

Summary
Vascular endothelial growth factor-a (VEGF-A) is a protein secreted by podocytes that is necessary for survival of endothelial cells, podocytes and mesangial cells. VEGF-A regulates slit-diaphragm signaling and podocyte shape via VEGFR2-nephrin-nck-actin interactions. Chronic hyperglycemia-induced excess podocyte VEGF-A and low endothelial nitric oxide drive the development and the progression diabetic nephropathy. The abnormal cross talk between VEGF-A and NO pathways is fueled by the diabetic milieu increased oxidative stress. Recent findings on these pathogenic molecular mechanisms provide new potential targets for therapy for diabetic renal disease.
Keywords: diabetes mellitus, diabetic nephropathy/complications, VEGF-A, endothelial nitric oxide synthase (eNOS), VEGF receptor 2 (VEGFR2), reactive oxygen species (ROS)
VEGF-A is a pleiotropic protein originally described as a vascular permeability and endothelial growth factor.1–3 VEGF-A is critical for endothelial cell differentiation, survival, proliferation and migration, as well as for vascular assembly, maintenance and remodeling.4 Deletion of the VEGF-A or its receptors genes is embryonic lethal.5–7 Conversely, excess VEGF-A drives pathological angiogenesis in multiple diseases, including tumors and diabetes, underscoring the importance and tight regulation of this pathway.8
Diabetes mellitus has increased to epidemic proportions in the last decade. Chronic complications of the disease are a major cause of morbidity and mortality. Diabetic nephropathy (DN) develops in approximately 30% of diabetic patients, representing the leading cause of end-stage renal disease worldwide.9 Although sustained hyperglycemia drives the clinical course and strict glycemic control decreases the incidence and progression of microvascular complications, genetic, metabolic, hemodynamic and environmental factors significantly influence DN.10–13 However, the precise determinants of who among diabetic patients will eventually develop DN are unknown, and the molecular mechanisms involved remain not fully defined. In this review we will focus on vascular endothelial growth factor A (VEGF-A) and podocytes, two central players in the pathogenesis of diabetic nephropathy.
VEGF-A expression and signaling in podocytes
At difference from other tissues that cease expressing VEGF-A at the completion of development, kidney podocytes and tubular cells express VEGF-A throughout life.14,15 Podocytes are the major source of VEGF-A in renal glomeruli. Podocytes synthesize three VEGF-A isoforms (VEGF121-165-189) by alternative mRNA splicing.16 VEGF-A isoforms differences in size, membrane and extracellular matrix binding properties (VEGF121 and VEGF165 are secreted) enable gradient formation and endothelial cell chemoattraction. VEGF165 is the most abundant isoform. Anti-angiogenic isoforms called VEGFxxxb have also been described.17 VEGF-A binds VEGF receptors 1 and 2 (VEGFR1 and VEGFR2), and co-receptors neuropilin 1 and 2 (NRP1 and NRP2). VEGF-A signals through VEGFR2, NRP1 and NRP2 amplifiy VEGFR2 signals, while VEGFR1 functions mostly as a decoy.8 All VEGF-A receptors are most abundant in endothelial cells but they are expressed by multiple cells, including podocytes and tubular cells (Figure 1).16,18,19 Hypoxia and high glucose upregulate podocyte VEGF-A protein expression.20
Figure 1. Expression of VEGFR2 in vivo.

Transmission electron microscopy from Flk-1-LacZ 6 kidney showing LacZ deposits en lieu of VEGFR2 localized to A) foot processes near slit-diaphragm, GBM and fenestrated endothelium; B) podocytes and endothelial cell. Scale bars= 200nm (A) and 1 μm (B). Sample processed as reported.26
During kidney organogenesis VEGF-A expressed in the metanephric mesenchyme acts as a chemoattractant for endothelial cells and directs their migration towards developing nephrons.21 VEGF-A is required for normal development of glomerular capillaries 22 and to acquire their fenestrated phenotype.23, 24 In addition, tightly regulated VEGF-A is necessary to establish and maintain normal podocyte phenotype, i.e. foot processes linked by slit-diaphragms, as revealed by both loss and gain of function mouse models.25–27 Moderate podocyte VEGF164 overexpression during organogenesis disrupts podocyte differentiation, and impairs slit-diaphragm development, resulting in congenital nephrotic syndrome in newborn mice.27 Massive VEGF164 overexpression in podocytes resulted in collapsing glomerulopathy in newborn mice.22 After birth podocyte VEGF-A gain of function induces reversible podocyte effacement associated with nephrin down-regulation (a minimal change-like disease) in mice.27 Notably, postnatal moderate podocyte VEGF-A gain of function did not induce glomerular endothelial damage, consistent with VEGF-A trophic effect on endothelial cells, while it caused extensive foot process effacement and proteinuria.26,27 These changes were reversible upon removal of the transgene expression, providing evidence of a cause-effect relationship between VEGF-A and foot process effacement.26,27 Surprisingly, in adult mice podocyte VEGF-A gain of function caused thickening of the glomerular basement membrane, glomerulomegaly and mesangial proliferation in addition to foot process effacement 26, a phenotype strikingly similar to early diabetic nephropathy or Class IIa DN.28 Thus, podocyte VEGF-A gain-of-function induces diverse glomerular phenotypes at specific developmental stages.
