

Abstract
Carbohydrates play a key role in the biological activity of numerous natural products. In many instances their biosynthesis requires radical mediated rearrangements, some of which are catalyzed by radical SAM enzymes. BtrN is one such enzyme responsible for the dehydrogenation of a secondary alcohol in the biosynthesis of 2-deoxystreptamine. DesII is another example that catalyzes a deamination reaction necessary for the net C4 deoxygenation of a glucose derivative en route to desosamine formation. BtrN and DesII represent the two most extensively characterized radical SAM enzymes involved in carbohydrate biosynthesis. In this review, we summarize the biosynthetic roles of these two enzymes, their mechanisms of catalysis, the questions that have arisen during these investigations and the insight they can offer for furthering our understanding of radical SAM enzymology. This article is part of a Special Issue entitled: Radical SAM enzymes and Radical Enzymology.
Keywords: Radical SAM enzymes, Biosynthesis, Unusual sugars, Enzyme mechanisms, DesII, BtrN
1. Introduction
Carbohydrates represent one of the key components of all biological systems. The members of this class of biomolecules are best known for their fundamental roles in energy handling; however, they also serve as the biosynthetic precursors for complex carbohydrates, nucleic acids and the post-translational modification of proteins and lipids. In addition to being essential for primary metabolism, sugars are also crucial for the biological activity of secondary metabolites such as bacterial lipopolysaccharides as well as small molecules exhibiting antibacterial and antiproliferative properties among others [1].
Considerable structural diversity among the sugar moieties in secondary metabolites is well known despite the majority of these sugars being derived from a relatively small collection of common precursors such as nucleotide diphosphate activated glucose (NDP-glucose) [2,3]. This process of “glycodiversification” starting from a common sugar precursor is achieved via complex biosynthetic transformations that often involve a few key steps of unique enzymatic chemistry to introduce the distinguishing structural feature(s). It has been demonstrated that enzyme bound radical intermediates are involved in some of the more novel and difficult transformations converting NDP-glucose to the final modified sugar species. More recently and for the purposes of this review, some of these diversification reactions have been established to involve radical S-adenosylmethionine (1,SAM) chemistry.
The radical SAM superfamily of enzymes is distinguished by the presence of a [4Fe–4S] cluster coordinated by the three cysteines of a conserved CxxxCxxC motif. The fourth or apical iron of the cluster interacts with the bound SAM to catalyze a reductive homolysis of the C–S bond at C5′ of SAM (1) to produce methionine (2) and a 5′-deoxyadenosyl radical (3, Fig. 1) [4,5], which is used as a radical initiator in the subsequent conversion of the nominal substrate to product. This aspect of radical SAM catalysis is reminiscent of that observed among the adenosylcobalamin-dependent enzymes [6,7]. Such chemistry was first discovered in the reaction catalyzed by lysine 2,3-aminomutase (LAM) from Clostridium subterminale in 1970 [8–12]. Since then, more than 2800 additional proteins have been predicted to be members of the radical SAM superfamily of enzymes based on sequence analysis [5]. Nevertheless, only a small fraction of these predicted enzymes have been purified and at least partially characterized, in large part due to the sensitivity of the catalytic iron–sulfur clusters to molecular oxygen. Thus, the biosynthesis of desosamine (5) and 2-deoxystreptamine (6) are presently the only two carbohydrate derivatives whose biosynthetic pathways have been shown definitively to utilize radical SAM enzymes, respectively identified as BtrN and DesII.
Fig. 1.

Electron transfer from an active site [4Fe–4S]1+ cluster leads to reductive homolysis of SAM (1) to generate a 5′-deoxyadenosyl radical (3) and methionine (2) during the catalytic cycle of radical SAM enzymes. The 5′-deoxyadenosyl radical can then be used as a radical initiator to abstract a hydrogen atom from the nominal substrate of the reaction.
In this review, a summary of the discovery and current mechanistic understanding of BtrN and DesII is presented. Consideration of the biosynthetic roles of these enzymes highlights the importance of radical mediated dehydrogenation reactions as well as the means by which sugars are regioselectively deoxygenated during processing to their final biologically active forms. Likewise, study of the mechanisms by which these enzymes employ radical SAM chemistry to effect their transformations provides general insight into how radical SAM enzymes are able to generate and control the highly reactive radical intermediates. Our discussion will focus on not only the comparison of the current biosynthetic and catalytic models but also the key questions which remain open to future investigation. Finally, the review is concluded with a selection of additional proteins hypothesized to function as radical SAM enzymes in other biosynthetic pathways of carbohydrate secondary metabolism.
2. Discovery and biological contexts of the DesII and BtrN catalyzed reactions
2.1. Biosynthesis of desosamine: DesII facilitates the C4 deoxygenation of TDP-6-deoxy-4-keto-d-glucose
A key avenue for achieving the broad structural diversity of unusual naturally occurring sugars is the regioselective deoxygenation of NDP-glucose (7) [2]. The mechanisms by which these reactions take place are generally conserved through homologous enzymes regardless of the overall biosynthetic pathway and the associated gene cluster in question (see Fig. 2) [2,13]. For example, deoxygenation at C6 of NDP-glucose (7) by NDP-hexose-4,6-dehydratase (Eod) employs NAD+ catalytically to effect an α,β-dehydration to eliminate the C6 hydroxyl group followed by reduction of the resulting enone to give the 6-deoxy-4-keto product 8 (see Fig. 2A) [13]. Deoxygenation at C2 similarly makes use of an α,β-dehydration reaction and the 4-keto derivative (8) of NDP-6-deoxyglucose generated after C6 deoxygenation (see Fig. 2B) [13]. In contrast, C3 deoxygenation involves the formation of a Δ3,4-glucoseen-PLP Schiff base intermediate (9) from 8 catalyzed by a PMP-dependent [2Fe–2S] containing enzyme (E1), followed by two one-electron reductions mediated by an NADH-dependent [2Fe–2S] flavoprotein (E3) (see Fig. 2C) [13–16]. This left the process by which C4 deoxygenation of a hexose takes place an open question.
Fig. 2.

A common biosynthetic mechanism of glycodiversification is the regioselective deoxygenation of nucleotide diphosphate activated glucose (NDP-glucose, 7). The enzyme Eod utilizes NAD+ catalytically to catalyze C6 deoxygenation (A). Deoxygenation at C2 is achieved via α,β-dehydration of the resulting 4-keto sugar (8) (B). Alternatively, the 4-keto sugar (8) can be deoxygenated at C3 via the coupled E1/E3 enzyme system to effect a net reduction of the substrate (C).
D-Desosamine (5) is a 4,6-dideoxysugar found in a number of macro-lide antibiotics including erythromycin, oleandomycin, mycinamicin, megalomicin, and methymycin/pikromycin, and is necessary for their biological activities [17,18]. As a 4-deoxysugar, its biosynthetic pathway in Streptomyces venezuelae[19] was chosen as the model system to investigate the mechanism of C4 deoxygenation, though the gene clusters responsible for its biosynthesis have also been identified in several other producing strains including Saccharopolyspora erythraea[20,21]. Previous sequence analyses showed that the desI gene product (DesI) from the desosamine biosynthetic gene cluster (see Fig. 3A) exhibits 24% sequence identity to E1[22]. Furthermore, a second protein encoded in the cluster, DesII, possesses a CxxxCxxC motif characteristic of [4Fe–4S] bearing enzymes [19,22]. These observations led to the initial hypothesis that C4 deoxygenation followed a mechanism similar to that of C3 deoxygenation. Thus, DesI and DesII were expected to function analogously to E1 and E3, respectively, and a third gene product, DesVIII, was proposed to prime the biosynthetic precursor TDP-6-deoxy-4-keto-d-glucose (8) by tautomerization to the corresponding 3-keto derivative (10, see Fig. 3B)[20,21,23,24].
Fig. 3.

Biosynthetic gene cluster for TDP-desosamine from Streptomyces venezuelae (A). Summary of results for the gene knockout experiment to determine the biosynthetic pathway of TDP-desosamine (5) as well as an early mechanistic hypotheses for the deamination reaction of DesII inspired by the mechanisms of the E1/E3 system and lysine 2,3-aminomutase (B). The currently accepted biosynthetic transformations of DesI and DesII (C). See text for additional details.
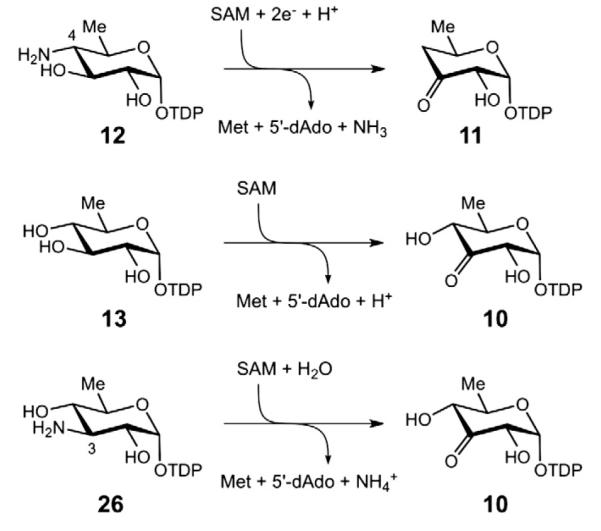
To test this hypothesis, desosamine biosynthesis in the ΔdesI and ΔdesII gene knock-out mutants was investigated. Had the C3/C4 deoxygenation analogy been correct, it was expected that both mutants would yield the same dead end product, namely TDP-6-deoxy-3-keto-d-glucose (10). What was observed instead was the production of TDP-4-amino-4,6-dideoxy-d-glucose (12) by the ΔdesII mutant and TDP-6-deoxy-d-glucose, i.e., TDP-d-quinovose (13), by the ΔdesI mutant (see Fig. 3B) [22]. Furthermore, when the putative substrate, TDP-6-deoxy-3-keto-d-glucose (10) was incubated with purified DesI in the presence of PLP and L-glutamate, neither substrate consumption nor product formation was observed [22]. In contrast, TDP-6-deoxy-4-keto-d-glucose (8) was recognized by DesI and converted to TDP-4-amino-4,6-dideoxy-d-glucose (12) under the same conditions [22]. These results indicated that DesI is specific for the 4-ketosugar and can act as a transaminase, thereby refuting the analogy with C3 deoxygenation.
