document susceptibility to RRV infection. Following acetone fixation, guinea pig anti-rotavirus IgG (1:1000) was added as the primary antibody and incubated at 37°C for 30 min. Wells were washed with PBS, and fluorescein isothiocyanate (FITC)-tagged goat anti-guinea pig IgG (1:500) was added as the secondary antibody and was incubated for 30 min at 37°C. Wells were washed twice and the cell nuclei were counterstained using DAPI.
In vitro assay for infectious virus in cholangiocyte cells
mCL, H69, and Hep G2 cells were seeded and grown to confluence in 24-well plates. Wells were washed with Earle’s balanced salt solution and inoculated in triplicate with all rotavirus strains described above at varying multiplicities of infection (MOI) at 37°C for 1 hr. At an MOI of 1, the amount of virus equals the number of cells. The cells were washed and incubated with serum-free DMEM plus 4μg trypsin/ml at 37°C for 24–48 hrs. Cultures were monitored for the cytopathic effect (CPE) which occurs during rotavirus infection when viral replication is complete and cells are lysed to release the replicated virus. After total cell lysis has occurred, virus can no longer replicate and may begin to die. Therefore, the optimal time to measure the amount of virus in a sample is at CPE of 4. CPE was determined by visual inspection of the cells and graded on a previously described scale.17 The resulting viral yield was assessed by a focus-forming assay (FFA) using MA104 cells as described below.
The primary human cholangiocytes were inoculated with RRV and TUCH using the same method described.
Focus-forming assay
Virus was quantified by formation of foci as described previously.18 In short, 96-well plates were seeded with MA104 cells and grown for 4 days. Once confluent, the cells were infected with serially diluted virus samples for 1.5 hrs. The cells were washed with DMEM and then incubated at 37°C for 14 to 16 hrs with DMEM containing 4μg of trypsin/ml. The medium was aspirated, and the cells were fixed with cold 80% acetone for 15 min at 20°C. Following a wash with PBS, guinea pig anti-rotavirus IgG (1:1000) was added as the primary antibody and was incubated for 30 min. Wells were washed with PBS, and fluorescein isothiocyanate (FITC)-tagged goat anti-guinea pig IgG (1:500) was added as the secondary antibody and was incubated for 30 min at 37°C. Wells were washed twice and were allowed to dry completely. Plates were scored using a UV microscope (10x objective), and quantities of infectious virus were reported as focus-forming units per milliliter.
Measurement of viral binding using an attachment assay
The ability of the virus to attach to mCL and H69 cells was assessed by binding assays as described previously. 13,17 Cultured cells were grown to confluence in 24-well plates. Attachment assays were performed in triplicate. At the time of assay, the cells, medium and inoculating virus were cooled to 4°C. Cells were inoculated with MOI of 0.5 and were incubated for 1 hr at 4°C. The inoculum was removed and the cells were washed twice to remove any unbound virus. The wash fluids and the residual inoculum were combined to account for all unbound viruses. The cells underwent 2 freeze-thaw cycles, and any virus found within the final cell fraction reflected bound virus. The amounts of bound and unbound virus were determined by FFA analysis. The amount of bound virus was expressed as a percentage of the total amount of virus used to inoculate the cells.
Results
Infection of mCL and H69 cells with multiple rotavirus strains
To ensure H69 cells maintained their biliary epithelial origin, cells underwent immunohistochemistry using pan-CK and CK-19 antibodies. The H69 cells showed green fluorescent staining for both cytokeratin and CK-19, markers consistent with biliary epithelium (Figure 1). The mCLs were well characterized as previously described.19
Figure 1. Immunohistochemistry for cytokeratin on H69 cells.

H69 cells stained with CK-19 antibody (A), pan-CK antibody (B) and counter-stained with DAPI showed green fluorescence staining cytoplasm with blue nucleus. Magnification 20X.
To demonstrate rotaviruses ability to infect H69, immunohistochemistry using an anti-rotavirus antibody was performed 24 hour post infection with RRV. The green staining represents RRV within the cells (Figure 2). To verify RRV replicates preferentially in cholangiocytes as opposed to hepatocytes, H69 and Hep G2 cells were infected with RRV. RRV replicated to a significantly higher titer (1.5 ± 0.3 × 107 ffu/ml) in H69 cells than in Hep G2 cells (5.2 ± 0.5 × 106 ffu/ml) (n=3, p<0.05). The difference in infectivity between the human immortalized cholangiocytes and hepatocyte-like cell lines with RRV mirrors the difference in infectivity that we have previously seen in the in vitro murine model.