VEGF-A is a survival factor for podocytes in culture and in vivo
Podocyte VEGF-A deletion in developing mice resulted in podocyte apoptosis, leading to glomerulosclerosis 22, whereas ablation of podocyte VEGF-A in mature mice resulted in thrombotic microangiopathy, associated with podocyte and endothelial cell damage.29 We found that acute podocyte VEGF-A knockdown in mature mice induces down-regulation of fibronectin and αvβ3 integrin, resulting in decreased αvβ3 integrin activity, endothelial cell damage, glomerular basement membrane lamination, foot process effacement and acute kidney injury.30 Upon exposure to VEGF-A murine and human podocytes significantly reduce apoptosis rate by activating Akt signaling.16,31,32 VEGF-A-induced protection from apoptosis and activation of the Akt survival pathway were abrogated by anti-VEGFR2 neutralizing antibody, a VEGFR2 kinase inhibitor and bevacizumab (Avastin, Genentech), demonstrating that VEGFR2 transduces VEGF-A autocrine survival signals in podocytes.16,32 VEGF-A also activates the PI3K-Akt survival pathway via VEGFR2 reducing apoptosis in endothelial and tubular epithelial cells.18,33 VEGF-A autocrine signaling is required for endothelial cell survival in vivo, a function that is not rescued by paracrine VEGF-A.34 Murine and human podocytes express VEGFR2 in culture and in vivo, as shown by our group and others.16, 26, 32, 35 VEGF165 induces nephrin and podocyte VEGFR2 tyrosine phosphorylation in vivo, causing foot process effacement without evidence of podocyte loss.26 These VEGF-A gain of function findings are consistent with a report demonstrating that phosphorylated nephrin interacts with the p85 subunit of PI3K and triggers Akt activation, Bad phosphorylation and inhibition, thereby promoting inhibition of detachment-induced podocyte apoptosis.36
VEGF-A regulates slit-diaphragm signaling and podocyte shape via VEGFR2
VEGF-A induces dose-dependent podocin upregulation, increases podocin-CD2AP interaction, downregulates and stimulates nephrin phosphorylation in cultured podocytes.16,31,37 Podocyte VEGF-A gain of function in mice causes reversible nephrin downregulation and phosphorylation, associated with foot process effacement and proteinuria.26,27 In this model we detected VEGFR2-nephrin interaction and VEGF-induced VEGFR2 phosphorylation, demonstrating a crosstalk between VEGF-A and nephrin signaling pathways in vivo.26 VEGFR2–nephrin interaction was confirmed in podocytes and COS-7 cells transfected with the corresponding plasmids by co-immunoprecipitation and mass spectrometry.37 A direct interaction between VEGFR2 and nephrin cytoplasmic domains was detected by GST-binding assay and overlay assay. VEGFR2-nephrin interaction is modulated by phosphorylation.37 Since nephrin expression in the kidney is limited to podocytes, these findings strongly implicate VEGFR2 signaling in this cell type. In one report VEGFR2 floxed mice carrying nephrin-Cre showed no glomerular phenotype.38 The absence of phenotype might be due to partial VEGFR2 knockdown, given that VEGFR2 heterozygotes are normal,6 or Cre-mediated recombination not resulting in protein loss.39 This cannot be ascertained because quantitation of Cre expression in podocytes was not performed to assess whether VEGFR2 ablation was complete.38,39 Our laboratory and others demonstrated podocyte VEGFR2 expression in vivo by immunoelectron microscopy and co-immunoprecipitation.26,35 We found that podocyte VEGFR2-nephrin binding partners form a multi-protein complex that includes nck and actin.37 Upon VEGF-A stimulation this complex induces significant cell shape change and decreases cell size 37, in agreement with the foot process effacement phenotype reported in mice.27, 40 Taken together, these findings identify VEGFR2 as a novel component of the slit-diaphragm signaling complex, and support the concept that extracellular VEGF-A signals are transduced to the slit-diaphragm and the podocyte cytoskeleton cell autonomously through the VEGFR2-nephrin-nck-actin complex, thereby regulating cell shape and function. Podocytes generate contractile forces via non-muscle myosins interaction with α-actinin-4 and actin, which are thought to enable dynamic shape adjustments in response to glomerular capillary pressure changes.41–43 In agreement with our findings in podocytes, VEGF-A signaling induces changes in endothelial cell morphology in vitro and in vivo.44,45
The precise mechanism of VEGF-induced nephrin downregulation is not fully defined downstream from VEGFR2. VEGFR2 Y1175 auto-phosphorylation mediates PLCγ binding and leads to PKC activation.46–48 Notably, the effect of hyperglycemia on nephrin-β-arrestin2 interaction, increasing nephrin endocytosis in podocytes, is mediated by PKCα.49,50 In endothelial cells PKCα is a negative regulator of VEGF signaling and eNOS phosphorylation.51 Collectively, these data raise the possibility that VEGFR2 signaling may play an important role in the regulation of nephrin endocytosis through PKCα.