This observation, along with the realization that DesVIII participates with DesVII in glycosyl transfer [25,26], indicated that C4 deoxygenation may instead take place via a direct interaction between DesI and DesII. Inspired by the mechanism of LAM [11], it was first suggested that deoxygenation may involve a radical mediated 1,2-amino shift of the internal aldimine (14) adduct of PMP and TDP-6-deoxy-4-keto-d-glucose (8) in the active site of DesI (see Fig. 3B) [22]. In this case DesII as a radical SAM enzyme would catalyze the hydrogen atom abstraction from 14 to initiate the rearrangement of the resulting radical intermediate 15 (15 → 16 → 17 → 18) to affor d11. However, this hypothesis was ruled out by the failure to demonstrate the formation of a DesI/DesII complex in vivo via a yeast two-hybrid assay and the observation that heterologously expressed and purified DesII can indeed convert TDP-4-amino-4,6-dideoxy-d-glucose (12) to the deoxygenation product (11) in the absence of DesI [24].
These findings implicated a novel mechanism for C4 deoxygenation that utilizes radical SAM chemistry to catalyze a deamination reaction. It is clear that the C4 deoxygenation of 8 proceeds in two discreet steps in which DesI first catalyzes the formation of a 4-amino sugar (12) from 8, and DesII carries out the C4 deamination of 12 to give TDP-4,6-dideoxy-3-keto-d-glucose (11, see Fig. 3C). However, the details of this deamination reaction are still obscure, and the current understanding of DesII catalysis will be described below.
2.2. Biosynthesis of 2-deoxystreptamine: BtrN catalyzes an unusual radical mediated dehydrogenation during aminoglycoside biosynthesis
The aminoglycosides are a class of antibiotics that have found clinical use in the treatment of Gram positive infections [17,27]. These bacteriocidal compounds are inhibitors of protein synthesis and are characterized by an aminocyclitol aglycone core decorated with various carbohydrate moieties. The aminoglycosides are divided into two classes depending on the nature of the aglycone core. The first class possesses a streptidine core and includes streptomycin and spectinomycin. In contrast, the second more broad class possesses a 2-deoxystreptamine (DOS) core (6) and includes the kanamycin and gentimicin families of aminoglycosides.
Butirosin (19) produced by Bacillus circulans is a member of the second class of aminoglycoside antibiotics possessing a DOS aminocyclitol core [27]. The butirosin biosynthetic pathway was thus chosen to investigate the biosynthesis of DOS (6), which was poorly understood in comparison to the alternative streptidine core. Significant progress was made when 2-deoxy-scyllo-inosose (DOI, 21) synthase was isolated from B. circulans[28]. This heterodimeric enzyme catalyzes the initial step of DOS biosynthesis in which glucose-6-phosphate (20) is converted to DOI (21). The gene encoding the larger catalytic subunit, btrC, was subsequently cloned [29], and the butirosin biosynthetic gene cluster was identified via chromosomal gene walking about the btrC gene (see Fig. 4A) [30,31].
Fig. 4.

Biosynthetic gene cluster for 2-deoxystreptamine (DOS, 6) from Bacillus circulans (A), and the associated biosynthetic pathway (B).
In the course of these studies, two adjacent genes, btrS and btrN, were uncovered. Expression and characterization of the btrS gene product led to its identification as a dual function PLP-dependent aminotransferase that converts DOI (21) to 2-deoxy-scyllo-inosamine (DOIA, 22) and amino-dideoxy-scyllo-inosose (amino-DOI, 23) to DOS (6, see Fig. 4B) [31–34]. The involvement of a second gene, btrN, in the dehydrogenation reaction linking DOIA (22) to amino-DOI (23) was also established using gene knock out mutants [35]. This observation was unexpected due to the lack of homology between BtrN and the nicotinamide- or flavin-dependent dehydrogenases [30,31], which are the enzymes involved in typical alcohol dehydrogenation reactions. Guided by the presence of a CxxxCxxC motif in its primary sequence, the overexpressed and purified BtrN enzyme was shown to be SAM-dependent and capable of catalyzing dehydrogenation of 22 in vitro following anaerobic reconstitution of the presumed [4Fe–4S] cluster [35]. These results effectively established BtrN as a radical SAM dehydrogenase, whose identity was further con-firmed by more detailed mechanistic investigations summarized below.
Interestingly, the BtrN homologue has not been found in any other gene cluster for the biosynthesis of DOS-containing aminoglycosides [36]. For example, NeoA and GacH are the dehydrogenases that catalyze the same reaction as BtrN in the biosynthetic pathways of neomycin in Streptomyces fradiae and gentamicin A2 in Micromonospora echinospora, respectively [37–39]. However, both utilize NAD(P)+ and Zn2+ instead of SAM and an iron–sulfur cluster for catalysis. The fact that NeoA homologues are found in all DOI derived amino-glycoside biosynthetic pathways strongly suggests that this key step to construct the DOS core is catalyzed by a nicotinamide-dependent dehydrogenase in the production of most DOS-type aminoglycosides[27,40,41]. This generalization even extends to the butirosin gene cluster, which also harbors a neoA-homologous gene btrE[30,37]. However, the corresponding gene product, which lacks the two zinc-binding motifs common to the other NeoA homologues, is incapable of catalyzing the nicotinamide-dependent dehydrogenation reaction [35,37]. Thus, the function of the btrE gene product remains a mystery, and it may be nothing more than an evolutionary remnant [41].
BtrN is thus one of the few enzymes known to catalyze radical mediated dehydrogenation. It is in fact one of the only two dehydrogenases utilizing radical SAM chemistry in a biologically relevant context. Together with the anaerobic sulfatase maturating enzymes[42,43], BtrN offers an excellent system for the study of this novel radical chemistry and its potential applicability in the biosynthesis of secondary metabolites. It is also of some interest as to why a unique mechanism of DOIA dehydrogenation has evolved only in the butirosin biosynthetic pathway.
3. Kinetic mechanisms and substrate specificities
The biosynthetic role of BtrN in vivo is the oxidation of DOIA (22) to amino-DOI (23) via dehydrogenation of the C3 hydroxyl group. This reaction proceeds in vitro with a kcat on the order of 2 min−1 and Michaelis constants for both SAM (1) and DOIA (22) in the micromolar range [35]. Investigation of the initial rates indicated a sequential mechanism, due to the significant uncompetitive substrate inhibition (Ki≈0.2 mM) observed with DOIA but not with SAM [35]. Such inhibition in a bisubstrate reaction is consistent with the basic paradigm in which binding of DOIA (22) forms a dead end quaternary complex with the reduction products of SAM, i.e., 5′-deoxyadensine (4) and methionine (2) [44]. Therefore, the current working model for this enzyme is an ordered mechanism in which DOIA (22) binds after SAM, and amino-DOI (23) is the first product to dissociate.
The substrate flexibility of BtrN has also been studied using an array of cyclitol and sugar compounds (see Fig. 5). Three of them, DOS (6), scyllo-inositol (24) and myo-inositol (25), were found to be substrates for the enzyme in the sense that 5′-deoxyadenosine (4) formation was observed [35]. It remains uncertain, however, whether these alternative substrates are indeed oxidized concomitant with the reduction of SAM (versus “uncoupling”) and whether the regiochemistry of the oxidation is as predicted. Nevertheless, the apparent competence of DOS (6) as a substrate is consistent with dehydrogenation of an amino functionality as well as an alcohol, similar to what has also been reported for DesII and the TDP-3-amino-3,6-dideoxy-d-glucose substrate (26) (see below).
Fig. 5.

While 2-deoxy-scyllo-inosamine (DOIA, 22) is the biological substrate of BtrN, additional substrate analogues have also been shown to facilitate reduction of SAM (1) by BtrN [35]. A collection of cyclitol and sugar analogues has also been demonstrated to show little to no activity with the enzyme.
Whereas the reaction catalyzed by BtrN is an oxidative dehydrogenation, that of DesII is the redox neutral deamination of a 3-hydroxy-4-amino substrate. However, when DesII is provided instead with a 3-hydroxy-4-hydroxy substrate or a 3-amino-4-hydroxy substrate, the reaction changes from redox neutral group elimination to oxidative dehydrogenation (see Fig. 6), in direct analogy to BtrN [45]. Both TDP-d-quinovose (13) and TDP-3-amino-3,6-dideoxy-d-glucose (26) react with DesII to form the common product TDP-6-deoxy-3-keto-d-glucose (10), the latter presumably through a Schiff base intermediate that is subsequently hydrolyzed. This switch to dehydrogenation is somewhat unexpected, given the precedence for radical mediated dehydration of 1,2-diols by the B12-dependent dioldehydratases [7]. Preliminary kinetic analyses of DesII-catalyzed deamination and dehydrogenation have yielded results similar to those for BtrN [45,46], and more detailed studies are currently in progress.
Fig. 6.
Reactions catalyzed by DesII in vitro.
4. Chemical mechanisms
Studies of BtrN and DesII have shown that these enzymes catalyze two related but different reactions using ostensibly similar substrates. The observation that DesII is capable of catalyzing both types of reactions suggests that similar chemical principles may underlie the mechanisms of these reactions. Therefore, both BtrN and DesII are excellent model systems for the study of radical transformations and their control in enzyme catalysis.