Figure 2. Immunohistochemistry for RRV infection of H69 cells.
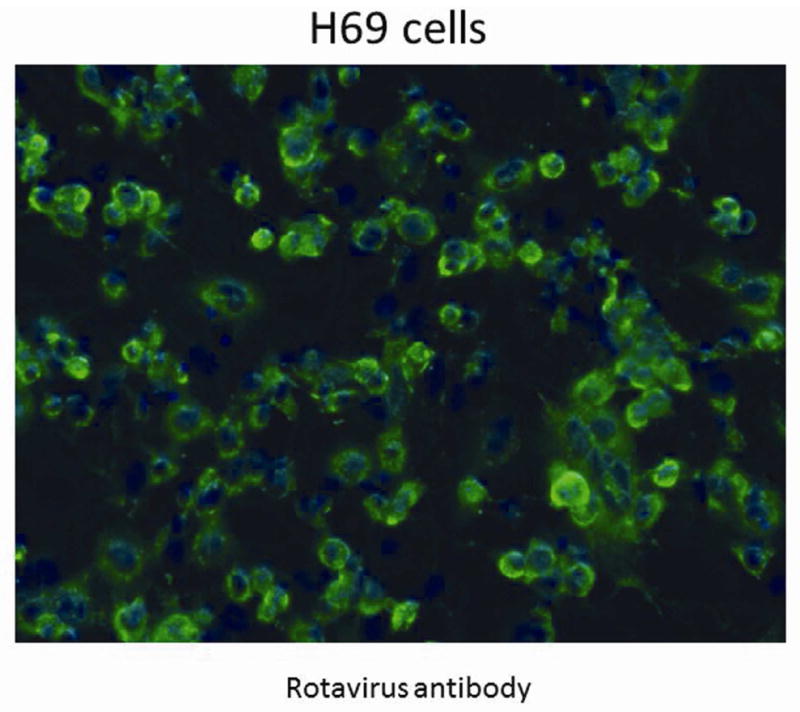
Staining with anti-rotavirus antibody, shown in green, demonstrates the presence of live RRV in the immortalized human cholangiocytes 24 hours post inoculation. Cell nuclei are counter stained with DAPI, shown in blue. Magnification 20X.
To validate parallels with in vivo models, six rotavirus strains were used to determine the infectivity profile of both the human and murine immortalized cholangiocyte cell lines. Cells were inoculated and observed for cell lysis, as measured by CPE at 24 hour intervals. An infection was determined to be complete when cell lysis was observed (CPE=4). We noted a CPE=4 at 24 hours in the H69 cells and at 48 hours in mCL cells (data not shown). The infection was halted and virus yield measured by focusforming assay at these time points. Both H69 and mCL cells demonstrated a strain specific infectivity pattern. In the mCL cell lines, RRV replicated to the highest titer (3.5±0.5 × 107 ffu/ml) followed by SA11- FM (1.3±0.2 × 107 ffu/ml) and SA11-SM (2.2±0.3 × 106 ffu/ml). RRV and SA11-FM cause the highest degree of symptoms and mortality in vivo while SA11-SM causes hepatobiliary injury but no obstruction as previously shown.14 TUCH, the non-model-inducing simian strain, Wa, and EDIM all had significantly decreased infectivity, as compared to RRV (Figure 3A).
Figure 3. Quantification of infectious rotavirus strains in mCL and H69 cells.

Yield of replicated virus 48 hours post infection with various rotavirus strains in mCL cells (A) and at 24 hours post infection in H69 cells (B). Quantity of infectious virus was measured using FFA. Values (n = 3) were expressed as mean FFU/ml with standard error, assay repeated two times. p<0.05
In H69 cells, RRV also exhibited the highest titer among the strains tested at 1.3±0.2 × 107 ffu/ml, followed by the two SA11 strains. TUCH was capable of replicating to a titer of 3.4±0.6 × 105 ffu/ml, which was 10 fold higher than in the mCLs, while Wa and EDIM only replicated to 103 (Figure 3B). When compared, the murine and human cholangiocytes demonstrated parallel, strain-specific infectivity patterns (Figure 3).