VEGF in diabetic nephropathy
Chronic hyperglycemia stimulates VEGF-A synthesis and secretion, and triggers a series of interconnected metabolic and hemodynamic effects that contribute to VEGF-A increase, and lead to DM microvascular complications (Figure 2). Circulating VEGF-A levels are elevated in adult diabetic nephropathy patients52 and in T1D children and adolescents.53 VEGFA polymorphisms have been associated with diabetic nephropathy and retinopathy.10 Since VEGF-A is considered a short-range morphogen, having mostly paracrine, autocrine and intracrine functions 54, tissue and cell-specific VEGF levels are thought to be important for DM complications. Human kidney biopsies showed high VEGF-A mRNA at early stages of DN, and lower VEGF-A expression in advanced DN, specifically in sclerotic glomeruli and mesangial nodules.55, 56 The decline in VEGF-A expression as overt DN progresses is thought to be due to podocyte dropout.57, 58 Urinary VEGF-A was elevated in type 2 diabetic patients.59 Similarly, in multiple rodent models of DN kidney VEGF-A was found increased at early and late stage of DN 57,60 Urine VEGF-A was elevated in diabetic mice too, bearing no correlation with their albuminuria.40
Figure 2. Pathways of VEGF-A increase in diabetes.
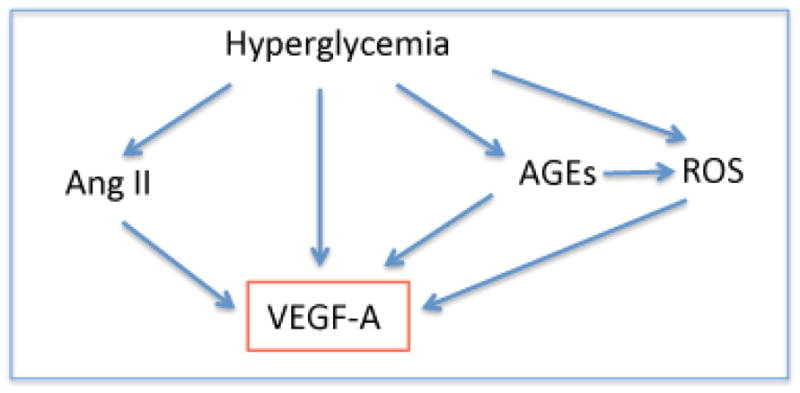
VEGF and renin-angiotensin system
The renin-angiotensin system plays a pivotal role in DN.61 In addition to Ang II -AT1-mediated glomerular hypertension and albuminuria, Ang II increases VEGF-A, TGFβ, and oxidative stress.62 Diabetes-induced increase in ACE enhances bradykinin degradation, which does not alter BP but significantly decreases NO availability.63 Transactivation of bradykinin receptor 2 and VEGFR2 induces eNOS activation.64, 65 Impairment of this mechanism likely contributes to the severity of DN in mice with bradykinin receptor deletion, and in humans carrying the ACE D allele.63, 66 By contrast ACE2, which cleaves Ang II into Ang1–7, is decreased in the diabetic kidney.67 ACE2 deletion enhances albuminuria and hypertension.68 In line with these findings, mice overexpressing ACE2 developed milder DN, had higher Ang1–7, lower Ang II, decreased VEGF-A, TGFβ, collagen IV deposition, oxidative stress and albuminuria.69
VEGF, reactive oxygen species and nitric oxide
Oxidative stress in diabetes mellitus results from excess reactive oxygen and nitrogen species (ROS/RNS) derived from the polyol pathway, glucose oxidation, advanced glycation, and mitochondrial electron transfer chain, which are not cleared by antioxidants (SOD, catalase, glutathione peroxidase).70,71 ROS/RNS increase VEGF-A by stabilizing HIF-1α72 and by activating notch signaling.73 VEGF-A activates eNOS via PI3K/Akt, and thereby stimulates nitric oxide (NO) generation.74–76 However, in the presence of superoxide (O2·−) NO· rapidly forms peroxynitrite (ONOO−), effectively increasing ROS rather than NO and stimulating guanylate cyclase.70 Accumulating evidence suggest that the crosstalk and positive feedback between VEGF-A and NO pathways plays a central role in the pathogenesis of diabetic complications. 40,77–79 Indeed, eNOS null mice express higher VEGF-A than wild type mice and develop advanced DN, whether diabetes is induced with STZ or by genetic mutations (Db/Db or Akita). 80,81,82 A modest decrease in eNOS (~30%), similar to that associated with human NOS3 polymorphisms linked to severe DN,83 is sufficient to worsen DN in mice.82 Surprisingly, oxidative stress decreased in diabetic eNOS+/− and −/− leading to exclude it as a factor in the exacerbation of DN.82 In the absence of diabetes, eNOS null mutants develop hypertension, micro and macrovascular disease, owing to endothelial damage caused by about 40% decrease in NO availability.82, 84
VEGF and advanced glycation end-products
Advanced glycation end products (AGEs), covalently bound glycosylated proteins and lipoproteins, increase in diabetic humans and animals and contribute to DN pathogenesis.85 AGEs induce increased VEGF-A in vitro and in vivo.86,87 AGEs bind to several receptors (RAGE and AGE-R1-3) located in multiple renal cell types, including podocytes.87,88 AGE-RAGE interaction activates NADPH oxidase, thereby increasing cytosolic ROS, and activates PKC and NFκB pathways leading to release of VEGF, TGFβ and CTGF.89,90 Inhibition of NADPH oxidase or PKCα in diabetic rats decreased VEGF-A, superoxide, collagen IV and fibronectin accumulation, albuminuria and glomerulosclerosis.90 Consistent with this, RAGE and aldose reductase null mice had lower VEGF and developed milder DN.91,92
VEGF downstream signals in DN
Irrespective of the mechanism driving VEGF-A in DM, its increase disregulates multiple signaling pathways and induces abnormalities that characterize diabetic glomerulopathy (Figure 3). Elevated VEGF-A associates with glomerulomegaly and excessive vessels in mice and humans with DN.40,52,93 Although high glucose and TGFβ are known to induce cell growth,94 several lines of evidence indicate that high local VEGF-A mediates the glomerulomegaly typically observed in DN. First, VEGF neutralizing antibodies block glomerular hypertrophy in diabetic rodents.95,96 Second, podocyte VEGF-A gain-of-function induces glomerulomegaly in the absence of diabetes and exacerbates the glomerular enlargement induced by diabetes.26,40 Third, VEGF-induced glomerulomegaly is reversible upon removal of the transgene induction.26 Fourth, podocyte VEGF knockdown decreases glomerular volume in non-diabetic mice, and prevents the development of glomerulomegaly in diabetic mice.97
Figure 3. Consequences of VEGF-A increase in diabetic nephropathy.