4.1. Initiation of radical chemistry: the [4Fe–4S] clusters
As with all radical SAM enzymes, the radical chemistry of both the BtrN and DesII catalytic cycles is initiated via a reduced [4Fe–4S]1+ cluster (see Fig. 1) [4,5]. The cluster is maintained in the active site via a coordination complex involving the three cysteine residues of the canonical CxxxCxxC motif [4]. Structural studies of LAM and pyruvate-formate lyase activating enzyme have led to a model in which the apical iron of the cluster is left free to coordinate SAM (1) via its α-amino and α-carboxylate groups [47,48]. Following electron transfer from the reduced iron–sulfur cluster to the sulfonium group of SAM and homolysis of the C–S bond at C5′ of SAM, the 5′-deoxyadenosyl radical (3) is produced along with a hexacoordinated apical iron involving the sulfide of the resulting methionine (2) [49,50]. Formation of the enzyme bound [4Fe–4S]1+• SAM complex as well as the favorable change in apical coordination upon oxidation of the cluster is believed to help compensate for the disparity in the redox potentials between the sulfonium of SAM and the reduced cluster [49,50].
The primary sequence of DesII from S. venezuelae contains only four cysteine residues: Cys15, Cys141, Cys145 and Cys148, the latter three constituting the CxxxCxxC motif [19]. Iron/sulfur titration of the reconstituted enzyme indicated four equivalents each, consistent with a single [4Fe–4S] cluster [45]. This cluster could be reduced to the active [4Fe–4S]1+ state with either sodium dithionite or the more biologically relevant flavodoxin/flavodoxin reductase/NADPH redox system [45,51]. Attempts to characterize the cluster by electron paramagnetic resonance spectroscopy (EPR) met with success only under reducing conditions and in the presence of SAM, consistent with the oxidized [4Fe–4S]2+ cluster being EPR silent [45]. The observed rhombic signal was characterized by g values of 2.01, 1.96 and 1.87 (gav = 1.95), similar to those reported for other radical SAM enzymes [52,53].
The inability to observe the DesII cluster in the absence of SAM was initially taken as evidence that either the [4Fe–4S] cluster of DesII is inherently unstable or requires SAM to sufficiently lower the redox potential of the cluster and facilitate reduction [45]. The latter hypothesis was suggested on the basis of voltamic measurements of the [4Fe–4S] cluster of LAM [54]. However, recent work with DesII demonstrated that the reconstituted enzyme is actually quite stable for extended periods of time in the absence of SAM and does not appear to require an overt presence of SAM for efficient reduction [51]. Therefore, the most likely explanation for the earlier results is the use of EDTA in the reconstitution procedure, which can help to solubilize adventitial and nonspecifically bound iron but might also lead to destruction of the cluster.
In contrast to DesII, characterization of the iron and sulfur content of BtrN has been somewhat more eventful. The primary sequence of BtrN from B. circulans possesses a total of eight cysteine residues of which only three, Cys16, Cys20 and Cys23, constitute a single CxxxCxxC motif [30]. This led to the early belief that the enzyme contained only one [4Fe–4S] complex, which was seemingly confirmed by iron and sulfur titration experiments [35,55]. EPR characterization of the cluster associated with the enzyme free of bound substrates revealed an axial signal with g-values of 1.92 and 2.04 [55], later assigned as g⊥ and g∥, respectively [56]. Upon binding SAM, the EPR signal was noted to broaden with the g-values assigned to 1.83 and 1.99. During turnover, a more complex rhombic signal with g-values of 1.87, 1.96 and 2.05 (gav = 1.96) was observed, following subtraction of signals from the free and SAM-bound enzyme as well as the substrate radical (see below) [55]. This latter result was unexpected, because under saturating conditions the cluster was expected to be largely in the EPR-silent [4Fe–4S]2+ oxidation state. The signal was therefore assigned to a BtrN•SAM•DOIA ternary Michaelis complex, because quaternary complexes with products showed only minor EPR perturbations compared to that of unbound BtrN [55].
The existence of eight cysteine residues in BtrN has also led to the suggestion that a second iron sulfur complex may be present. Indeed, a repeat of the iron/sulfur titrations and their correlation with results of Mössbauer spectroscopy indicated the presence of 1.7 [4Fe–4S] clusters per BtrN monomer [56]. Furthermore, when the three cysteines of the CxxxCxxC motif were all mutated to alanine, the resulting C16A/C20A/C23A triple mutant was found to still contain 0.9 [4Fe–4S] cluster per monomer, bolstered by the combined approach of iron/ sulfur titration and Mössbauer spectroscopy [56]. These results provided strong evidence that some subset of the remaining five cysteine residues coordinate a second [4Fe–4S] cluster. Systematic mutation of the remaining five cysteines to alanine yielded soluble enzyme only in the case of C69A and C235A mutants, though the activity of the latter mutant was reduced in comparison to the wild type enzyme [56]. In contrast, all mutations with the Cys169/Cys187/Cys232 subgroup produced insoluble proteins that could not be characterized [56]. This led the authors to propose that this latter subgroup was responsible for binding the second [4Fe–4S] cluster in such a way so as to also provide an uncoordinated apical iron center.
A second EPR characterization of BtrN within the context of two catalytic [4Fe–4S] clusters allowed the assignment of an axial rather than a rhombic signal during turnover. Furthermore, the observed g⊥ and g∥ values of 1.99 and 1.83 implicated a quaternary BtrN•5′-dAdo•Met•amino-DOI complex with the second cluster in the reduced [4Fe–4S]1+ state. The unprecedented inversion of the g-values (g⊥>g∥) was also noted [56]. While the origin of the discrepancies between the two EPR characterizations remains unclear, it does appear that a second iron–sulfur cluster is present and participates in the BtrN catalytic cycle. Furthermore, when the cluster coordinated by the CxxxCxxC motif was removed by mutation, no EPR signal was observed even under reducing conditions, despite the fact that the presence of the second complex was confirmed by Mössbauer spectroscopy [56]. These results suggest that the second cluster likely remains in the oxidized [4Fe–4S]2+ state in the free enzyme and is either inaccessible to reduction by reagents in the bulk medium or has a particularly low redox potential.
4.2. Formation of the substrate radical
In the BtrN and DesII reactions, the 5′-deoxyadenosyl radical (3) produced from the reductive homolysis of SAM acts to abstract a hydrogen atom from the nominal substrate. The locus of hydrogen atom abstraction from DOIA (22) was identified as the C3 carbon (see Fig. 7), which bears the hydroxyl group to be oxidized [35]. This determination was made by observing deuterium transfer from [3-2H]-DOIA to the methyl group of 5′-deoxyadenosine (4) using a combination of NMR and mass spectrometry. A similar experiment with DesII was also performed using TDP-4-amino-4,6-dideoxy-d-[3-2H] glucose [45] and TDP-d-[3-2H]quinovose (unpublished results) (see Figs. 8 and 9). In both cases the presence of monodeuterated 5′-deoxyadenosine (4) was detected and formation of dideuterated SAM was also noted in the deamination reaction. The observation of dideuterated SAM in the reaction of DesII with 12 was initially taken as evidence for the catalytic involvement of SAM during deamination. However, this conclusion has since been shown to be incorrect (see below). Nevertheless, multiple incorporation of deuterium into the 5′-deoxyadenosine product was also reported in the BtrN study [35]. While these results have been interpreted as evidence for significant kinetic reversibility of the initial hydrogen abstraction by 5′-deoxyadenosyl radical (3), further experiments may be necessary to clarify the details underlying this process.
Fig. 7.

The current hypothesis for the radical mediated dehydrogenation of DOIA (22) by BtrN involves an auxiliary [4Fe–4S] cluster that acts to polarize the C3 hydroxyl group and accept the single reducing equivalent following hydrogen atom abstraction by the 5′-deoxyadenosyl radical [35,55,56].
Fig. 8.

The current hypothesis for DesII-catalyzed dehydrogenation of TDP-d-quinovose (13) involves deprotonation of the intermediary α-hydroxylalkyl radical (28) followed by direct electron transfer from 29 back to the oxidized [4Fe–4S]2+ cluster [51,57].
Fig. 9.

A possible mechanism for the DesII-catalyzed deamination of TDP-4-amino-4,6-dideoxy-d-glucose (12) analogous to the working model of ethanolamine ammonia-lyase [78–81] involves a 1,2-amino shift to form a radical carbinolamine intermediate (31). A second hypothesis that proposes direct elimination of the amine is shown in Fig. 10.
The substrate radicals themselves have also been characterized using EPR. Following incubation of BtrN with a 30-fold excess of both DOIA (22) and SAM (1), an organic radical was trapped by manual freeze quench following a 10 min incubation [55]. The organic radical was noted to exhibit a ddd splitting pattern with hyperfine splitting constants of 36, 36 and 7.7 G. This splitting pattern is consistent with the aforementioned deuterium transfer experiments placing the unpaired electron at C3 of 27. Confirmation as well as assignment of the hyperfine coupling constants was made as the substrate radical generated from [2,2-2H2]-DOIA displayed a collapse of the ddd splitting pattern to a doublet with a hyperfine splitting constant of 36 G [55]. This constraint places the p-orbital harboring the lone electron at the sp2-hybridized C3 center periplanar to both the C4–H and pro-R C2–H bonds (36 G, each) leaving it clinal with respect to the pro-S C2–H bond (7.7 G).
A similar EPR characterization of the DesII-catalyzed dehydroge-nation of TDP-d-quinovose (13) has also been performed [57]. In this case a 3-fold substrate to enzyme ratio was employed with the reaction being effectively complete within approximately 1 min. The organic radical (28) observed approximately 10 s after mixing exhibits a dd pattern with hyperfine splitting constants of 34 G each, which collapses to a doublet split by 34 G with the monodeuterated isotopologue TDP-d-[4-2H]quinovose. Analogous to BtrN, the magnitude of the splitting constants implies a periplanar arrangement of the partially filled C3 centered p-orbital versus the C–H bonds at both C2 and C4. Unfortunately, efforts to trap an organic radical intermediate during the deamination of TDP-4-amino-4,6-dideoxy-d-glucose (12) were unsuccessful [57]. The discrepancy between the results with the two reactions likely stems from differences in the redox processes characterizing the dehydrogenation versus the deamination reactions (see below).
