

Abstract
Leukotrienes are pro-inflammatory mediators that are locally produced in coronary atherosclerotic plaques. The response induced by cysteinyl leukotrienes (CysLT) in human coronary arteries may be altered under pathological conditions, such as atherosclerosis. The aim of the present study was to elucidate cysteinyl leukotriene signaling in vascular smooth muscle cells (SMCs) and the effects of inflammation on this process. Immunohistochemical analysis of human carotid endarterectomy samples revealed that the CysLT1 leukotriene receptor was expressed in areas that also stained positive for α-smooth muscle actin. In human coronary artery smooth muscle cells, lipopolysaccharide significantly upregulated the CysLT1 receptor and significantly enhanced the changes in intracellular calcium induced by leukotriene C4 (LTC4). In these cells, the CysLT1 receptor exhibited a perinuclear expression, and LTC4 stimulation predominantly enhanced nuclear calcium increase, which was significantly inhibited by the CysLT1 receptor antagonist MK-571. Microarray analysis revealed, among a number of significantly upregulated genes after 24 h stimulation of human coronary artery smooth muscle cells with LTC4, a 5-fold increase in mRNA levels for plasminogen activator inhibitor (PAI)-2. The LTC4-induced increase in PAI-2 expression was confirmed by real-time quantitative PCR and ELISA and was inhibited by the CysLT1 receptor antagonist MK-571 and by calcium chelators. In summary, pro-inflammatory stimulation of vascular SMCs upregulated a perinuclear CysLT1 receptor expression coupled to nuclear calcium signaling and changes in gene expression, such as upregulation of PAI-2. Taken together, these findings suggest a role of nuclear CysLT1 receptor signaling in vascular SMCs inducing gene expression patterns associated with atherosclerosis.
Electronic supplementary material
The online version of this article (doi:10.1007/s00109-012-0904-1) contains supplementary material, which is available to authorized users.
Keywords: Atherosclerosis, Eicosanoids, Inflammation, Lipoxygenase, PAI-2
Introduction
Although initially identified as targets in the treatment of asthma, recent findings have brought attention to leukotrienes (LTs) as potential mediators of cardiovascular disease [1], such as atherosclerosis [2], abdominal aortic aneurysms [3], and aortic stenosis [4]. Human coronary artery atherosclerotic lesions are a source of cysteinyl LTs (i.e., LTC4, D4, and E4) [5], and urinary levels of LTE4 are increased in acute coronary syndromes [6], implicating these mediators in coronary atherosclerosis and plaque instability. The notion of cysteinyl LTs as potential effectors of atherosclerosis has also received support from animal models showing beneficial effects on atherosclerosis burden [7, 8] and intimal hyperplasia [9] by specific antagonists of the leukotriene CysLT1 receptor.
The cysteinyl LTs induce their action through G-protein-coupled receptors referred to as CysLT1 and CysLT2, and the existence of further subclasses of CysLT receptors also has been suggested [10]. In human carotid atherosclerotic lesions, a 3-fold higher CysLT1 receptor expression compared with the CysLT2 receptor has been reported [11], although the cellular localization of these receptors has not yet been resolved. The signaling through the CysLT1 receptor subtypes have been widely studied in the context of bronchoconstriction and asthma [10]. Recently, the CysLT1 receptor antagonist montelukast was shown to be associated with a decreased risk of ischemic stroke and a decreased risk of myocardial infarction in males [12]. Although the latter report provides a first indication of beneficial effects of clinically used anti-leukotriene drugs, the exact role of CysLT receptor signaling in atherosclerosis remains to be established.
In addition to being bronchoconstrictors, cysteinyl LTs are also potent vasoconstrictors in the human lung [13]. Their role in the coronary vasculature, however, can be said to be contextually antithetical, as healthy human coronary arteries are unresponsive to cysteinyl LTs, but a contractile response to either LTC4 or LTD4 is observed in atherosclerotic coronaries [14, 15]. Although no previous study has addressed the mechanism for this differential sensitivity between healthy and atherosclerotic vessels, it is interesting to note that these leukotriene-induced contractions are inhibited by CysLT1 receptor antagonists [14]. The latter observation is, however, in contrast to the dominant CysLT2 receptor expression in human coronary artery smooth muscle cells (SMCs) [16].
With the notion in mind that a local production of cysteinyl LTs within the atherosclerotic lesion [5] could potentially activate CysLT receptors within the vascular wall, we engaged in this study with the hypothesis that the inflammatory environment of atherosclerosis could lead to an upregulation of CysLT1 receptors on vascular SMCs. Here, we have investigated this idea, with the aim to quantify and describe the nature of the increased CysLT1 signaling. Further, we have explored the potential downstream effects of this phenomenon on both intracellular signaling and gene expression in vascular SMCs in an effort to link CysLT1 receptor signaling to atherogenic properties of SMCs in the context of inflammation and leukotriene stimulation.
Methods
Cell culture
Human coronary artery SMC purchased from Clonetics (Cambrex Bio Science, Walkersville, MD, USA) were cultured in SmGM2 kit medium as previously described [17] and harvested for experiments between passages 5 and 8. Cells were seeded in six-well plates (105 cells/well) containing SmGM2 48 h before the respective treatment, which was replaced with Dulbecco’s modified Eagle’s medium supplemented with 2 % fetal calf serum (starvation medium) 24 h before experiments. Subsequently, cells were incubated in the absence or presence of lipopolysaccharide (LPS; from Sigma, 10 μg/mL), interleukin-6 (IL-6; from Peprotech, 20 ng/mL), interferon-γ (IFN-γ; from Peprotech, 20 ng/mL), or tumor necrosis factor-α (TNF-α; from Peprotech, 10 ng/mL). Duration of treatment was experiment-dependent, as described in detail below. Cells were treated with various substances across experiments, including LTC4, LTD4, and LTE4 (from Cayman Chem, Ann Arbor, MI, USA; 1 μM), the CysLT1 receptor antagonist MK571 (from Cayman; 1 μM), ethylene glycol tetraacetic acid (EGTA; from Sigma, 5 mM), and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM; from Invitrogen, 10 μM).