Binding assays of mCl and H69 cells
The rotavirus replication cycle begins with binding of the virus particle to cell-surface receptors on the host target cells. Therefore, the ability of each strain of rotavirus to replicate in a host cell is first influenced by its ability to bind to the cells. To determine the binding capability of virus strains, binding assays were performed. Binding assays were focused on the two simian strains, RRV and TUCH. RRV replicates to higher degree than does its simian relation, TUCH, and it induces the murine model of biliary atresia while mice infected with TUCH do not show symptoms.15 As expected, the differences in the strains were reflected in their binding ability to cholangiocytes as well. RRV binds to mCL at 11.6±0.4% while TUCH binds at 5.9±0.5% (p<0.05) (Figure 4A). This pattern is similar in H69 cells with RRV binding at a significantly higher percentage than TUCH; 36.4±0.4% vs. 19.3±1.0% respectively p<0.05 (Figure 4B).
Figure 4. In vitro binding assay of RRV and TUCH strains to mCL and H69 cells.

The percentage of RRV and TUCH binding to mCL (A) and H69 cells (B) at 4°C. Quantity of bound virus was measured using FFA. Values (n = 3) were expressed as mean percentage of bound virus with standard error, assay performed three times. In both cell lines RRV bound at a higher percentage than TUCH. * p<0.05
Isolation, characterization, and infection of primary human cholangiocytes
After isolation, primary human cholangiocytes were characterized by biliary epithelium specific cytokeratin staining. The primary human cholangiocytes demonstrated a positive pan-cytokeratin staining pattern similar to that seen in the H69 cell line (Figure 5).
Figure 5. Immunohistochemistry for cytokeratin on primary human cholangiocytes.
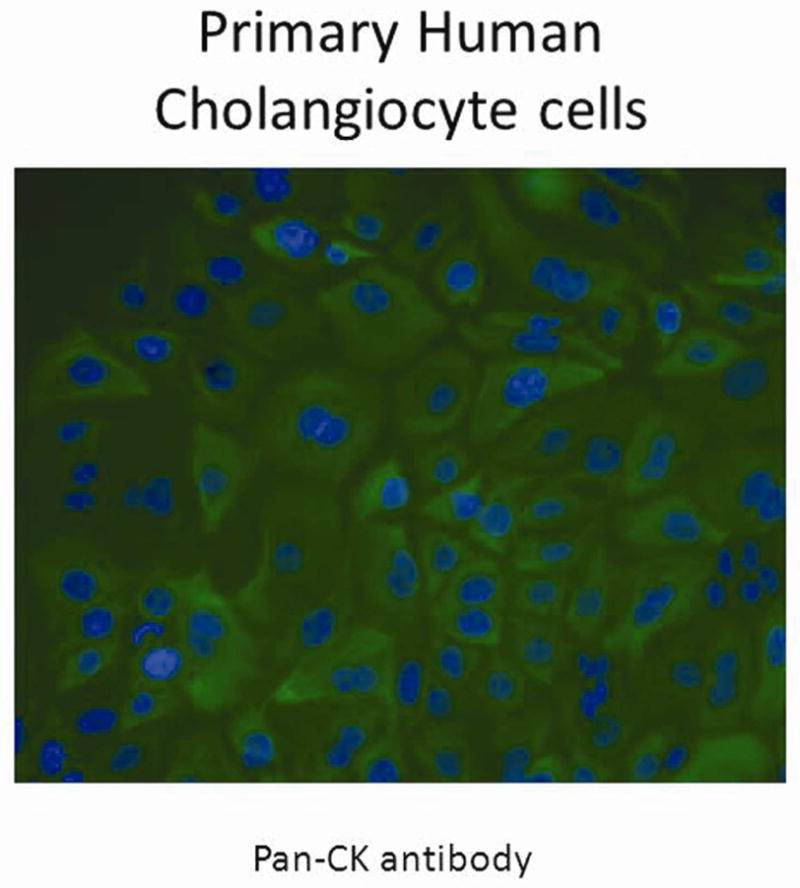
Primary human cholangiocytes stained with pan-CK antibody and counter stained with DAPI showed green fluorescence staining cytoplasm with blue nucleus. Magnification 40X.
Immortalization of a cell line often changes the inherent characteristics of the cell. Isolation of primary cells allowed us to simulate the in vivo behavior of the cell. RRV and TUCH infections in the primary cells produce higher viral yields of RRV than TUCH, with RRV mean viral titers of 2.04 ± 0 .09 × 107 ffu/ml and TUCH mean titers of 2.14 ± 0.4 × 104 ffu/ml (n= 3, p<0.05). While these results are solely descriptive, they illustrate that human cholangiocytes are susceptible to RRV infection. In addition, the character of the cells and the pattern of infectivity are consistent with that seen in both the H69 and mCL cell lines. These results further illustrate the similarities between the murine and human in vitro models.