The substrate radical (28) generated by DesII from TDP-d-quinovose (13) has also been shown to be an α-hydroxyalkyl radical rather than the corresponding conjugate base, i.e., a ketyl radical (29) [57]. This conclusion was drawn based on the decrease in the EPR line width when the reaction was run in D2O (5.0 G) versus H2O (7.2 G). In the •C3–OH radical, the hydroxyl hydrogen has a weak hyperfine interaction with the unpaired electron at the adjacent carbon that is observed as an inhomogenous broadening. This broadening is reduced when the hydroxyl hydrogen is replaced with deuterium on account of its smaller magnetogyric ratio [58]. These results imply that deprotonation of the hydroxyl group prior to or concerted with hydrogen atom abstraction is not required. Nevertheless, in the case of BtrN a hypothesis has been advanced suggesting that the apical iron of the second [4Fe–4S] cluster may serve as a Lewis acid to coordinate the C3 hydroxyl group of DOIA (22) [56]. Such an interaction would help to polarize and even deprotonate the hydroxyl group so as to lower the redox potential and facilitate the hydrogen atom transfer step.
4.3. Rearrangement of the substrate radical: dehydrogenation
In contrast to the formation of the substrate radical, much less is presently known in regards to its subsequent rearrangement. The BtrN and DesII catalyzed dehydrogenation reactions are both believed to proceed via one electron transfer from the substrate/product radical back to the oxidized [4Fe–4S]2+ cluster (see Figs. 7 and 8). Thus, SAM serves as a net two-electron oxidant [5,59] while the reduced [4Fe–4S]1+ cluster is regenerated with each catalytic cycle. This is consistent with the 1:1 consumption of SAM versus dehydrogenation substrate observed for both BtrN and DesII-catalyzed dehydrogenation reactions [35,51]. Furthermore, experimental evidence for the catalytic redox cycling of the [4Fe–4S]1+ cluster was provided for the DesII catalyzed dehydrogenation reaction, where DesII prereduced with sodium dithionite was observed to oxidize TDP-d-quinovose (13) even after allowing sufficient time for all the reductant to decompose via disprortionation [51,60]. Demonstration of multiple turnovers by the pre-reduced enzyme, but not the oxidized enzyme treated with filtrate from the pre-reduction, indicated that an external source of reductant is not required. While direct evidence for catalytic cycling of the primary [4Fe–4S]1+ cluster in BtrN has not been reported, the same type of chemistry is expected [35].
Redox cycling of the reduced cluster during the dehydrogenation reactions requires that a single reducing equivalent be returned from the organic radical back to the oxidized [4Fe–4S]2+ cluster. The [4Fe–4S]2+,1+ redox couple in aprotic solvents exhibits a midpoint potential more negative than −1V[61–63], though this may be significantly more positive within the enzyme active site [49,54]. In contrast, the redox potential for the (CH3)2C•–OH α-hydroxyalkyl radical is more positive than −0.9 V, and the deprotonated (CH3)2–C•–O− ketyl radical is more negative than −1.5 V [64,65]. Therefore, a second auxiliary [4Fe–4S]2+ cluster could serve as a Lewis acid to help equalize the redox potentials by polarizing the C3 α-hydroxyalkyl radical or even promoting complete deprotonation in BtrN [56].
Direct inner sphere electron transfer to an active site metal complex is also believed to operate in other radical mediated dehydrogenases. A good example is the well-studied galactose oxidase which utilizes a CuII complex coordinated to a unusual protein-based 3′-(S-cysteinyl)tyro-sine (TyrCys) radical to dehydrogenate the C6 hydroxyl group of galactose [66–68]. In the free enzyme, a water or acetate ligand coordinates the copper adjacent to the TyrCys residue and is replaced by the C6 hydroxyl group of galactose in the Michaelis complex [69,70]. Coordination to the metal helps to polarize the hydroxyl group facilitating its deprotonation. The redox potential of the resulting copper-coordinated alkoxide would then be sufficiently reduced allowing hydrogen atom abstraction by the TyrCys radical. Direct reduction of CuII to CuI stepwise or concerted with hydrogen atom abstraction would then complete the oxidation [71]. Another example is the anaerobic sulfatase maturating enzymes (anSMEs), such as AtsB from Klebsiella pneumonia[42,72], anSMEcpe from Clostridium perfringes[43,73,74], and anSMEbt from Bacteriodes thetaiotaomicron[73]. These enzymes employ radical SAM chemistry to activate sulfatase enzymes via dehydrogenation of an active site cysteine or serine residue to a Cα-formylglycine in the zymogen substrates [43,72]. AtsB contains 13 cysteine residues but only one canonical CxxxCxxC motif [72] and has been shown to possess three [4Fe–4S] clusters [75]. Likewise, anSMEcpe and anSMEbt possess 18 and 15 cysteine residues, respectively [76,77], and are also expected to contain multiple [4Fe–4S] clusters. As in the case of BtrN, the auxiliary [4Fe–4S] clusters may act as inner or outer sphere electron acceptors to provide rapid and controlled return of the reducing equivalent back to the primary cluster.
These results have led to the proposal of a consensus mechanism for the radical SAM dehydrogenases involving auxiliary [4Fe–4S]2+ clusters in electron transfer [56]. However, the observations made with DesII suggest that such a mechanism does not appear to be strictly required. The unit stoichiometries, lack of obvious reaction side products, and demonstrable recycling of the [4Fe–4S]1+ cluster indicate that the radical mediated dehydrogenation is well controlled by DesII, despite the fact that TDP-d-quinovose (13) and TDP-3-amino-3,6-dideoxy-d-glucose (26) are not the natural substrates[45,51]. Detection of the protonated α-hydroxyalkyl radical (28) by EPR indicates that deprotonation of the C3 hydroxyl group is not a prerequisite for C3 hydrogen atom abstraction [57]. Furthermore, electron return to the [4Fe–4S]2+ cluster may also take place via an outersphere tunneling event. Nevertheless, the unfavorable difference in redox potentials between the [4Fe–4S]2+ cluster and the α-hydroxyalkyl radical must be overcome. It has therefore been proposed that electron transfer may follow deprotonation of the α-hydroxyalkyl radical (to 29) via an active site base. Such a deprotonation event is reasonable, because the pKa of an α-hydroxylalkyl radical (e.g., 28) is typically reduced by approximately five units compared to the corresponding alcohol [64]. This would place it within the range of known basic protein residues. Further studies are needed to gain more insight into the underlying processes.
4.4. Rearrangement of the substrate radical: deamination
The deamination reaction of TDP-4-amino-4,6-dideoxy-d-glucose (12) catalyzed by DesII is mechanistically more complex than the dehydrogenation reaction. Deuterium tracer experiments revealed that hydrogen atom abstraction occurs at C3 analogous to that observed during the dehydrogenation of TDP-d-quinovose (13) [45]. However, in this case direct electron transfer back to the [4Fe–4S]2+ cluster, as happens with TDP-quinovose, does not take place. Rather, the reaction flux is channeled into elimination of the C4 amino group. How the deamination reaction proceeds is unclear, and two working hypotheses are presently under consideration. The first, which is inspired by the currently accepted mechanisms for B12-dependent ethanolamine ammonia-lyase (EAL) [78–81], and the dioldehydratases [7], involves migration of the C4 amino group to C3 to form a carbinolamine radical (31) that subsequently undergoes elimination of ammonia as shown in Fig. 9. The second hypothesis, reminiscent of the ribonucleotide reductases [82] and 2-hydroxyacyl-CoA dehydratase [83], involves an E1cb-type elimination of ammonia at C4 following or concerted with deprotonation of the C3 α-hydroxyalkyl radical (30) to yield a stabilized enol radical (32) as shown in Fig. 10.
Fig. 10.

A possible mechanism for the DesII-catalyzed deamination of TDP-4-amino-4,6-dideoxy-d-glucose (12) involves a direct E1cb-type elimination of the amino group at C4 by a ketyl radical intermediate to yield a stabilized enol radical (32). A second hypothesis that proposes a carbinolamine radical intermediate is shown in Fig. 9.
Whether a carbinolamine or enol radical species occurs during the catalytic cycle, the redox neutral nature of the deamination reaction requires that a single reducing equivalent be returned to the product radical. In analogy with lysine amino mutase [8–10] and sporephoto-product lyase [84,85], it was originally thought that SAM as well as the reduced [4Fe–4S]1+ cluster in DesII would be regenerated with each turnover [45]. This possibility was later ruled out when careful stoichiometric measurements of SAM versus TDP-4,6-dideoxy-3-keto-d-glucose (11) production yielded a ratio of 0.96 ± 0.05 in the presence of sodium dithionite [51]. It is conceivable that the unexpected 1:1 stoichiometry may be a result of an “uncoupling” phenomenon, also noted to various degrees with other radical SAM enzymes [74,86–92], in which SAM is reduced to 5′-deoxyadenosine (4) and methionine (2) in excess of stoichiometric consumption of the substrate. This interpretation, however, was disputed when no reduction of SAM was observed in the absence of substrate. Furthermore, the stoichiometry was constant as a function of turnover, i.e., there was no change in the 1:1 stoichiometry from early time points when product concentration was low to late time points when product concentration was elevated. A second explanation is that reduction of the [4Fe–4S]2+ cluster occurs mid-way through the catalytic cycle by Na2S2O4. Such a premature reduction would effectively prevent a 5′-deoxyadenosyl radical (3) from recombining oxidatively with methionine (2) to regenerate SAM (1), i.e., the extra electron would have no place to go. This “abortive” hypothesis was recently ruled out by two observations [51]. First, replacing Na2S2O4 with the more biologically relevant flavodoxin/flavodoxin reductase/NADPH reducing system again yielded a unit stoichiometry of 1.05 ± 0.05. Second, when the cluster was pre-reduced with Na2S2O4 and then incubated to allow all the Na2S2O4 to decompose by disportionation, no more than single turnover of the amine was observed.