Immunostainings
Atherosclerotic vascular tissue was collected from eight patients undergoing carotid endarterectomy. All experiments were performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and were approved by the local ethics committee (reference number 02/147). All persons gave their informed consent prior to their inclusion in the study. Immunofluorescent stainings were performed on acetone-fixed frozen sections of carotid endarterectomies and on human coronary artery SMCs cultured in LabTek slides after fixation and permeabilization with acetone–methanol. Rabbit anti-human CysLT1 receptor (from Cayman Chem) and mouse anti-human α-smooth muscle actin (from DAKO) were used as primary antibodies. Isotype-specific either DyLight 594 or DyLight 488-conjugated secondary antibodies (from Vector) were used, and the nuclei were counterstained with 4′, 6-diamino-2-phenylindol (DAPI; Vector). Images were captured with confocal microscope Leica DMI.
Calcium signaling experiments
SMCs incubated for 48 h in the absence or presence of LPS (10 μg/mL) were washed and loaded with the fluorescent Ca2+ indicator fluo-3. The cells were subsequently stimulated with LTC4 (1 μM, 30 min at room temperature) in either Tyrode’s solution (composition, millimolars: NaCl 121, KCl 5.0, NaHCO3 24, CaCl2 0.5, MgCl2 0.4, NaH2PO4 0.4, EDTA0.1, and glucose 5.5) gassed with 5 % CO2 in O2 or Tyrode’s solution containing the CysLT1 receptor antagonist MK571 (1 μM). Changes in [Ca2+] were recorded using a BioRad MRC 1024 confocal microscopy unit attached to a Nikon Diaphot 200 inverted microscope with a Nikon Plan Apo ×20 or ×60 oil immersion objective (N.A.1.3), as previously described [18].
RNA extraction, cDNA synthesis, and TaqMan real-time PCR
Total RNA was extracted using RNeasy Mini kit (from Qiagen, Hilden, Germany) with an on-column DNase digestion step. RNA quantity was assessed using a Nanodrop ND-1000 microvolume spectrophotometer (Thermo Fisher Scientific), and RNA quality was assessed by a Bioanalyzer capillary electrophoresis system (Agilent Technologies, Palo Alto, CA, USA). First-strand cDNA was synthesized using Superscript II (Invitrogen, Carlsbad, CA, USA) with random hexamers according to the manufacturer’s instructions. Quantitative TaqMan PCR was performed on a 7900HT Fast Real-Time PCR system (Applied Biosystems) with primer/probe pairs that were obtained using Assay-on-demand™ from Applied Biosystems for human CysLT1 (Hs00272624_s1) and Plasminogen Activator Inhibitor 2 (PAI-2/SERPIN B2; Hs01010736_m1). Levels of mRNA were normalized to expression levels of cyclophilin A (Hs99999904_m1), which previously has been determined as an appropriate housekeeping gene in these cells [17]
Microarray analysis
Microarray analysis experiments were performed on RNA derived from three separate SMC culture experiments, using either Agilent one-color whole human genome (44 K) kit (Agilent Technologies, Redwood City, CA, USA; n = 2) or the Affymetrix Human Genome U133 Plus 2.0 array (n = 1). Microarrays were analyzed with the Agilent high-resolution microarray scanner. Data were subsequently uploaded to GeneSpring GX10 (Agilent technologies) and analyzed using advanced analysis workflow for the Agilent one-color arrays. The set of data was normalized according to recommendations by GeneSpring for one-color arrays. (http://www.chem.agilent.com/cag/bsp/products/gsgx/manuals/GeneSpring-manual.pdf). Variability between chips was accounted for by applying a shift to the 75th percentile (dividing all measured signals by a 75th percentile value). Per-gene normalization was performed by bringing the baseline to the median of all samples. Probe sets were firstly filtered by confidence of detection, where genes that were not confidently detected in any sample were excluded from further analysis. Further filtering based on expression discarded any genes where less than 100 % of samples in either the relapse or diagnosis condition had expression values below the 20th percentile. The most differentially expressed genes, defined as those with an uncorrected P value of <0.05 and demonstrating a fold change in expression of 2.0 or greater, were selected for analysis. The list of 90 genes generated was subsequently compared to data from the Affymetrix arrays, and the genes of interest were verified in terms of direction of regulation. Genes meeting all these criteria are presented in Table 1. The 45 genes listed were submitted to Ingenuity® pathway analysis for prediction of canonical pathways and functional gene networks affected by the significant differential expression of these genes.
Table 1.