Discussion
Neonates with BA present clinically with jaundice, acholic stools, and bilirubinuria. As the disease progresses, sequelae of biliary obstruction and liver failure are evident. Histologically, the livers from affected neonates demonstrate evidence of BA with portal fibrosis and ductular proliferation.20 The pathogenesis of human disease is unfortunately, still not understood.
In the murine model of BA, affected neonatal mice display jaundice, bilirubinuria, and acholic stools.11 Bile duct obstruction is evident on cholangiography14 while histologic evaluation demonstrates inflammation of portal triads and proliferation of bile ducts.12 As previously reported, the induction of the murine model is dependent on the strain of rotavirus. RRV infection produces the well-defined symptoms of biliary atresia. Injection with other viruses results in a range of symptoms from none to bilirubinuria and jaundice without full biliary obstruction.14 Our work with viral reassortants (using RRV and TUCH) led to the identification of viral gene segment 4 (VP4) of RRV as a determinant of RRV infection leading to the development of murine biliary atresia. TUCH, while related to RRV as a simian strain, expresses a different VP4 protein and does not induce the model.15
With its similarities to human BA, the murine model is an invaluable tool in determining mechanisms and pathogenesis of this devastating disease. To aid in the study, we have developed an in vitro system to study murine biliary atresia.13,17 In this system, immortalized murine cholangiocytes can be inoculated with rotavirus and the infectivity measured. In addition, RRV preferentially infects murine cholangiocytes over hepatocytes in vitro.13 The infectivity of the cholangiocyte is dependent on the strain of rotavirus and correlates with the symptoms noted in the in vivo system. The pattern of strain dependent infectivity provides a predictable, reproducible model to perform mechanistic studies.
Understanding the mechanism through which rotavirus infection induces the murine model of biliary atresia is the first step in helping to elucidate the pathogenesis of the human disease process. Through manipulation of an in vitro murine model, we found that integrins and MAPK pathways played a role governing susceptibility to RRV infection.13,17 To correlate findings in murine BA to the human disease we developed a novel human in vitro model. Using H69 cells, a previously established human cholangiocyte cell line we found that the binding of rotavirus strains was in a pattern similar to the murine model with RRV binding at 1.8 times greater than TUCH in both cell lines. Furthermore, the H69 cells were susceptible to rotavirus infection in a strain dependent pattern similar to the murine model. The H69 cells produced a greater viral replication yield than the Hep G2 cells, another parallel to the findings in the murine in vitro system. To ensure immortalization did not affect susceptibility to rotaviral infection, primary cholangiocytes were isolated from human donor livers. The primary cholangiocytes had a pattern of cytokeratin staining consistent with H69 cells. Initial infections with the simian strains RRV and TUCH showed infectivity in a consistent pattern.
This study demonstrates novel evidence that human biliary epithelium is susceptible to rotavirus infection. The similarities described here, between the infectivity of murine and human cholangiocytes, are the first evidence to suggest that the human in vitro model provides a reliable tool to further study the pathogenic mechanisms involved in human BA. The availability of such an in vitro human model will provide opportunities to enhance the fundamental understanding of the pathogenesis of BA and will establish a scientific basis for exploring treatment and prevention strategies. We will use this model to study the biology of rotavirus and other viral infection, pathogenesis of diseases, and host defense immune responses.
Acknowledgments
GRANTS
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases grant R03-DK-087974 and R01-DK-091566 to G. Tiao.