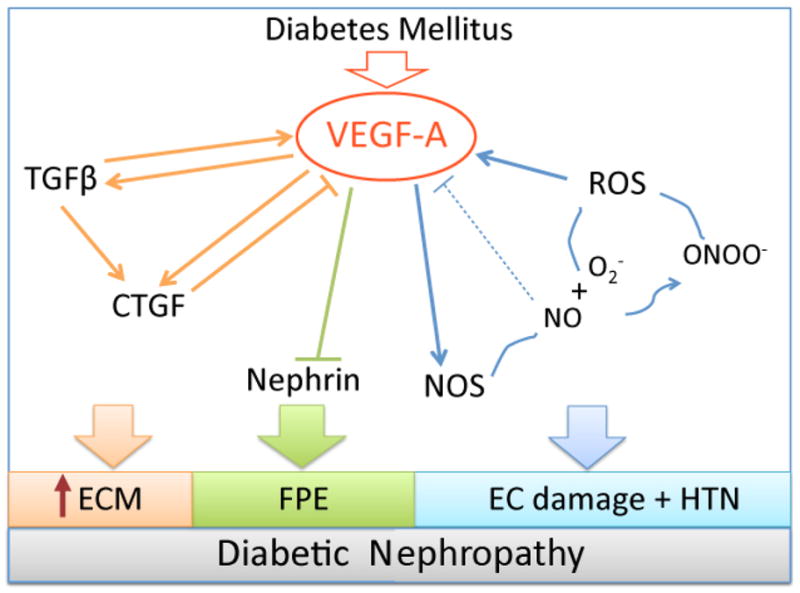
VEGF-A increases TGFβ, CTGF and established a positive feedback loop, leading to extracellular matrix (ECM) accumulation and GBM thickening (orange); VEGF-A induces nephrin downregulation and foot process effacement (FPE) (green); VEGF-A stimulates eNOS, which in the setting of high ROS leads to peroxinitrite (ONOO−) and further ROS generation, a positive feedback loop (blue), the dashed line represents the normal negative feedback regulation of VEGF-A by NO, not operative in DN. Low NO and high ROS damage endothelial cells and induce HTN.
Podocyte VEGF-A gain-of function in diabetic mice increases albuminuria ten-fold above control diabetics, by disrupting the glomerular filtration barrier via foot process effacement (Figure 4), nephrin downregulation,40 and probably by increasing endothelial fenestration and disrupting cell-cell interactions.98 VEGF-A-induced nephrin disregulation is mediated by VEGFR2 signaling 37, likely via PKCα activation,49,50 even in the absence of podocyte loss.26 However, additional angiogenic factors, such as semaphorin3a, PDGF B, and angiopoietin 2, which are also increased in DN, may contribute to VEGF-A-induced proteinuria in diabetic mice.40, 99,100 In the context of high VEGF, angiopoietin2 destabilizes vessels and induces angiogenesis78,100, while excess sema3a causes podocyte effacement and endothelial damage per se.101,102 Consistent with this possibility, Ang1 deletion worsened DN,103 while PDGFRβ deletion 99 and anti-angiogenic factors, such as angiostatin, tumstatin, endostatin and VEGF blockers (neutralizing antibody, kinase inhibitor or sVEGFR1) decreased albuminuria in rodent DN models.35,78,95,96,104–108
Figure 4.