Most significantly differentially expressed genes in response to LTC4 (1 μM) in LPS-primed human coronary artery SMCs (sorted by fold change)
| Gene ID | Probe ID | Gene name | Mean fold change | Direction | P value |
|---|---|---|---|---|---|
| CSF3 | A_23_P501754 | Colony stimulating factor 3 | 15.88 | Up | 0.04128 |
| IL24 | A_23_P51951 | Interleukin 24 | 15.11 | Up | 0.04918 |
| SERPINB2 | A_23_P153185 | Serine proteinase inhibitor 2 (plasminogen activator inhibitor 2) | 5.46 | Up | 0.03882 |
| NEFM | A_24_P264832 | Neurofilament medium polypeptide | 4.89 | Up | 0.01537 |
| GJA1 | A_23_P93591 | Gap junction protein. Alpha 1 | 4.74 | Up | 0.00931 |
| IL1A | A_23_P72096 | Interleukin 1 alpha | 4.15 | Up | 0.01096 |
| CXCR4 | A_23_P102000 | Chemokine (C-X-C motif) receptor 4 | 3.94 | Up | 0.00170 |
| TFPI2 | A_24_P95070 | Tissue factor pathway inhibitor 2 | 3.90 | Up | 0.02599 |
| CCND1 | A_24_P124550 | Cyclin D1 | 3.28 | Up | 0.04980 |
| CXCL3 | A_24_P183150 | Chemokine (C-X-C motif) ligand 3 | 3.27 | Up | 0.02149 |
| ABCG1 | A_23_P166297 | ATP-binding cassette. Sub-family G. Member 1 | 3.03 | Up | 0.00178 |
| HAS1 | A_23_P27400 | Hyaluronan synthase 1 | 2.85 | Up | 0.03572 |
| MMP3 | A_23_P161698 | Matrix metallopeptidase 3 | 2.70 | Up | 0.01282 |
| PITPNC1 | A_24_P772103 | Phosphatidylinositol transfer protein. Cytoplasmic 1 | 2.55 | Up | 0.03758 |
| KITLG | A_23_P204654 | KIT ligand | 2.49 | Up | 0.00730 |
| FUBP3 | A_23_P435833 | Far upstream element (FUSE) binding protein 3 | 2.46 | Up | 0.00619 |
| NPTX1 | A_23_P124905 | Neuronal pentraxin I | 2.45 | Up | 0.00504 |
| PCSK1 | A_23_P213508 | Proprotein convertase subtilisin/kexin type 1 | 2.41 | Up | 0.04376 |
| ADSS | A_23_P859 | Adenylosuccinate synthase | 2.40 | Up | 0.01023 |
| TFPI2 | A_23_P393620 | Tissue factor pathway inhibitor 2 | 2.38 | Up | 0.01401 |
| NPR3 | A_23_P327451 | Natriuretic peptide receptor C/guanylate cyclase C | 2.33 | Up | 0.01309 |
| SPRED1 | A_23_P54460 | Sprouty-related. Evh1 domain containing 1 | 2.30 | Up | 0.02536 |
| PITPNC1 | A_23_P84189 | Phosphatidylinositol transfer protein. Cytoplasmic 1 | 2.29 | Up | 0.01626 |
| NKAIN1 | A_23_P51376 | Na+/K+ transporting ATPase interacting 1 | 2.26 | Up | 0.02838 |
| SOCS4 | A_24_P90637 | Suppressor of cytokine signaling 4 | 2.23 | Up | 0.00918 |
| TMTC3 | A_24_P944222 | Transmembrane and tetratricopeptide repeat containing 3 | 2.22 | Up | 0.04956 |
| CACNG8 | A_32_P61693 | Calcium channel. Voltage-dependent. Gamma subunit 8 | 2.19 | Up | 0.00480 |
| RRN3 | A_23_P206877 | RNA polymerase I-specific transcription initiation factor RRN3 | 2.15 | Up | 0.00613 |
| APOBEC3F | A_23_P369966 | Apolipoprotein B mRNA editing enzyme. Catalytic polypeptide-like 3F | 2.12 | Up | 0.03633 |
| NFE2L3 | A_24_P136653 | Nuclear factor (erythroid-derived 2)-like 3 | 2.08 | Up | 0.00912 |
| MYH11 | A_24_P70183 | Myosin heavy chain 11 (smooth muscle) | 10.44 | Down | 0.02218 |
| MYH11 | A_23_P206920 | Myosin heavy chain 11 (smooth muscle) | 9.66 | Down | 0.01751 |
| SCRG1 | A_23_P167159 | Stimulator of chondrogenesis 1 | 4.73 | Down | 0.01807 |
| TPD52L1 | A_23_P31143 | Tumor protein D52-like 1 | 4.64 | Down | 0.01830 |
| CARD9 | A_23_P500433 | Caspase recruitment domain family. Member 9 | 3.54 | Down | 0.00009 |
| CLIC3 | A_23_P254654 | Chloride intracellular channel 3 | 3.53 | Down | 0.00765 |
| KRT7 | A_23_P381945 | Keratin 7 | 3.37 | Down | 0.03569 |
| PCK2 | A_23_P128817 | Phosphoenolpyruvate carboxykinase 2 (mitochondrial) | 2.66 | Down | 0.02696 |
| CTAGE1 | A_24_P124805 | Cutaneous T cell lymphoma-associated antigen 1 | 2.57 | Down | 0.01412 |
| GLI1 | A_23_P105251 | GLI family zinc finger 1 | 2.56 | Down | 0.01105 |
| SLC1A7 | A_23_P325562 | Solute carrier family 1. Member 7 | 2.53 | Down | 0.00758 |
| TGM1 | A_23_P65618 | Transglutaminase 1 | 2.45 | Down | 0.00203 |
| IL17RD | A_32_P188860 | Interleukin 17 receptor D | 2.39 | Down | 0.01570 |
| ZNF467 | A_23_P59470 | Zinc finger protein 467 | 2.19 | Down | 0.00619 |
| LIMS2 | A_23_P142796 | LIM and senescent cell antigen-like domains 2 | 2.05 | Down | 0.01940 |
Note that some genes may be listed twice due to significant differences detected by independent probes
ELISA
PAI-2 ELISA was carried out on supernatants from untreated SMCs and SMCs treated LTC4 (1 μM) for 24 h using IMUBIND® PAI-2 ELISA kit (from American Diagnostica GmBH, Pfungstadt, Germany) according to manufacturer’s protocol.