These results indicated that SAM is indeed consumed during the DesII-catalyzed deamination in vitro. Since the substrate itself is not supplying the necessary two reducing equivalents, as is the case during the dehydrogenation, they must come from a second “external” source, such as NADPH or Na2S2O4. Results with the dehydrogenation reaction imply that electron transfer between the [4Fe–4S] cluster and the product radical is possible; however, re-abstraction of a hydrogen atom from 5′-deoxyadenosine (4) without regeneration of SAM cannot presently be excluded. How exactly the product radical is reduced and whether it takes place in vivo is unclear and thus offers considerable fodder for future investigations.
4.5. Control of the radical
In the cases of both BtrN and DesII, the possibility exists for the radical intermediates to react via alternative pathways. The precedence set by the dioldehydratases [7,93] raises the question as to how the enzymes channel reaction flux into a dehydrogenation rather than a dehydration reaction, because TDP-d-quinovose (13), TDP-3-amino-3,6-dideoxy-d-glucose (26), and DOIA (22) all possess hydroxyl groups at carbon centers adjacent to the unpaired electron. This question becomes even more interesting when one also takes into account TDP-4-amino-4,6-dideoxy-d-glucose (12), which specifically undergoes elimination of the C4 amino group rather than eliminating the C2 hydroxyl group or oxidizing the C3 hydroxyl group.
In the case of BtrN, there appear to be two properties of the reaction that promote oxidation of the C3 hydroxyl group over elimination of the C4 hydroxyl group. The first already mentioned above is the proposed coordination of the C3 hydroxyl moiety to the apical iron of the auxiliary cluster [56]. This would facilitate a more rapid innersphere resolution of the organic radical [94,95]. Secondly, the clinal orientation of the partially filled C3 p-orbital and the C–OH bond at C4 would inhibit any productive interaction whether it be a migration or an E1cb-type elimination (see Fig. 11A) [55]. This arrangement is to be contrasted with that observed for the substrate radical intermediates of EAL [96] and dioldehydratase [97], where the migrating/breaking C–NH3+ or C–OH bond is periplanar to the partially filled p-orbital as shown in Fig. 11C and D, respectively. Thus, in the case of BtrN, radical control may be a combination of inhibiting interactions with the substituent at C4 while concomitantly accelerating the electron transfer to a more stable [4Fe–4S] electron sink.
Fig. 11.

The stereochemistry of radical intermediates in the BtrN and DesII catalyzed dehydrogenation reactions has been predicted based on EPR hyperfine coupling constants. (A) The C3 substrate radical of 2-deoxy-scyllo-inosamine (DOIA) (27) during BtrN catalyzed dehydrogenation [55]. (B) The C3 substrate radical of TDP-d-quinovose (30) during DesII catalyzed dehydrogenation [57]. Similar geometries were also observed along the C2–C3 bonds (see text) for both BtrN and DesII. (C) The C1 substrate radical of S-2-aminopropanol during EAL catalyzed deamination [96]. (D) The C1 substrate radical of S-2-propanediol during dioldehydratase catalyzed dehydration [97]. The geometry in (D) is based on the authors’ observation that “the hyperfine coupling constant for the β-proton is small”.
A similar argument can also be made for the DesII-catalyzed dehydrogenation of TDP-d-quinovose (13). While there is no evidence for a coordinated metal, the p-orbital harboring the unpaired electron at C3 is again roughly orthogonal to the C–OH bonds at both C2 and C4 as shown in Fig. 11B [57]. During deamination, however, a productive interaction of the C3 centered radical with the C–NH3+ bond must take place. While structural data is awaited to address this issue, elimination of the C4 amine may result from some combination of a different binding conformation as well as the ability of ammonia to serve as a better leaving group versus hydroxide. It should be pointed out, however, that this latter hypothesis would be suggestive of an E1cb-type elimination mechanism.
5. Evidence for the involvement of radical SAM chemistry in the biosynthesis of other biological carbohydrate derivatives
While BtrN and DesII are the only two radical SAM enzymes of sugar biosynthesis that have been studied at the mechanistic level, several others have also been implicated to be radical SAM-dependent. Examples include TunB and OxsB along with the putative methyl-cobalamin-dependent radical SAM methyltransferases MoeK5, GenK and ForK.
The tunicamycins (33) represent a class of antibiotics that target bacterial cell wall biosynthesis by inhibiting translocase I, an enzyme required for the biosynthesis of peptidoglycan [98]. The defining feature of these natural products is an 11-carbon dialdose moiety, tunicamine, derived from the covalent adduct of uridine (36) and UDP-N-acetyl-galactoseamine-5,6-ene (38), which is derived from UDP-N-acetylgalactosamine (35) [99]. Cloning of the gene cluster responsible for the biosynthesis of tunicamycin from Streptomyces chartreusis by Chen and coworkers has identified 12 genes that are necessary and sufficient for its production [100]. During the course of this work, the TunB gene product was annotated as a putative radical SAM protein based on sequence analysis. This gene product is proposed to catalyze a radical coupling reaction in which a 5′-uridyl radical (37) undergoes radical addition at C6 of N-acetylgalactoseamine-5,6-ene (38) to yield an α-alkoxyalkyl radical (39) as shown in Fig. 12. Subsequent hydrogen atom abstraction from 5′-deoxyadenosine (4) could lead to UDP-N-acetyl-tunicamine-uracil (34) and the regeneration of SAM (1). This mechanistic proposal for TunB is of some interest, because it corresponds to a radical SAM C–C bond coupling reaction reminiscent of the reverse reaction of sporephotoproduct lyase [85].
Fig. 12.

Possible mechanism for the formation of UDP-N-acetyl-tunicamine-uracil (34) by TunB involving radical SAM chemistry in the biosynthesis of tunicamycins (33).
Oxetanocin A (40) from Bacillus megaterium is an antiviral agent[101,102] possessing an unusual four-member oxetane ring (see Fig. 13) [103,104]. The oxetanocin biosynthetic gene cluster is plasmid encoded and possesses four genes believed to be responsible for its production [105]. Three of these genes are annotated to encode phosphatase-like proteins (oxsA, osrA, and osrB), while the fourth (oxsB) has been identified as a putative radical SAM enzyme (OxsB) [106]. The biosynthetic pathway of oxetanocin A is at the present time unknown; however, it is hypothesized that OxsB catalyzes a radical mediated ring contraction of adenosine or one of its derivatives to generate the oxetane ring.
Fig. 13.

The oxsB gene product from the biosynthetic gene cluster for oxetanocin A (40) is believed to utilize radical SAM chemistry to generate an oxetane intermediate from aden-osine or one of its derivatives.
Methyl-cobalamin-dependent radical SAM methyltransferases have also been implicated among the reactions of glycodiversification. This class of enzymes is believed to utilize both methylcobalamin as well as radical SAM chemistry in order to facilitate methylation at unactivated carbon centers [107,108]. In the biosynthetic pathway of moenomycin (41), gene knock out experiments have identified MoeK5 as a putative radical SAM methyltransferase required for the C4 methylation of ring F (see Fig. 14) [109]. Similarly, GenK [110,111] and ForK [112] from the gentamicin C1 (42) and fortimicin A (43) biosynthetic gene clusters, respectively, have also been proposed to operate as radical SAM methyltransferases to introduce a methyl substituent at the C6 position of the 2,6-dideoxy-2,6-diaminoglucosyl residues (see Fig. 14). Presently no radical SAM methyltransferase has been characterized, and this class of enzymes and the reactions they catalyze represent an exciting new avenue of future research.
Fig. 14.

The moeK5, genK and forK gene products are believed to act as methyl-cobalamin-dependent radical SAM methyltransferases in the biosynthesis of moenomycin A (41), gentamicin C1 (42) and fortimicin A (43), respectively.
Acknowledgements
This work was supported in part by grants from the National Institutes of Health (GM035906), Welch Foundation (F-1511), and a Fellowship from the National Institute of Allergy and Infectious Diseases (F32AI082906) to M.W.R.
Abbreviations
- amino-DOI
amino-dideoxy-scyllo-inosose
- Cys
cysteine
- 5′-dAdo
5′-deoxyadenosine
- DOI
2-deoxy-scyllo-inosose
- DOIA
2-deoxy-scyllo-inosamine
- DOS
2-deoxystreptamine
- EAL
ethanolamine ammonia-lyase
- EDTA
ethylenediaminetetraacetic acid
- EPR
electron paramagnetic resonance
- FAD
flavin adenine dinucleotide
- LAM
lysine 2,3-aminomutase
- Met
methionine
- NAD(P)+
oxidized nicotinamide adenine dinucleo-tide (phosphate)
- NAD(P)H
reduced nicotinamide adenine dinucleotide (phosphate)
- NDP
nucleotide diphosphate
- PLP
pyridoxal 5′-phosphate
- PMP
pyridoxamine 5′-phosphate
- SAM
S-adenosylmethionine
- TyrCys
3′-(S-cysteinyl)tyrosine
Footnotes
This article is part of a Special Issue entitled: Radical SAM enzymes and Radical Enzymology.