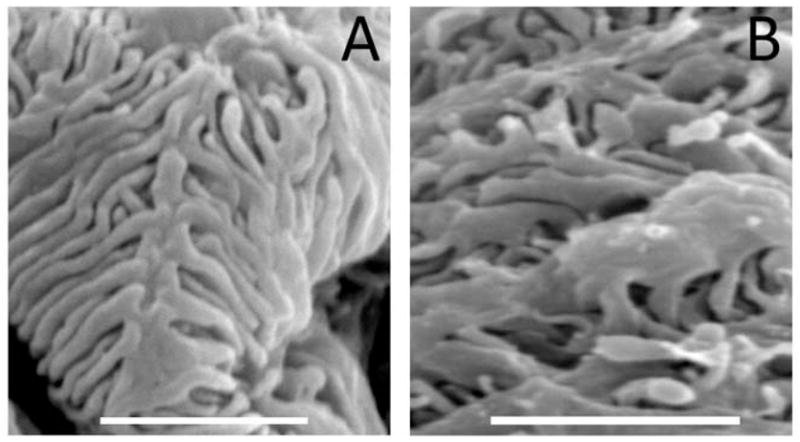
VEGF-induced thickening and distortion of podocyte foot processes in diabetic mice, observed by scanning electron microscopy. A) Control diabetic glomerulus; B) Vegf164 overexpressing glomerulus showing wider foot processes. Scale bars=2 μm.
VEGF-A induces mesangial proliferation and stimulates TGFβ activation, TGFβRII and α3 collagen IV synthesis by podocytes and mesangial cells.109–112 Accordingly, podocyte VEGF-A gain-of-function in diabetic mice resulted in Kimmelsteil-Wilson-like nodular glomerulosclerosis.40 High glucose and VEGF-A induce rapid activation of the TGFβ latent complex into active TGFβ, mediated by MMP2 and MMP9, triggering cell growth and proliferation.99 TGFβ1 signaling is thought to be responsible for mesangial cell proliferation and extracellular matrix expansion observed in human and rodent DN.28,58,113 VEGF-A and TGFβ induce connective tissue growth factor (CTGF), a secreted protein of the CCN family and downstream mediator of TGFβ, critically important in fibrosis.114 CTGF is increased in human DN, in rodent models of DN and in mesangial cells exposed to high glucose, TGFβ or VEGF-A.115 CTGF is mitogenic for mesangial and endothelial cells, induces synthesis of collagen IV and fibronectin and associates with the extracellular matrix. Interestingly, CTGF binds VEGF165 and inhibits VEGF signaling, an interaction cleaved by MMP2 and MMP9.116,117 Nitric oxide is a strong repressor of CTGF in mesangial cells, and thereby inhibits mesangial cell proliferation, their adhesion to extracellular matrix, collagen IV and fibronectin synthesis.118 In the context of DN, hyperglycemia, high VEGF, MMP activation and low NO availability establish a positive feedback loop between TGFβ and CTGF, likely to drive glomerular basement membrane thickening and mesangial matrix deposition (Figure 3).
Although ample evidence indicates that ROS, AGEs, Ang II and low NO play pathogenic roles in DN, none of these factors induces a DN phenotype per se, in the absence of diabetes, suggesting they act in concert with the diabetic milieu. By contrast, we showed that excess podocyte Vegf164 in adult mice causes glomerular abnormalities remarkably similar to human early DN and rodent DN models, i.e the triad of glomerulomegaly, mesangial expansion and thickened GBM associated with albuminuria, in the absence of diabetes.26 Reversibility of the phenotype upon removal of the transgene induction provided proof of causality,26 and recapitulated human early DN.93 Notably, excess circulating VEGF164 resulting from tubular overexpression stimulates chaotic capillary growth and mesangial proliferation within the glomerular tuft, a characteristic feature of mesangial diabetic nodules.119 Moreover, podocyte Vegf164 overexpressing mice harboring eNOS deletion developed an advanced DN-like phenotype with glomerulosclerosis, massive proteinuria and renal insufficiency in the absence of diabetes (Veron and Tufro, unpublished results), suggesting that excess podocyte VEGF-A signals and low NO may be sufficient to induce advanced DN-like phenotype. Interestingly, deletion of the insulin receptor in podocytes or mesangial Glut1 overexpression induced mild DN-like phenotype and glomerulosclerosis in the absence of diabetes, suggesting that loss of insulin signaling in podocytes or increased intracellular glucose may play a role in DN pathogenesis.120,121 The downstream mechanisms mediating regulation of podocyte function by insulin signaling remain to be established.
For many years, our understanding of DN pathogenesis was centered on mesangial cells, their proliferation and expansion of the extracellular matrix, as a major driving force for the progression of the disease. Endothelial damage was thought to result from hypertension, and podocyte loss was considered a mere consequence of the progression of the disease. The recently recognized pivotal role of VEGF-A and nitric oxide driving the development of typical DN lesions and the progression of the disease has restored some balance to the contributions of the three glomerular cell types to DN. In this paradigm, the diabetic podocyte produces excessive VEGF in the setting of low endothelial NO, stimulates growth and proliferation of mesangial and endothelial cells, leading to increased extracellular matrix accumulation, hyperfiltration and proteinuria. As glomerular damage progresses VEGF may decrease leading to mesangiolysis and endothelial cell damage, exacerbated by disregulated RAS-driven hypertension and by elevated reactive oxygen species. Based on the pleiotropic effects of VEGF-A and NO, this view of DN pathogenesis fits the vast majority of previous and recent findings in DN animal models, the natural course of the disease in humans, and the known polymorphisms associated with severe DN. Several potential targetable steps in DN emerge from this view, including the regulation of VEGF secretion or receptor binding, bradykinin, NO availability and critical downstream signals such as PKCα and CTGF.