Data analysis
All results are expressed as mean±SE. Statistically significant differences were determined by either a Student’s t test (for pair-wise comparisons) or a one-way analysis of variances, followed by Holm–Sidak post hoc test, for multiple comparisons, using Sigma Stat software. A P value of less than 0.05 was considered significant.
Results
CysLT1 receptor expression on vascular SMC
Immunohistochemical staining showed colocalization of the CysLT1 receptor protein with markers for SMC (α-smooth muscle actin) in human atherosclerotic lesions (Fig. 1). In human coronary artery SMCs, the transcriptional levels of the CysLT1 receptor were time-dependently increased by LPS, IL-6, and IFN-γ (Fig. 2). Fluorescent immunostainings revealed a predominantly perinuclear localization of the CysLT1 receptor in human coronary artery SMCs compared with α-smooth muscle actin, which stained positive in the whole cytoplasm (Fig. 3). The CysLT1 receptor in some cases demonstrated nuclear inclusions, as indicated by arrows in Fig. 3.
Fig. 1.
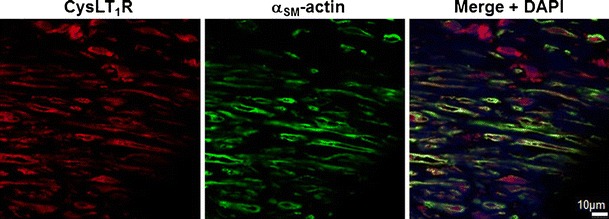
CysLT1 receptor expression in human atherosclerotic lesions. Representative immunofluorescent staining of human atherosclerotic plaques from carotid artery showing colocalization of the CysLT1 with α-smooth muscle actin-positive vascular smooth muscle cells. Original magnification, ×40
Fig. 2.
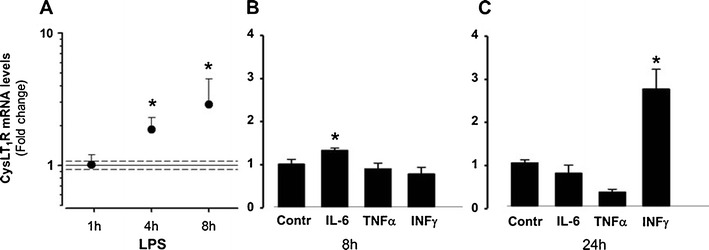
CysLT1 receptor expression in human coronary artery smooth muscle cells is upregulated by pro-inflammatory stimuli. Real-time quantitative TaqMan RT-PCR for CysLT receptor mRNA in SMCs incubated in the absence and presence of LPS (10 μg/mL) for 1, 4, and 8 h (a) and IL-6 (20 ng/mL), TNF-α (10 ng/mL), or IFN-γ (20 ng/mL) for either 8 h (b) or 24 h (c). Results are expressed as fold increase compared with untreated cells (n = 3–5). *P < 0.05 vs. time-matched control
Fig. 3.
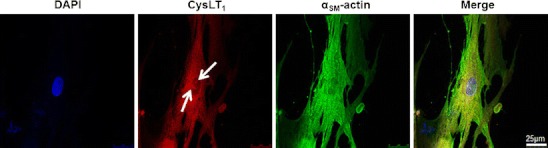
Perinuclear CysLT1 receptor expression in human coronary artery smooth muscle cells. Fluorescent labeling of CysLT1 receptor protein (DyLight 594 red chromogen) and αSM-actin (DyLight 488 green chromogen) in SMCs. Nuclei were stained with DAPI. Arrows indicate nuclear inclusions. Original magnification, ×63
LTC4-induced nuclear calcium signaling in vascular SMCs
To evaluate whether CysLT1 receptors expressed on vascular SMC were functional, calcium changes in human coronary artery SMC were studied using the fluorescent Ca2+ indicator fluo-3 (Fig. 4a). LTC4 induced a dose-dependent increase in intracellular calcium, which was predominantly located in the nucleus (Fig. 4b). The LTC4-induced calcium increase was significantly higher in LPS-treated cells compared with untreated cells (Fig. 4c). In LPS-treated cells, the LTC4-induced increase in nuclear calcium was significantly inhibited by the CysLT1 receptor antagonist MK571 (Fig. 4c). The time course of the LTC4-induced calcium increase in the nuclear and cytosolic compartments is shown in Fig. 4d. The increase in nuclear calcium preceded the increase in cytosolic calcium (Fig. 4d).
Fig. 4.