References
- [1].Thorson JS, Hosted TJ, Jr., Jiang J, Biggins JB, Ahlert J. Nature’s carbohydrate chemists: the enzymatic glycosylation of bioactive bacterial metabolites. Curr. Org. Chem. 2001;5:139–167. [Google Scholar]
- [2].Thibodeaux CJ, Melancon CE, Liu H-W. Unusual sugar biosynthesis and natural product glycodiversification. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. [DOI] [PubMed] [Google Scholar]
- [3].Thibodeaux CJ, Melancon CE, III, Liu H-W. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew. Chem. Int. Ed. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Frey P, Magnusson O.Th. S-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chem. Rev. 2003;103:2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- [5].Frey PA, Hegeman AD, Ruzicka FJ. The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- [6].Banerjee R, Ragsdale SW. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- [7].Toraya T. Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem. Rev. 2003;103:2095–2127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- [8].Chirpich TP, Zappia V, Costilow RN, Barker HA. Lysine 2,3-aminomutase: purification and properties of a pyridoxal phosphate and S-adenosylmethionine-activated enzyme. J. Biol. Chem. 1970;245:1778–1789. [PubMed] [Google Scholar]
- [9].Moss M, Frey PA. The role of S-adenosylmethionine in the lysine 2,3-aminomutase reaction. J. Biol. Chem. 1987;262:14859–14862. [PubMed] [Google Scholar]
- [10].Baraniak J, Moss ML, Frey PA. Lysine 2,4-aminomutase: support for a mechanism of hydrogen transfer involving S-adenosylmethionine. J. Biol. Chem. 1989;264:1357–1360. [PubMed] [Google Scholar]
- [11].Ballinger MD, Frey PA, Reed GH. Structure of a substrate radical intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry. 1992;31:10782–10789. doi: 10.1021/bi00159a020. [DOI] [PubMed] [Google Scholar]
- [12].Magnusson O.Th., Reed GH, Frey PA. Spectroscopic evidence for the participation of an allylic analogue of the 5′-deoxyadenosyl radical in the reaction of lysine 2,3-aminomutase. J. Am. Chem. Soc. 1999;121:9764–9765. [Google Scholar]
- [13].He X, Agnihotri G, Liu H-W. Novel enzymatic mechanisms in carbohydrate metabolism. Chem. Rev. 2000;100:4615–4661. doi: 10.1021/cr9902998. [DOI] [PubMed] [Google Scholar]
- [14].Thorson JS, Liu H-W. Coenzyme B6 as a redox cofactor: a new role for an old coenzyme? J. Am. Chem. Soc. 1993;115:12177–12178. [Google Scholar]
- [15].Johnson DA, Gassner GT, Bandarian V, Ruzicka FJ, Ballou DP, Reed GH, Liu H-W. Kinetic characterization of an organic radical in the ascarylose biosynthetic pathway. Biochemistry. 1996;35:15846–15856. doi: 10.1021/bi961370w. [DOI] [PubMed] [Google Scholar]
- [16].Chang C.-w.T., Johnson DA, Bandarian V, Zhou H, LoBrutto R, Reed GH, Liu H.-w. Characterization of a unique coenzyme B6 radical in the ascarylose biosynthetic pathway. J. Am. Chem. Soc. 2000;122:4239–4240. [Google Scholar]
- [17].Chambers HF. Antimicrobial agents: the aminoglycosides. In: Hardman JG, Limbird LE, Gilman A. Goodman, editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 10th ed McGraw-Hill; New York: 2001. pp. 1219–1238. [Google Scholar]
- [18].Schlünzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- [19].Xue Y, Zhao L, Liu H.-w., Sherman DH. A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: architecture of metabolic diversity. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12111–12116. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gaisser S, Böhm GA, Cortés J, Leadlay PF. Analysis of seven genes from the eryAI–eryK region of the erythromycin biosynthetic gene cluster in Saccharopolyspora erythraea. Mol. Gen. Genet. 1997;256:239–251. doi: 10.1007/s004380050566. [DOI] [PubMed] [Google Scholar]
- [21].Summers RG, Donadio S, Staver MJ, Wendt-Pienkowski E, Hutchinson CR, Katz L. Sequencing and mutagenesis of genes from the erythromycin biosynthetic gene cluster of Saccharopolyspora erythraea that are involved in l-mycarose and d-desosamine production. Microbiology. 1997;143:3251–3262. doi: 10.1099/00221287-143-10-3251. [DOI] [PubMed] [Google Scholar]
- [22].Zhao L, Borisova S, Yeung S-M, Liu H-W. Study of C-4 deoxygenation in the biosynthesis of desosamine: evidence implicating a novel mechanism. J. Am. Chem. Soc. 2001;123:7909–7910. doi: 10.1021/ja010587x. [DOI] [PubMed] [Google Scholar]
- [23].Zhao L, Sherman DH, Liu H-W. Biosynthesis of desosamine: molecular evidence suggesting β-glucosylation as a self-resistance mechanism in methymycin/-neomethymycin producing strain, Streptomyces venezuelae. J. Am. Chem. Soc. 1998;120:9374–9375. [Google Scholar]
- [24].Szu P.-h., He X, Zhao L, Liu H.-w. Biosynthesis of TDP-d-desosamine: identification of a strategy for C4 deoxygenation. Angew. Chem. Int. Ed. 2005;44:6742–6746. doi: 10.1002/anie.200501998. [DOI] [PubMed] [Google Scholar]
- [25].Borisova SA, Zhao L, Melancon CE, III, Kao C-L, Liu H-W. Characterization of the glycosyltransferase activity of DesVII: analysis of and implications for the biosynthesis of macrolide antibiotics. J. Am. Chem. Soc. 2004;126:6534–6535. doi: 10.1021/ja049967j. [DOI] [PubMed] [Google Scholar]
- [26].Hong JSJ, Kim WS, Lee SK, Koh HS, Park HS, Park SJ, Kim YS, Yoon YJ. The role of a second protein (DesVIII) in glycosylation for the biosynthesis of hybrid macrolide antibiotics in Streptomyces venezuelae. J. Microbiol. Biotechnol. 2005;15:640–645. [Google Scholar]
- [27].Llewellyn NM, Spencer JB. Biosynthesis of 2-deoxystreptamine-containing aminoglycoside antibiotics. Nat. Prod. Rep. 2006;23:864–874. doi: 10.1039/b604709m. [DOI] [PubMed] [Google Scholar]
- [28].Kudo F, Hosomi Y, Tamegai H, Kakinuma K. Purification and characterization of 2-deoxy-scyllo-inosose synthase derived from Bacillus circulans. A crucial carbocyclization enzyme in the biosynthesis of 2-deoxystreptamine-containing aminoglycoside antibiotics. J. Antibiot. 1999;52:81–88. doi: 10.7164/antibiotics.52.81. [DOI] [PubMed] [Google Scholar]
- [29].Kudo F, Tamegai H, Fujiwara T, Tagami U, Hirayama K, Kakinuma K. Molecular cloning of the gene for the key carbocycle-forming enzyme in the biosynthesis of 2-deoxystreptamine-containing aminocyclitol antibiotics and its comparison with dehydroquinate synthase. J. Antibiot. 1999;52:559–571. doi: 10.7164/antibiotics.52.559. [DOI] [PubMed] [Google Scholar]
- [30].Ota Y, Tamegai H, Kudo F, Kuriki H, Koike-Takeshita A, Eguchi T. Butirosinbiosynthetic gene cluster from Bacillus circulans. J. Antibiot. 2000;53:1158–1167. doi: 10.7164/antibiotics.53.1158. [DOI] [PubMed] [Google Scholar]
- [31].Tamegai H, Nango E, Kuwahara M, Yamamoto H, Ota Y, Kuriki H, Eguchi T, Kakinuma K. Identification of L-glutamine:2-deoxy-scyllo-inosose aminotransferase required for the biosynthesis of butirosin in Bacillus circulans. J. Antibiot. 2002;55:707–714. doi: 10.7164/antibiotics.55.707. [DOI] [PubMed] [Google Scholar]
- [32].Lucher LA, Chen Y-M, Walker JB. Reactions catalyzed by purified L-glutamine: keto-scyllo-inositol aminotransferase, an enzyme required for biosynthesis of aminocyclitol antibiotics. Antimicrob. Agents Chemother. 1989;33:452–459. doi: 10.1128/aac.33.4.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huang F, Li Y, Yu J, Spencer JB. Biosynthesis of aminoglycoside antibiotics: cloning, expression and characterisation of an aminotransferase involved in the pathway to 2-deoxystreptamine. Chem. Commun. 2002:2860–2861. doi: 10.1039/b209799k. [DOI] [PubMed] [Google Scholar]
- [34].Yokoyama K, Kudo F, Kuwahara M, Inomata K, Tamegai H, Eguchi T, Kakinuma K. Stereochemical recognition of doubly functional aminotransferase in 2-deoxystreptamine biosynthesis. J. Am. Chem. Soc. 2005;127:5869–5874. doi: 10.1021/ja0445948. [DOI] [PubMed] [Google Scholar]
- [35].Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. Characterization and mechanistic study of a radical SAM dehydrogenase in the biosynthesis of butirosin. J. Am. Chem. Soc. 2007;129:15147–15155. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- [36].Kudo F, Eguchi T. Biosynthetic genes for aminoglycoside antibiotics. J. Antibiot. 2009;62:471–481. doi: 10.1038/ja.2009.76. [DOI] [PubMed] [Google Scholar]
- [37].Kudo F, Yamamoto Y, Yokoyama K, Eguchi T, Kakinuma K. Biosynthesis of 2-deoxystreptamine by three crucial enzymes in Streptomyces fradiae NBRC 12773. J. Antibiot. 2005;58:766–774. doi: 10.1038/ja.2005.104. [DOI] [PubMed] [Google Scholar]
- [38].Kurumbang NP, Oh T-J, Liou K, Sohng JK. Heterologous production and detection of recombinant directing 2-deoxystreptamine (DOS) in the nonaminoglycoside-producing host Streptomyces venezuelae YJ003. J. Microbiol. Biotechnol. 2008;18:866–873. [PubMed] [Google Scholar]