Acknowledgments
Grant support: This work was supported by NIH-NIDDK RO1-059333 (A.T.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–09. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 2.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–12. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- 3.Senger DR. Vascular endothelial growth factor: much more than an angiogenesis factor. Mol Biol Cell. 2010;21:377–9. doi: 10.1091/mbc.E09-07-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–45. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 6.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 7.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 8.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–04. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 9.US Renal Data System: USRDS 2008 Annual Data Report. http://www.usrds.org.
- 10.Rossing K, Christensen PK, Hovind P, Parving HH. Remission of nephrotic-range albuminuria reduces risk of end-stage renal disease and improves survival in type 2 diabetic patients. Diabetologia. 2005;48:2241–47. doi: 10.1007/s00125-005-1937-6. [DOI] [PubMed] [Google Scholar]
- 11.Hovind P, Tarnow L, Rossing P, Carstensen B, Parving HH. Improved survival in patients obtaining remission of nephrotic range albuminuria in diabetic nephropathy. Kidney Int. 2004;66:1180–86. doi: 10.1111/j.1523-1755.2004.00870.x. [DOI] [PubMed] [Google Scholar]
- 12.Hovind P, Tarnow L, Rossing K, Rossing P, Eising S, Larsen N, et al. Decreasing incidence of severe diabetic microangiopathy in type 1 diabetes. Diabetes Care. 2003;26:1258–64. doi: 10.2337/diacare.26.4.1258. [DOI] [PubMed] [Google Scholar]
- 13.McKnight AJ, Maxwell AP, Patterson CC, Brady HR, Savage DA. Association of VEGF-1499C-->T polymorphism with diabetic nephropathy in type 1 diabetes mellitus. J Diab Compl. 2007;21:242–5. doi: 10.1016/j.jdiacomp.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Simon M, Grone HJ, Johren O, Kullmer J, Plate KH, Risau W, et al. Expression of vascular endothelial growth factor and its receptors in human renal ontogenesis and in adult kidney. Am J Physiol Renal Fluid Electrolyte Physiol. 1995;268:F240–F250. doi: 10.1152/ajprenal.1995.268.2.F240. [DOI] [PubMed] [Google Scholar]
- 15.Tufro A, Norwood VF, Carey RM, Gomez RA. Vascular endothelial growth factor induces nephrogenesis, and vasculogenesis. J Am Soc Nephrol. 1999;10:2125–34. doi: 10.1681/ASN.V10102125. [DOI] [PubMed] [Google Scholar]
- 16.Guan F, Villegas G, Teichman J, Mundel P, Tufro A. Autocrine VEGF-A system in podocytes regulates podocin and its interaction with CD2AP. Am J Physiol Renal Physiol. 2006;291:F422–428. doi: 10.1152/ajprenal.00448.2005. [DOI] [PubMed] [Google Scholar]
- 17.Cui T, Foster RR, Saleem M, Mathieson PW, Gillatt DA, Bates DO, et al. Differentiated human podocytes endogenously express an inhibitory isoform of vascular endothelial growth factor (VEGF165b) mRNA and protein. Am J Physiol Renal Physiol. 2004;286:F767–F773. doi: 10.1152/ajprenal.00337.2003. [DOI] [PubMed] [Google Scholar]
- 18.Villegas G, Lange-Sperandio GB, Tufro A. Autocrine and paracrine functions of vascular endothelial growth factor (VEGF) in renal tubular epithelial cells. Kidney Int. 2005;67:449–57. doi: 10.1111/j.1523-1755.2005.67101.x. [DOI] [PubMed] [Google Scholar]
- 19.Kanellis J, Fraser S, Katerelos M, Power DA. Vascular endothelial growth factor is a survival factor for renal tubular epithelial cells. Am J Physiol Renal Physiol. 2000;278:F905–F915. doi: 10.1152/ajprenal.2000.278.6.F905. [DOI] [PubMed] [Google Scholar]
- 20.Kim BS, Chen J, Weinstein T, Noiri E, Goligorsky MS. VEGF expression in hypoxia and hyperglycemia: reciprocal effect on branching angiogenesis in epithelial-endothelial co-cultures. J Am Soc Nephrol. 2002;8:2027–36. doi: 10.1097/01.asn.0000024436.00520.d8. [DOI] [PubMed] [Google Scholar]
- 21.Tufro A. VEGF spatially directs angiogenesis during metanephric development in vitro. Dev Biol. 2000;227:558–566. doi: 10.1006/dbio.2000.9845. [DOI] [PubMed] [Google Scholar]
- 22.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts WG, Palade GE. Neovasculature induced by vascular endotheli al growth factor is fenestrated. Cancer Res. 199;757:765–772. [PubMed] [Google Scholar]
- 24.Kamba T, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol. 2006;290:H560–H576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- 25.Eremina V, Cui S, Gerber H, Ferrara N, Haigh J, Nagy A, et al. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17:724–735. doi: 10.1681/ASN.2005080810. [DOI] [PubMed] [Google Scholar]
- 26.Veron D, Reidy K, Villegas G, Kopp J, Thomas D, Tufro A. Induction of podocyte VEGF-A overexpression in adult mice causes glomerular disease. Kidney Int. 2010;77:989–999. doi: 10.1038/ki.2010.64. [DOI] [PubMed] [Google Scholar]