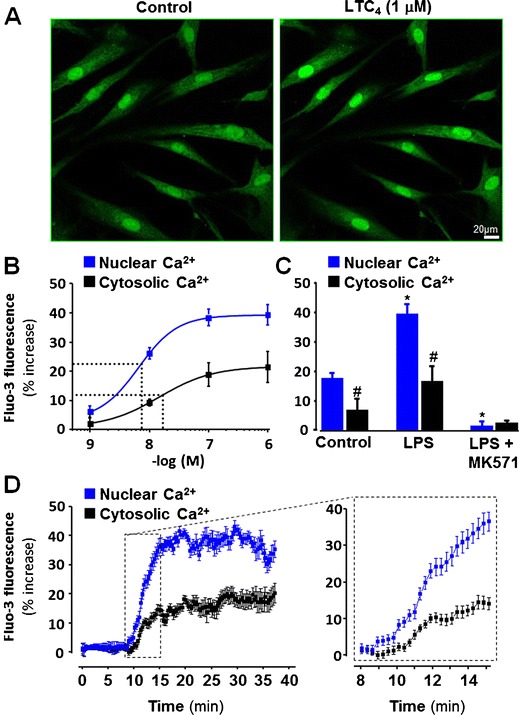
LTC4-induced calcium signaling in human coronary artery SMC. a Representative micrographs of Ca2+ fluorescence in the absence and presence of LTC4 (1 μM). b Concentration–response curves for Ca2+ fluorescence in nuclei (blue symbols) and cytosol (black symbols) of SMCs incubated for 48 h in the presence of LPS (10 μg/ml). c Ca2+ fluorescence in nuclei (blue bars) and cytosol (black bars) of SMCs incubated for 48 h in the absence (control) or presence of LPS (10 μg/ml) prior to stimulation with LTC4 (1 μM, 30 min). d The time course of the LTC4-induced calcium increase shows that the increase in nuclear calcium (blue symbols) preceded the increase in cytosolic calcium (black symbols). *P < 0.05 vs. controls, # P < 0.05 vs Nuclear Ca2+
LTC4-induced gene expression in vascular SMCs
The genes most significantly differentially expressed in response to LTC4 (1 μM) in LPS-primed human coronary artery SMCs are presented in Table 1. PAI-2 (SERPIN B2), a member of the serine protease inhibitor superfamily, presented as one of the most significantly upregulated genes in the microarray analysis. This finding was confirmed by quantitative PCR (Fig. 5a), and in addition, increased PAI-2 protein levels were detected in the supernatant derived from LTC4-stimulated human coronary artery SMCs, compared with unstimulated cells (Fig. 5b). The increased mRNA levels of PAI-2 induced by LTC4 (1 μM) were mimicked by LTD4 (1 μM) but not LTE4 (1 μM) and significantly inhibited by the CysLT1 receptor antagonist MK 571 (1 μM; Fig. 5a). In addition, the LTC4-induced increase in PAI-2 mRNA was abolished by the removal of intra- and extracellular calcium, through experiments performed in the presence of BAPTA-AM and EGTA (Fig. 5c).
Fig. 5.

LTC4-induced upregulation of PAI-2 in human coronary artery SMC. a Real-time quantitative RT-PCR for PAI-2 mRNA in human coronary artery SMC incubated in the absence and presence of LTC4, LTD4 or LTE4 (1 μM) for 24 h. In some experiments, cells were pretreated with the CysLT1 receptor antagonist MK571 (1 μM) for 1 h before addition of LTC4. *P < 0.05 vs. non-LTC4 stimulated Contr (n = 3–6). b PAI-2 concentrations in supernatants from human coronary artery SMC incubated 24 h in the absence (Contr) or presence of LTC4 (1 μM). *P < 0.05 vs. Contr (n = 7). c Increase in PAI-2 mRNA levels induced by LTC4 (1 μM) in human coronary artery SMC incubated in the absence (Contr) or presence of either BAPTA-AM or EGTA. *P < 0.05 vs. Contr (n = 3–6)
Ingenuity pathway analysis identified several functional gene networks predicted to be significantly affected by LTC4 stimulation as based on the 45 genes determined to be differentially expressed through our microarray analysis (Table 1). The highest-scoring network (with a score of 35, equating to a fishers’ exact test score of 1 × 10−35) containing 15 genes from Table 1 is suggested to be implicated in cellular movement and hematopoietic system function and development. An outline of the network is shown in Supplementary Fig. 1.
Discussion
The results of the present study showed an upregulation of predominantly perinuclear CysLT1 receptors in vascular SMCs under inflammatory conditions which was associated with increased nuclear calcium signaling and changes in gene expression. Taken together, these findings suggest a role of nuclear CysLT1 receptor signaling in vascular SMCs inducing gene expression patterns associated with atherosclerosis.
Previous studies have suggested a dominant expression of the leukotriene CysLT2 receptor subtype in human coronary arteries [16]. In the present study, priming of vascular SMCs with LPS upregulated CysLT1 receptor mRNA and enhanced LTC4-induced effects. Similar findings have been reported in endothelial cells, which under resting conditions exhibit a dominant CysLT2 receptor, but in which prolonged exposure to LPS or pro-inflammatory cytokines upregulate CysLT1 receptor expression [19]. In the present study, CysLT1 receptor expression was also upregulated in SMCs by IL-6 and by prolonged exposure to IFN-γ. Taken together, these observations suggest that a pro-inflammatory environment, such as atherosclerosis, may induce CysLT1 receptor expression within the vascular wall. In support of the latter notion, it has been shown that LTC4 induces contractions of atherosclerotic but not healthy coronary arteries [14, 15] and that CysLT1 receptor signaling, but not CysLT2 receptor signaling, is coupled to vasoconstriction in isolated systemic vessels [20]. LTC4 has also been associated with SMC proliferation and the shift of SMCs into a synthetic phenotype [21], which is in line with findings that CysLT1 receptor antagonism inhibits intimal hyperplasia after vascular injury in mice [9].