- [39].Park JW, Hong JSJ, Parajuli N, Jung WS, Park SR, Lim S-K, Sohng JK, Yoon YJ. Genetic dissection of the biosynthetic route to gentamicin A2 by heterologous expression of its minimal gene set. Proc. Nat. Acad. Sci. U.S.A. 2008;105:8399–8404. doi: 10.1073/pnas.0803164105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Huang F, Haydock SF, Mironenko T, Spiteller D, Li Y, Spencer JB. The neomycin biosynthetic gene cluster of Streptomyces fradiae NCIMB 8233: characterisation of an aminotransferase involved in the formation of 2-deoxystreptamine. Org. Biomol. Chem. 2005;3:1410–1419. doi: 10.1039/b501199j. [DOI] [PubMed] [Google Scholar]
- [41].Kudo F, Eguchi T. Biosynthetic enzymes for the aminoglycosides butirosin and neomycin. Method. Enzymol. 2009;459:493–519. doi: 10.1016/S0076-6879(09)04620-5. [DOI] [PubMed] [Google Scholar]
- [42].Fang Q, Peng J, Dierks T. Post-translational formylglycine modification of bacterial sulfatases by the radical S-adenosylmethionine protein AtsB. J. Biol. Chem. 2004;279:14570–14578. doi: 10.1074/jbc.M313855200. [DOI] [PubMed] [Google Scholar]
- [43].Benjdia A, Leprince J, Guillot A, Vaudry H, Rabot S, Berteau O. Anaerobic sulfatase-maturating enzymes: radical SAM enzymes able to catalyze in vitro sulfatase post-translational modification. J. Am. Chem. Soc. 2007;129:3462–3463. doi: 10.1021/ja067175e. [DOI] [PubMed] [Google Scholar]
- [44].Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. Taylor & Francis; New York: 2007. [Google Scholar]
- [45].Szu P-H, Ruszczycky MW, Choi S-H, Liu H-W. Characterization and mechanistic studies of DesII: a radical S-adenosyl-l-methionine enzyme involved in the biosyn-thesis of TDP-d-desosamine. J. Am. Chem. Soc. 2009;131:14030–14042. doi: 10.1021/ja903354k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Szu P-H. Ph.D. Thesis. University of Texas; Austin: 2008. The Biosynthesis of TDP-d-desosamine: Characterization and mechanistic studies of DesII, a radical S-adenosylmethionine-dependent enzyme. [Google Scholar]
- [47].Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe–4S] cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- [48].Chen D, Walsby C, Hoffman BM, Frey PA. Coordination and mechanism of reversible cleavage of S-adenosylmethionine by the [4Fe–4S] center in lysine 2,3-aminomutase. J. Am. Chem. Soc. 2003;125:11788–11789. doi: 10.1021/ja036120z. [DOI] [PubMed] [Google Scholar]
- [49].Wang SC, Frey PA. Binding energy in the one-electron reductive cleavage of S-adenosylmethionine in lysine 2,3-aminomutase, a radical SAM enzyme. Biochemistry. 2007;46:12889–12895. doi: 10.1021/bi701745h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Duschene KS, Veneziano SE, Silver SC, Broderick JB. Control of radical chemistry in the AdoMet radical enzymes. Curr. Opin. Chem. Biol. 2009;13:74–83. doi: 10.1016/j.cbpa.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ruszczycky MW, Choi S.-h., Liu H.-w. Stoichiometry of the redox neutral deamination and oxidative dehydrogenation reactions catalyzed by the radical SAM enzyme DesII. J. Am. Chem. Soc. 2010;132:2359–2369. doi: 10.1021/ja909451a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu A, Gräslund A. Electron paramagnetic resonance evidence for a novel inter-conversion of [3Fe–4S]+ and [4Fe–4S]+ clusters with endogenous iron and sul-fide in anaerobic ribonucleotide reductase activase in vitro. J. Biol. Chem. 2000;275:12367–12373. doi: 10.1074/jbc.275.17.12367. [DOI] [PubMed] [Google Scholar]
- [53].Petrovich RM, Ruzicka FJ, Reed GH, Frey PA. Characterization of iron–sulfur clusters in lysine 2,3-aminomutase by electron paramagnetic resonance spectroscopy. Biochemistry. 1992;31:10774–10781. doi: 10.1021/bi00159a019. [DOI] [PubMed] [Google Scholar]
- [54].Hinckley GT, Frey P. Cofactor dependence of reduction potentials for [4Fe–4S]2 +/1+ in lysine-2,3-aminomutase. Biochemistry. 2006;45:3219–3225. doi: 10.1021/bi0519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yokoyama K, Ohmori D, Kudo F, Eguchi T. Mechanistic study on the reaction of a radical SAM dehydrogenase BtrN by electronic paramagnetic resonance spectroscopy. Biochemistry. 2008;47:8950–8960. doi: 10.1021/bi800509x. [DOI] [PubMed] [Google Scholar]
- [56].Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ. A consensus mechanism for radical SAM-dependent dehydrogenation? BtrN contains two [4Fe–4S] clusters. Biochemistry. 2010;49:3783–3785. doi: 10.1021/bi9022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ruszczycky MW, Choi S.-h., Mansoorabadi SO, Liu H.-w. Mechanistic studies of the radical S-adenosyl-l-methionine enzyme DesII: EPR characterization of a radical intermediate generated during its catalyzed dehydrogenation of TDP-d-quinovose. J. Am. Chem. Soc. 2011;133:7292–7295. doi: 10.1021/ja201212f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Weil JA, Bolton JR, Wertz JE. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. John Wiley & Sons; New York: 1994. [Google Scholar]
- [59].Wang SC, Frey PA. S-Adenosylmethionine as an oxidant: the radical SAM superfamily. Trends Biochem. Sci. 2007;32:101–110. doi: 10.1016/j.tibs.2007.01.002. [DOI] [PubMed] [Google Scholar]
- [60].de Carvalho LM, Schwedt G. Polarographic determination of dithionite and its decomposition products: kinetic aspects, stabilizers, and analytical application. Analytica Chimica Acta. 2001;436:293–300. [Google Scholar]
- [61].Daley CJA, Holm RH. Reactivity of [Fe4S4(SR)4]2−,3− clusters with sulfonium cations: analogue reaction systems for the initial step in biotin synthase catalysis. Inorg. Chem. 2001;40:2785–2793. doi: 10.1021/ic010039k. [DOI] [PubMed] [Google Scholar]
- [62].Daley CJA, Holm RH. Reactions of site-differentiated [Fe4S4]2+,1+ clusters with sulfonium cations: reactivity analogues of biotin synthase and other members of the S-adenosylmethionine enzyme family. J. Inorg. Biochem. 2003;97:287–298. doi: 10.1016/s0162-0134(03)00280-0. [DOI] [PubMed] [Google Scholar]
- [63].Rao PV, Holm RH. Synthetic analogues of the active sites of iron–sulfur proteins. Chem. Rev. 2004;104:527–559. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- [64].Hayon E, Simic M. Acid–base properties of free radicals in solution. Acc. Chem. Res. 1974;7:114–121. [Google Scholar]
- [65].Rao PS, Hayon E. Correlation between ionization constants of organic free radicals and electrochemical properties of parent compounds. Anal. Chem. 1976;48:564–568. [Google Scholar]
- [66].Ito N, Phillips SEV, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KDS, Knowles PF. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature. 1991;350:87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- [67].Whittaker MM, Whittaker JW. A tyrosine-derived free radical in apogalactose oxidase. J. Biol. Chem. 1990;265:9610–9613. [PubMed] [Google Scholar]
- [68].Lee Y-K, Whittaker MM, Whittaker JW. The electronic structure of the Cys-Tyr• free radical in galactose oxidase determined by EPR spectroscopy. Biochemistry. 2008;47:6637–6649. doi: 10.1021/bi800305d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ito N, Phillips SEV, Yadev KDS, Knowles PF. Crystal structure of a free radical enzyme, galactose oxidase. J, Mol. Biol. 1994;238:794–814. doi: 10.1006/jmbi.1994.1335. [DOI] [PubMed] [Google Scholar]
- [70].Whittaker JW. Free radical catalysis by galactose oxidase. Chem. Rev. 2003;103:2347–2363. doi: 10.1021/cr020425z. [DOI] [PubMed] [Google Scholar]
- [71].Whittaker JW. The radical chemistry of galactose oxidase. Arch. Biochem. Biophys. 2005;433:227–239. doi: 10.1016/j.abb.2004.08.034. [DOI] [PubMed] [Google Scholar]
- [72].Szameit C, Miech C, Balleininger M, Schmidt B, von Figura K, Dierks T. The iron sulfur protein AtsB is required for posttranslational formation of formylglycine in the Klebsiella sulfatase. J, Biol. Chem. 1999;274:15375–15381. doi: 10.1074/jbc.274.22.15375. [DOI] [PubMed] [Google Scholar]
- [73].Benjdia A, Subramanian S, Leprince J, Vaudry H, Johnson MK, Berteau O. Anaerobic sulfatase-maturating enzymes, first dual substrate radical S-adenosylmethionine enzymes. J. Biol. Chem. 2008;283:17815–17826. doi: 10.1074/jbc.M710074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Benjdia A, Leprince J, Sandström C, Vaudry H, Berteau O. Mechanistic investigations of anaerobic sulfatase-maturating enzyme: direct Cβ H-atom abstraction catalyzed by a radical AdoMet enzyme. J. Am. Chem. Soc. 2009;131:8348–8349. doi: 10.1021/ja901571p. [DOI] [PubMed] [Google Scholar]
- [75].Grove TL, Lee K-H, Clair J. St., Krebs C, Booker SJ. In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three [4Fe– 4S] clusters. Biochemistry. 2008;47:7523–7538. doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Berteau O, Guillot A, Benjdia A, Rabot S. A new type of bacterial sulfatase reveals a novel maturation pathway in prokaryotes. J. Biol. Chem. 2006;281:22464–22470. doi: 10.1074/jbc.M602504200. [DOI] [PubMed] [Google Scholar]
- [77].Cheng Q, Hwa V, Salyers AA. A locus that contributes to colonization of the intestinal tract by Bacteroides thetaiotaomicron contains a single regulatory gene (chuR) that links two polysaccharide utilization pathways. J. Bacteriol. 1992;174:7185–7193. doi: 10.1128/jb.174.22.7185-7193.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Babior BM. The mechanism of action of ethanolamine deaminase: I. Studies with isotopic hydrogen and oxygen. J. Biol. Chem. 1969;244:449–456. [PubMed] [Google Scholar]