- 27.Veron D, Reidy K, Marlier A, Villegas G, Kashgarian M, Tufro A. Induction of podocyte VEGF164 overexpression at different stages of development causes congenital nephrosis or steroid resistant nephrotic syndrome. Am J Pathol. 2010;177:2225–33. doi: 10.2353/ajpath.2010.091146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tervaert TW, Mooyaart AL, Amann K, Society RP, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 29.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–36. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veron D, Villegas G, Aggarwal P, Moeckel G, Kashgarian M, Tufro A. Acute podocyte VEGF-A knockdown disrupts αVβ3 integrin signaling in the glomerulus. J Am Soc Nephrol. 2011;22:9A. doi: 10.1371/journal.pone.0040589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foster RR, Saleem MA, Mathieson PW, Bates DO, Harper SJ. Vascular endothelial growth factor and nephrin interact and reduce apoptosis in human podocytes. Am J Physiol Renal Physiol. 2005;288:F48–57. doi: 10.1152/ajprenal.00146.2004. [DOI] [PubMed] [Google Scholar]
- 32.Müller-Deile J, Worthmann K, Saleem M, Tossidou I, Haller H, Schiffer M. The balance of autocrine VEGF-A and VEGF-C determines podocyte survival. Am J Physiol Renal Physiol. 2009;297:F1656–67. doi: 10.1152/ajprenal.00275.2009. [DOI] [PubMed] [Google Scholar]
- 33.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–43. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 34.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ku CH, White KE, Dei Cas A, Hayward A, Webster Z, Bilous R, et al. Inducible overexpression of sFlt-1 in podocytes ameliorates glomerulopathy in diabetic mice. Diabetes. 2008;57:2824–33. doi: 10.2337/db08-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huber TB, Hartleben B, Kim J, Schmidts M, Schermer B, Keil A, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol. 2003;23:4917–28. doi: 10.1128/MCB.23.14.4917-4928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertuccio C, Veron D, Aggarwal P, Holzman L, Tufro A. Vascular endothelial growth factor receptor 2 direct interaction with nephrin links VEGF-A signals to actin in kidney podocytes. J Biol Chem. 2011;286:39933–44. doi: 10.1074/jbc.M111.241620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol. 2010;21:1691–01. doi: 10.1681/ASN.2010030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turlo KA, Gallaher SD, Vora R, Laski FA, Iruela-Arispe ML. When Cre-mediates recombination does not result in protein loss. Genetics. 2010;186:959–67. doi: 10.1534/genetics.110.121608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veron D, Bertuccio CA, Marlier A, Reidy K, Garcia AM, Jimenez J, Velazquez H, Kashgarian M, Moeckel GW, Tufro A. Podocyte vascular endothelial growth factor (Vegf164) overexpression causes severe nodular glomerulosclerosis in a mouse model of type 1 diabetes. Diabetologia. 2011;54:1227–41. doi: 10.1007/s00125-010-2034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faul C, Asanuma K, Yanagida-Asanuma E, Kimand K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–37. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Saleem MA, Zavadil J, Bailly M, McGee K, Witherden IR, Pavenstadt H, et al. The molecular and functional phenotype of glomerular podocytes reveals key features of contractile smooth muscle cells. Am J Physiol Renal Physiol. 2008;295:F959–970. doi: 10.1152/ajprenal.00559.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med. 2011;365:295–306. doi: 10.1056/NEJMoa1101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsukawa M, Sakamoto H, Kawasuji M, Furuyama T, Ogawa M. Different roles of Foxo1 and Foxo3 in the control of endothelial cell morphology. Genes Cells. 2009;14:1167–1181. doi: 10.1111/j.1365-2443.2009.01343.x. [DOI] [PubMed] [Google Scholar]
- 45.Strilić B, Kucera T, Eglinger J, Hughes MR, McNagny KM, Tsukita S, et al. The molecular basis of vascular lumen formation in the developing mouse aorta. Dev Cell. 2009;17:505–515. doi: 10.1016/j.devcel.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 46.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 47.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–95. [PubMed] [Google Scholar]
- 48.Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 2001;20:2768–78. doi: 10.1093/emboj/20.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quack I, Woznowski M, Potthoff SA, Palmer R, Königshausen E, Sivritas S, et al. PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia. J Biol Chem. 2011;286:12959–70. doi: 10.1074/jbc.M110.204024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tossidou I, Teng B, Menne J, Shushakova N, Park JK, Becker JU, et al. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS One. 2010;5 (4):e10185. doi: 10.1371/journal.pone.0010185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rask-Madsen C, King GL. Differential regulation of VEGF signaling by PKC-alpha and PKC-epsilon in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:919–924. doi: 10.1161/ATVBAHA.108.162842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hovind P, Tarnow L, Oestergaard PB, Parving HH. Elevated vascular endothelial growth factor in type 1 diabetic patients with diabetic nephropathy. Kidney Int. 2000;75:S56–61. [PubMed] [Google Scholar]