The present study is the first demonstration of a perinuclear localization of functional CysLT1 receptors in vascular SMCs and a leukotriene-induced nuclear calcium signaling in these cells. These findings are nevertheless consistent with other G-protein-coupled receptors in vascular SMCs. For example, the ETA endothelin receptor and the AT1 angiotensin II receptor exhibit a perinuclear localization in vascular SMCs coupled to nuclear calcium signaling [22–24]. In addition, we have recently demonstrated a perinuclear localization of the CysLT1 receptor in valvular interstitial cells derived from human aortic valves [4], corroborating prior observations of CysLT1 receptor expression at the outer nuclear membrane in intestinal epithelial cells [25]. In both of these cell types, leukotriene stimulation induces an increase in nuclear calcium [4, 25]. The present study extends those findings by demonstrating an enhanced nuclear calcium increase in response to LTC4 after priming of cells with LPS and that the increase in nuclear calcium preceded the increase in cytosolic calcium. Whereas the present study cannot definitely conclude whether the subcellular CysLT1 receptor localization represents an internalization process, previous studies support a translocation of CysLT1 receptors between different cellular compartments, including nuclear inclusions [26].
Nuclear calcium is a key regulator of gene expression [27], and in line with this notion, LTC4 induced significant changes in expression of several genes in the present study. Of particular interest was the appearance of PAI-2 as one of the most significantly upregulated genes in the microarray analysis. This was confirmed with qPCR analysis, and ELISA measures in addition showed that PAI-2 protein secretion from human coronary artery SMCs was increased by LTC4 stimulation. PAI-1 and PAI-2 are members of the serine proteinase inhibitor family and act as important inhibitors of fibrinolysis by interfering with the plasminogen system. PAI-1 is induced by atherogenic stimuli in vascular SMCs and may participate in cell growth and matrix degradation associated with atherosclerosis [28]. In addition, using cDNA representational difference analysis, PAI-2 has previously been identified as one of the most differentially expressed genes in atherosclerotic lesions compared with normal vessels, with elevated PAI-2 expression preferentially observed in unstable carotid plaques [29]. In addition, another study using serial analysis of gene expression in human vascular SMCs also identified PAI-2 as one of the most upregulated genes in response to conditioned media derived from macrophages activated by oxidized low-density lipoprotein [30]. Finally, immunohistochemical analysis of human atherosclerotic lesions has also confirmed that vascular SMCs stain positive for PAI-2 [29, 30]. In addition to acting as a plasminogen activator inhibitor, PAI-2 may serve as a regulator of Th1 immune responses through the modulation of cytokine-induced responses [31]. Furthermore, PAI-2 may be associated with the process of wound healing post-plaque rupture [29].
The LTC4-induced increase in PAI-2 mRNA was abolished when experiments were performed in the presence of calcium chelators, suggesting a calcium dependent upregulation of PAI-2. The latter notion is supported by previous studies showing that angiotensin II is a potent inducer of PAI-2 in vascular SMCs through the AT1 receptor [32], which is in line with a perinuclear AT1 receptor localization and an EGTA-sensitive nuclear calcium signaling induced by angiotensin II [33].
Ingenuity pathway analysis revealed a significant number of LTC4-upregulated genes to be implicated in a functional gene network linked to hematopoietic system function and cellular movement. Of note in this network was the involvement of cAMP response element binding protein (CREB), a transcription factor and member of the leucine zipper family of DNA binding proteins, which is known to be activated by nuclear calcium [27]. Furthermore, the PAI-2 gene promoter region contains a binding site for CREB (−1,319 bp), and this transcription factor has been shown to be associated with the induction of PAI-2 expression [34]. Taken together, these findings suggest that LTC4-induced changes in gene expression may be induced through an increase in nuclear calcium leading to CREB activation. However, the pathway analysis also revealed other pathways that may be involved in LTC4-induced gene expression, such as the NF-κB signaling pathway which has been previously shown to be activated after CysLT1 receptor ligation in leukocytes [10].
In summary, we have shown that pro-inflammatory stimulation of vascular SMCs enhances perinuclear CysLT1 receptor expression coupled to nuclear calcium signaling and results in changes in gene expression, such as upregulation of PAI-2. Since cysteinyl-LT production is increased in atherosclerosis [5] and acute coronary syndromes [6], an altered vascular sensitivity to leukotriene-induced SMC gene expression secretion may further enhance the inflammatory response. As such, targeting CysLT1 receptors could potentially be of therapeutic interest in atherosclerosis.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 194 kb)
Acknowledgments
The authors would like to Professor Håkan Westerblad for helpful advice on the study. This work was supported by the Swedish Heart and Lung Foundation, the Swedish Research Council (grant numbers 06816 and 2011-2988), and the French–Swedish Foundation. AE was supported by a fellowship from Mach-Gaensslen Foundation of Canada.