- [79].Bandarian V, Reed GH. Ethanolamine ammonia-lyase. In: Banerjee R, editor. Chemistry and Biochemistry of B12. John Wiley & Sons; New York: 1999. pp. 811–833. [Google Scholar]
- [80].Semialjac M, Schwarz H. Computational exploration of rearrangements related to the vitamin B12-dependent ethanolamine ammonia lyase catalyzed transformation. J. Am. Chem. Soc. 2002;124:8974–8983. doi: 10.1021/ja020101s. [DOI] [PubMed] [Google Scholar]
- [81].Warncke K, Canfield JM. Direct determination of product radical structure reveals the radical rearrangement pathway in a coenzyme B12-dependent enzyme. J. Am. Chem. Soc. 2004;126:5930–5931. doi: 10.1021/ja031569d. [DOI] [PubMed] [Google Scholar]
- [82].Stubbe J, van der Donk WA. Protein radicals in enzyme catalysis. Chem. Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- [83].Buckel W. Radical and electron recycling in catalysis. Angew. Chem. Int. Ed. 2009;48:6779–6787. doi: 10.1002/anie.200900771. [DOI] [PubMed] [Google Scholar]
- [84].Buis JM, Cheek J, Kalliri E, Broderick JB. Characterization of an active spore photoproduct lyase, a DNA repair enzyme in the radical S-adenosylmethionine superfamily. J. Biol. Chem. 2006;281:25994–26003. doi: 10.1074/jbc.M603931200. [DOI] [PubMed] [Google Scholar]
- [85].Cheek J, Broderick JB. Direct, H atom abstraction form spore photoproduct C-6 initiates DNA repair in the reaction catalyzed by spore photoproduct lyase: evidence for a reversibly generated adenosyl radical intermediate. J. Am. Chem. Soc. 2002;124:2860–2861. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- [86].Padovani D, Thomas F, Trautwein AX, Mulliez E, Fontecave M. Activation of class III ribonucleotide reductase from E. coli. The electron transfer from the iron–sulfur center to S-adenosylmethionine. Biochemistry. 2001;40:6713–6719. doi: 10.1021/bi002936q. [DOI] [PubMed] [Google Scholar]
- [87].Rebeil R, Nicholson WL. The subunit structure and catalytic mechanism of the Bacillus subtilis DNA repair enzyme spore photoproduct lyase. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9038–9043. doi: 10.1073/pnas.161278998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Ollagnier-de Choudens S, Sanakis Y, Hewitson KS, Roach P, Münck E, Fontecave M. Reductive cleavage of S-adenosylmethionine by biotin synthase from Escherichia coli. J. Biol. Chem. 2002;277:13449–13454. doi: 10.1074/jbc.M111324200. [DOI] [PubMed] [Google Scholar]
- [89].Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Tu L, Booker SJ. Lipoyl synthase requires two equivalents of S-adenosyl-l-methionine to synthesize one equivalent of lipoic acid. Biochemistry. 2004;43:6378–6386. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- [90].Lotierzo M, Raux E, Bui B. Tse Sum, Goasdoue N, Libot F, Florentin D, Warren MJ, Marquet A. Biotin synthase mechanism: mutagenesis of the YNHNLD conserved motif. Biochemistry. 2006;45:12274–12281. doi: 10.1021/bi060662m. [DOI] [PubMed] [Google Scholar]
- [91].Pieck JC, Hennecke U, Pierik AJ, Friedel MG, Carell T. Characterization of a new thermophilic spore photoproduct lyase from Geobacillus stearothermophilus (SplG) with defined lesion containing DNA substrates. J. Biol. Chem. 2006;281:36317–36326. doi: 10.1074/jbc.M607053200. [DOI] [PubMed] [Google Scholar]
- [92].Taylor AM, Farrar CE, Jarrett JT. 9-Mercaptodethiobiotin is formed as a competent catalytic intermediate by Escherichia coli biotin synthase. Biochemistry. 2008;47:9309–9317. doi: 10.1021/bi801035b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Buckel W. Unusual dehydrations in anaerobic bacteria: considering ketyls (radical anions) as reactive intermediates in enzymatic reactions. FEBS Lett. 1996;389:20–24. doi: 10.1016/0014-5793(96)00530-3. [DOI] [PubMed] [Google Scholar]
- [94].Kochi JK. Electron transfer and charge transfer: twin themes in unifying the mechanisms of organic and organometallic reactions. Angew. Chem. Int. Ed. Engl. 1988;27:1227–1388. [Google Scholar]
- [95].Eberson L, Shaik S. Electron-transfer reactions of radical anions: do they follow outer- or inner-sphere mechanisms. J. Am. Chem. Soc. 1990;112:4484–4489. [Google Scholar]
- [96].Bandarian V, Reed GH. Analysis of the electron paramagnetic resonance spectrum of a radical intermediate in the coenzyme B12-dependent ethanolamine ammonia-lyase catalyzed reaction of S-2-aminopropanol. Biochemistry. 2002;41:8580–8588. doi: 10.1021/bi0201217. [DOI] [PubMed] [Google Scholar]
- [97].Yamanishi M, Ide H, Murakami Y, Toraya T. Identification of the 1,2-propane-diol-1-yl radical as an intermediate in adenosylcobalamin-dependent diol dehydratase reaction. Biochemistry. 2005;44:2113–2118. doi: 10.1021/bi0481850. [DOI] [PubMed] [Google Scholar]
- [98].Kimura K-I, Bugg TDH. Recent advances in antimicrobial nucleoside antibiotics targeting cell wall biosynthesis. Nat. Prod. Rep. 2003;20:252–273. doi: 10.1039/b202149h. [DOI] [PubMed] [Google Scholar]
- [99].Tsvetanova BC, Kiemle DJ, Price NPJ. Biosynthesis of tunicamycin and metabolic origin of the 11-carbon dialdose sugar, tunicamine. J. Biol. Chem. 2002;277:35289–35296. doi: 10.1074/jbc.M201345200. [DOI] [PubMed] [Google Scholar]
- [100].Chen W, Qu D, Zhai L, Tao M, Wang Y, Lin S, Price NPJ, Deng Z. Characterization of the tunicamycin gene cluster unveiling unique steps involved in its biosynthesis. Protein Cell. 2010;1:1093–1105. doi: 10.1007/s13238-010-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Nagahata T, Ueda K, Tsurimoto T, Chisaka O, Matsubara K. Anti-hepatitis B virus activities of purine derivatives of oxetanocin A. J. Antibiot. 1989;42:644–646. doi: 10.7164/antibiotics.42.644. [DOI] [PubMed] [Google Scholar]
- [102].Hoshino H, Shimizu N, Shimada N, Takita T, Takeuchi T. Inhibition of infectivity of human immunodeficiency virus by oxetanocin. J. Antibiot. 1987;40:1077–1078. doi: 10.7164/antibiotics.40.1077. [DOI] [PubMed] [Google Scholar]
- [103].Shimada N, Hasegawa S, Harada T, Tomisawa T, Fujii A, Takita T. Oxetanocin, a novel nucleoside from bacteria. J. Antibiot. 1986;39:1623–1625. doi: 10.7164/antibiotics.39.1623. [DOI] [PubMed] [Google Scholar]
- [104].Nakamura H, Hasegawa S, Shimada N, Fujii A, Takita T, Iitaka Y. The X-ray structure determination of oxetanocin. J. Antibiot. 1986;39:1626–1629. doi: 10.7164/antibiotics.39.1626. [DOI] [PubMed] [Google Scholar]
- [105].Morita M, Tomita K, Ishizawa M, Takagi K, Kawamura F, Takahashi H, Morino T. Cloning of oxetanocin A biosynthetic and resistance genes that reside on a plasmid of Bacillus megaterium strain NK84-0128. Biosci. Biotechnol. Biochem. 1999;63:563–566. doi: 10.1271/bbb.63.563. [DOI] [PubMed] [Google Scholar]
- [106].Morita M, Tomita K, Ishizawa M, Takagi K, Kawamura F, Takahashi H, Morino T. Molecular cloning of oxetanocin A biosynthetic and resistance genes which reside on a plasmid of Bacillus megaterium strain NK84-0128, Unpublished direct submission to NCBI Data Base. 2007 http://www.ncbi.nlm.nih.gov. Accession No. AB005787.
- [107].Woodyer RD, Li G, Zhao H, van der Donk WA. New insight into the mechanism of methyl transfer during the biosynthesis of fosfomycin. Chem. Commun. 2007:359–361. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- [108].Booker SJ. Anaerobic functionalization of unactivated C–H bonds. Curr. Opin. Chem. Biol. 2009;13:58–73. doi: 10.1016/j.cbpa.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ostash B, Doud EH, Lin C, Ostash I, Perlstein DL, Fuse S, Wolpert M, Kahne D, Walker S. Complete characterization of the seventeen step moenomycin bio-synthetic pathway. Biochemistry. 2009;48:8830–8841. doi: 10.1021/bi901018q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Park JW, Hong JSJ, Parajuli N, Koh HS, Park SR, Lee M-O, Lim S-K, Yoon YJ. Analytical profiling of biosynthetic intermediates involved in the gentamicin pathway of Micromonospora echinospora by high-performance liquid chromatography using electrospray ionization mass spectrometric detection. Anal. Chem. 2007;79:4860–4869. doi: 10.1021/ac070028u. [DOI] [PubMed] [Google Scholar]
- [111].Unwin J, Standage S, Alexander D, Hosted T, Jr., Horan AC, Wellington EMH. Gene cluster in Micromonospora echinospora ATCC15835 for the biosynthesis of the gentamicin C complex. J. Antibiot. 2004;57:436–445. doi: 10.7164/antibiotics.57.436. [DOI] [PubMed] [Google Scholar]
- [112].Kuzuyama T, Seki T, Dairi T, Hidaka T, Seto H. Nucleotide sequence of fortimicin KL1 methyltransferase gene isolated from Micromonospora olivasterospora, and comparison of its deduced amino acid sequence with those of methyltransferases involved in the biosynthesis of bialaphos and fosfomycin. J. Antibiot. 1995;48:1191–1193. doi: 10.7164/antibiotics.48.1191. [DOI] [PubMed] [Google Scholar]