Footnotes
Conflict of interest statement
No conflicts of interest exist
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bessho K, Bezerra JA. Biliary atresia: will blocking inflammation tame the disease? Annual review of medicine. 2011;62:171–85. doi: 10.1146/annurev-med-042909-093734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sokol RJ, Shepherd RW, Superina R, Bezerra JA, Robuck P, Hoofnagle JH. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology. 2007;46:566–81. doi: 10.1002/hep.21790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sokol RJ, Mack C. Etiopathogenesis of biliary atresia. Seminars in liver disease. 2001;21:517–24. doi: 10.1055/s-2001-19032. [DOI] [PubMed] [Google Scholar]
- 4.Glaser JH, Balistreri WF, Morecki R. Role of reovirus type 3 in persistent infantile cholestasis. The Journal of pediatrics. 1984;105:912–5. doi: 10.1016/s0022-3476(84)80076-1. [DOI] [PubMed] [Google Scholar]
- 5.Morecki R, Glaser JH, Cho S, Balistreri WF, Horwitz MS. Biliary atresia and reovirus type 3 infection. The New England journal of medicine. 1984;310:1610. doi: 10.1056/NEJM198406143102418. [DOI] [PubMed] [Google Scholar]
- 6.Morecki R, Glaser JH, Johnson AB, Kress Y. Detection of reovirus type 3 in the porta hepatis of an infant with extrahepatic biliary atresia: ultrastructural and immunocytochemical study. Hepatology. 1984;4:1137–42. doi: 10.1002/hep.1840040608. [DOI] [PubMed] [Google Scholar]
- 7.Fischler B, Ehrnst A, Forsgren M, Orvell C, Nemeth A. The viral association of neonatal cholestasis in Sweden: a possible link between cytomegalovirus infection and extrahepatic biliary atresia. Journal of pediatric gastroenterology and nutrition. 1998;27:57–64. doi: 10.1097/00005176-199807000-00010. [DOI] [PubMed] [Google Scholar]
- 8.Drut R, Drut RM, Gomez MA, Cueto Rua E, Lojo MM. Presence of human papillomavirus in extrahepatic biliary atresia. Journal of pediatric gastroenterology and nutrition. 1998;27:530–5. doi: 10.1097/00005176-199811000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Fjaer RB, Bruu AL, Nordbo SA. Extrahepatic bile duct atresia and viral involvement. Pediatric transplantation. 2005;9:68–73. doi: 10.1111/j.1399-3046.2005.00257.x. [DOI] [PubMed] [Google Scholar]
- 10.Riepenhoff-Talty M, Gouvea V, Evans MJ, et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. The Journal of infectious diseases. 1996;174:8–15. doi: 10.1093/infdis/174.1.8. [DOI] [PubMed] [Google Scholar]
- 11.Riepenhoff-Talty M, Schaekel K, Clark HF, et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatric research. 1993;33:394–9. doi: 10.1203/00006450-199304000-00016. [DOI] [PubMed] [Google Scholar]
- 12.Shivakumar P, Campbell KM, Sabla GE, et al. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN-gamma in experimental biliary atresia. The Journal of clinical investigation. 2004;114:322–9. doi: 10.1172/JCI21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jafri M, Donnelly B, Allen S, et al. Cholangiocyte expression of alpha2beta1-integrin confers susceptibility to rotavirus-induced experimental biliary atresia. American journal of physiology Gastrointestinal and liver physiology. 2008;295:G16–G26. doi: 10.1152/ajpgi.00442.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen SR, Jafri M, Donnelly B, et al. Effect of rotavirus strain on the murine model of biliary atresia. Journal of virology. 2007;81:1671–9. doi: 10.1128/JVI.02094-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Donnelly B, Bondoc A, et al. The rhesus rotavirus gene encoding VP4 is a major determinant in the pathogenesis of biliary atresia in newborn mice. Journal of virology. 2011;85:9069–77. doi: 10.1128/JVI.02436-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blutt SE, Kirkwood CD, Parreno V, et al. Rotavirus antigenaemia and viraemia: a common event? Lancet. 2003;362:1445–9. doi: 10.1016/S0140-6736(03)14687-9. [DOI] [PubMed] [Google Scholar]
- 17.Jafri M, Donnelly B, McNeal M, Ward R, Tiao G. MAPK signaling contributes to rotaviral-induced cholangiocyte injury and viral replication. Surgery. 2007;142:192–201. doi: 10.1016/j.surg.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 18.McNeal MM, Broome RL, Ward RL. Active immunity against rotavirus infection in mice is correlated with viral replication and titers of serum rotavirus IgA following vaccination. Virology. 1994;204:642–50. doi: 10.1006/viro.1994.1579. [DOI] [PubMed] [Google Scholar]
- 19.Jafri M, Donnelly B, Bondoc A, Allen S, Tiao G. Cholangiocyte secretion of chemokines in experimental biliary atresia. Journal of pediatric surgery. 2009;44:500–7. doi: 10.1016/j.jpedsurg.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet. 2009;374:1704–13. doi: 10.1016/S0140-6736(09)60946-6. [DOI] [PubMed] [Google Scholar]