Conflict of interest
None declared.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
References
- 1.Poeckel D, Funk CD. The 5-lipoxygenase/leukotriene pathway in preclinical models of cardiovascular disease. Cardiovasc Res. 2010;86:243–253. doi: 10.1093/cvr/cvq016. [DOI] [PubMed] [Google Scholar]
- 2.Bäck M, Hansson GK. Leukotriene receptors in atherosclerosis. Ann Med. 2006;38:493–502. doi: 10.1080/07853890600982737. [DOI] [PubMed] [Google Scholar]
- 3.Houard X, Ollivier V, Louedec L, Michel JB, Bäck M. Differential inflammatory activity across human abdominal aortic aneurysms reveals neutrophil-derived leukotriene B4 as a major chemotactic factor released from the intraluminal thrombus. FASEB J. 2009;23:1376–1383. doi: 10.1096/fj.08-116202. [DOI] [PubMed] [Google Scholar]
- 4.Nagy E, Andersson DC, Caidahl K, Eriksson MJ, Eriksson P, Franco-Cereceda A, Hansson GK, Bäck M. Upregulation of the 5-lipoxygenase pathway in human aortic valves correlates with severity of stenosis and leads to leukotriene-induced effects on valvular myofibroblasts. Circulation. 2011;123:1316–1325. doi: 10.1161/CIRCULATIONAHA.110.966846. [DOI] [PubMed] [Google Scholar]
- 5.Piomelli D, Feinmark SJ, Cannon PJ. Leukotriene biosynthesis by canine and human coronary arteries. J Pharmacol Exp Ther. 1987;241:763–770. [PubMed] [Google Scholar]
- 6.Carry M, Korley V, Willerson JT, Weigelt L, Ford-Hutchinson AW, Tagari P. Increased urinary leukotriene excretion in patients with cardiac ischemia. In vivo evidence for 5-lipoxygenase activation. Circulation. 1992;85:230–236. doi: 10.1161/01.CIR.85.1.230. [DOI] [PubMed] [Google Scholar]
- 7.Mueller CF, Wassmann K, Widder JD, Wassmann S, Chen CH, Keuler B, Kudin A, Kunz WS, Nickenig G. Multidrug resistance protein-1 affects oxidative stress, endothelial dysfunction, and atherogenesis via leukotriene C4 export. Circulation. 2008;117:2912–2918. doi: 10.1161/CIRCULATIONAHA.107.747667. [DOI] [PubMed] [Google Scholar]
- 8.Jawien J, Gajda M, Wolkow P, Zuranska J, Olszanecki R, Korbut R. The effect of montelukast on atherogenesis in apoE/LDLR-double knockout mice. J Physiol Pharmacol. 2008;59:633–639. [PubMed] [Google Scholar]
- 9.Kaetsu Y, Yamamoto Y, Sugihara S, Matsuura T, Igawa G, Matsubara K, Igawa O, Shigemasa C, Hisatome I. Role of cysteinyl leukotrienes in the proliferation and the migration of murine vascular smooth muscle cells in vivo and in vitro. Cardiovasc Res. 2007;76:160–166. doi: 10.1016/j.cardiores.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 10.Bäck M, Dahlen SE, Drazen JM, Evans JF, Serhan CN, Shimizu T, Yokomizo T, Rovati GE. International union of basic and clinical pharmacology. LXXXIV: leukotriene receptor nomenclature, distribution, and pathophysiological functions. Pharmacol Rev. 2011;63:539–584. doi: 10.1124/pr.110.004184. [DOI] [PubMed] [Google Scholar]
- 11.Lotzer K, Spanbroek R, Hildner M, Urbach A, Heller R, Bretschneider E, Galczenski H, Evans JF, Habenicht AJ. Differential leukotriene receptor expression and calcium responses in endothelial cells and macrophages indicate 5-lipoxygenase-dependent circuits of inflammation and atherogenesis. Arterioscler Thromb Vasc Biol. 2003;23:e32–e36. doi: 10.1161/01.ATV.0000082690.23131.CB. [DOI] [PubMed] [Google Scholar]
- 12.Ingelsson E, Yin L, Bäck M (2012) Nationwide cohort study of the leukotriene receptor antagonist montelukast and incident or recurrent cardiovascular disease. J Allergy Clin Immunol. doi:10.1016/j.jaci.2011.11.052 [DOI] [PubMed]
- 13.Bäck M. Functional characteristics of cysteinyl-leukotriene receptor subtypes. Life Sci. 2002;71:611–622. doi: 10.1016/S0024-3205(02)01733-2. [DOI] [PubMed] [Google Scholar]
- 14.Allen SP, Dashwood MR, Chester AH, Tadjkarimi S, Collins M, Piper PJ, Yacoub MH. Influence of atherosclerosis on the vascular reactivity of isolated human epicardial coronary arteries to leukotriene C4. Cardioscience. 1993;4:47–54. [PubMed] [Google Scholar]
- 15.Allen S, Dashwood M, Morrison K, Yacoub M. Differential leukotriene constrictor responses in human atherosclerotic coronary arteries. Circulation. 1998;97:2406–2413. doi: 10.1161/01.CIR.97.24.2406. [DOI] [PubMed] [Google Scholar]
- 16.Kamohara M, Takasaki J, Matsumoto M, Matsumoto S, Saito T, Soga T, Matsushime H, Furuichi K. Functional characterization of cysteinyl leukotriene CysLT(2) receptor on human coronary artery smooth muscle cells. Biochem Biophys Res Commun. 2001;287:1088–1092. doi: 10.1006/bbrc.2001.5695. [DOI] [PubMed] [Google Scholar]
- 17.Bäck M, Bu DX, Branström R, Sheikine Y, Yan ZQ, Hansson GK. Leukotriene B4 signaling through NF-kappaB-dependent BLT1 receptors on vascular smooth muscle cells in atherosclerosis and intimal hyperplasia. Proc Natl Acad Sci U S A. 2005;102:17501–17506. doi: 10.1073/pnas.0505845102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavi P, Hansson A, Zhang SJ, Larsson NG, Westerblad H. Abnormal Ca(2+) release and catecholamine-induced arrhythmias in mitochondrial cardiomyopathy. Hum Mol Genet. 2005;14:1069–1076. doi: 10.1093/hmg/ddi119. [DOI] [PubMed] [Google Scholar]
- 19.Gronert K, Martinsson-Niskanen T, Ravasi S, Chiang N, Serhan CN. Selectivity of recombinant human leukotriene D(4), leukotriene B(4), and lipoxin A(4) receptors with aspirin-triggered 15-epi-LXA(4) and regulation of vascular and inflammatory responses. Am J Pathol. 2001;158:3–9. doi: 10.1016/S0002-9440(10)63937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mechiche H, Candenas L, Pinto FM, Nazeyrollas P, Clément C, Devillier P. Characterization of cysteinyl leukotriene receptors on human saphenous veins: antagonist activity of montelukast and its metabolites. J Cardiovasc Pharmacol. 2004;43:113–120. doi: 10.1097/00005344-200401000-00017. [DOI] [PubMed] [Google Scholar]
- 21.Palmberg L, Claesson HE, Thyberg J. Effects of leukotrienes on phenotypic properties and growth of arterial smooth muscle cells in primary culture. J Cell Sci. 1989;93(Pt 3):403–408. doi: 10.1242/jcs.93.3.403. [DOI] [PubMed] [Google Scholar]
- 22.Bkaily G, Choufani S, Hassan G, El-Bizri N, Jacques D, D'Orleans-Juste P. Presence of functional endothelin-1 receptors in nuclear membranes of human aortic vascular smooth muscle cells. J Cardiovasc Pharmacol. 2000;36:S414–S417. doi: 10.1097/00005344-200036051-00121. [DOI] [PubMed] [Google Scholar]
- 23.Bkaily G, Sleiman S, Stephan J, Asselin C, Choufani S, Kamal M, Jacques D, Gobeil F, Jr, D'Orleans-Juste P. Angiotensin II AT1 receptor internalization, translocation and de novo synthesis modulate cytosolic and nuclear calcium in human vascular smooth muscle cells. Can J Physiol Pharmacol. 2003;81:274–287. doi: 10.1139/y03-007. [DOI] [PubMed] [Google Scholar]
- 24.Haller H, Lindschau C, Erdmann B, Quass P, Luft FC. Effects of intracellular angiotensin II in vascular smooth muscle cells. Circ Res. 1996;79:765–772. doi: 10.1161/01.RES.79.4.765. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen CK, Campbell JI, Ohd JF, Morgelin M, Riesbeck K, Landberg G, Sjolander A. A novel localization of the G-protein-coupled CysLT1 receptor in the nucleus of colorectal adenocarcinoma cells. Cancer Res. 2005;65:732–742. [PubMed] [Google Scholar]
- 26.Jiang Y, Borrelli LA, Kanaoka Y, Bacskai BJ, Boyce JA. CysLT2 receptors interact with CysLT1 receptors and down-modulate cysteinyl leukotriene dependent mitogenic responses of mast cells. Blood. 2007;110:3263–3270. doi: 10.1182/blood-2007-07-100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardingham GE, Bading H. Nuclear calcium: a key regulator of gene expression. Biometals. 1998;11:345–358. doi: 10.1023/A:1009257909785. [DOI] [PubMed] [Google Scholar]
- 28.Dichtl W, Stiko A, Eriksson P, Goncalves I, Calara F, Banfi C, Ares MP, Hamsten A, Nilsson J. Oxidized LDL and lysophosphatidylcholine stimulate plasminogen activator inhibitor-1 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:3025–3032. doi: 10.1161/01.ATV.19.12.3025. [DOI] [PubMed] [Google Scholar]
- 29.Tyson KL, Weissberg PL, Shanahan CM. Heterogeneity of gene expression in human atheroma unmasked using cDNA representational difference analysis. Physiol Genomics. 2002;9:121–130. doi: 10.1152/physiolgenomics.00116.2001. [DOI] [PubMed] [Google Scholar]
- 30.Beauchamp NJ, van Achterberg TA, Engelse MA, Pannekoek H, de Vries CJ. Gene expression profiling of resting and activated vascular smooth muscle cells by serial analysis of gene expression and clustering analysis. Genomics. 2003;82:288–299. doi: 10.1016/S0888-7543(03)00127-7. [DOI] [PubMed] [Google Scholar]
- 31.Schroder WA, Le TT, Major L, Street S, Gardner J, Lambley E, Markey K, MacDonald KP, Fish RJ, Thomas R, Suhrbier A. A physiological function of inflammation-associated SerpinB2 is regulation of adaptive immunity. J Immunol. 2010;184:2663–2670. doi: 10.4049/jimmunol.0902187. [DOI] [PubMed] [Google Scholar]
- 32.Feener EP, Northrup JM, Aiello LP, King GL. Angiotensin II induces plasminogen activator inhibitor-1 and -2 expression in vascular endothelial and smooth muscle cells. J Clin Invest. 1995;95:1353–1362. doi: 10.1172/JCI117786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haller H, Lindschau C, Quass P, Distler A, Luft FC. Nuclear calcium signaling is initiated by cytosolic calcium surges in vascular smooth muscle cells. Kidney Int. 1994;46:1653–1662. doi: 10.1038/ki.1994.465. [DOI] [PubMed] [Google Scholar]
- 34.Park JM, Greten FR, Wong A, Westrick RJ, Arthur JS, Otsu K, Hoffmann A, Montminy M, Karin M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis—CREB and NF-kappaB as key regulators. Immunity. 2005;23:319–329. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 194 kb)