- 53.Chiarelli F, Spagnoli A, Basciani F, Tumini S, Mezzetti A, Cipollone F, et al. Vascular endothelial growth factor (VEGF) in children, adolescents and young adults with Type 1 diabetes mellitus: relation to glycaemic control and microvascular complications. Diabet Med. 2000;17:650–6. doi: 10.1046/j.1464-5491.2000.00350.x. [DOI] [PubMed] [Google Scholar]
- 54.Stieger N, Worthmann K, Schiffer M. The role of metabolic and haemodynamic factors in podocyte injury in diabetes. Diabetes Metab Res Rev. 2011;27:207–15. doi: 10.1002/dmrr.1164. [DOI] [PubMed] [Google Scholar]
- 55.Shulman K, Rosen S, Tognazzi K, Manseau EJ, Brown LF. Expression of vascular permeability factor (VPF/VEGF) is altered in many glomerular diseases. J Am Soc Nephrol. 1996;7:661–66. doi: 10.1681/ASN.V75661. [DOI] [PubMed] [Google Scholar]
- 56.Hohenstein B, Hausknecht B, Boehmer K, Riess R, Brekken RA, Hugo CP. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int. 2006;69:1654–61. doi: 10.1038/sj.ki.5000294. [DOI] [PubMed] [Google Scholar]
- 57.Brosius FC, 3rd, Alpers CE, Bottinger EP, et al. Animal models of diabetic complications consortium. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–12. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brosius FC, Khoury CC, Buller CL, Chen S. Abnormalities in signaling pathways in diabetic nephropathy. Expert Rev Endocrinol Metab. 2010;5:51–64. doi: 10.1586/eem.09.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim NH, Kim KB, Kim DL, Kim SG, Choi KM, Baik SH, et al. Plasma and urinary vascular endothelial growth factor and diabetic nephropathy in Type 2 diabetes mellitus. Diabet Med. 2004;21:545–51. doi: 10.1111/j.1464-5491.2004.01200.x. [DOI] [PubMed] [Google Scholar]
- 60.Cooper ME, Vranes D, Youssef S, Stacker SA, Cox AJ, Rizkalla B, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48:2229–39. doi: 10.2337/diabetes.48.11.2229. [DOI] [PubMed] [Google Scholar]
- 61.Gurley SB, Coffman TM. The renin-angiotensin system and diabetic nephropathy. Semin Nephrol. 2007;27:144–52. doi: 10.1016/j.semnephrol.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 62.Wolf G, Ziyadeh FN. Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol. 2007;106:26–31. doi: 10.1159/000101797. [DOI] [PubMed] [Google Scholar]
- 63.Kakoki M, Sullivan KA, Backus C, Hayes JM, Oh SS, Hua K, et al. Lack of both bradykinin B1 and B2 receptors enhances nephropathy, neuropathy, and bone mineral loss in Akita diabetic mice. Proc Natl Acad Sci U S A. 2010;107:10190–5. doi: 10.1073/pnas.1005144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanchez de Miguel L, Neysari S, Jakob S, Petrimpol M, Butz N, Banfi A, et al. B2-kinin receptor plays a key role in B1-, angiotensin converting enzyme inhibitor-, and vascular endothelial growth factor-stimulated in vitro angiogenesis in the hypoxic mouse heart. Cardiovasc Res. 2008;80:106–13. doi: 10.1093/cvr/cvn170. [DOI] [PubMed] [Google Scholar]
- 65.Thuringer D, Maulon L, Frelin C. Rapid transactivation of the vascular endothelial growth factor receptor KDR/Flk-1 by the bradykinin B2 receptor contributes to endothelial nitric-oxide synthase activation in cardiac capillary endothelial cells. J Biol Chem. 2002;18(277):2028–32. doi: 10.1074/jbc.M109493200. [DOI] [PubMed] [Google Scholar]
- 66.Rigat B, Hubert C, Alhenc-Gelas F, et al. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–6. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bodin S, et al. Kallikrein protects against microalbuminuria in experimental type I diabetes. Kidney Int. 2009;76:395–403. doi: 10.1038/ki.2009.208. [DOI] [PubMed] [Google Scholar]
- 68.Tikellis C, Bialkowski K, Pete J, Sheehy K, Su Q, Johnston C, et al. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018–25. doi: 10.2337/db07-1212. [DOI] [PubMed] [Google Scholar]
- 69.Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY, Feng JB, et al. Angiotensin-converting enzyme (ACE) 2 overexpression ameliorates glomerular injury in a rat model of diabetic nephropathy: a comparison with ACE inhibition. Mol Med. 2011;17:59–69. doi: 10.2119/molmed.2010.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 71.Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. 2005;4:5. doi: 10.1186/1475-2840-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin CL, Wang FS, Hsu YC, Chen CN, Tseng MJ, Saleem MA, et al. Modulation of notch-1 signaling alleviates vascular endothelial growth factor-mediated diabetic nephropathy. Diabetes. 2010;59:1915–25. doi: 10.2337/db09-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Richard DE, Berra E, Pouyssegur J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J Biol Chem. 2000;275:26765–71. doi: 10.1074/jbc.M003325200. [DOI] [PubMed] [Google Scholar]
- 74.Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsurumi Y, Murohara T, Krasinski K, Chen D, Witzenbichler B, Kearney M, et al. Reciprocal relation between VEGF and NO in the regulation of endothelial integrity. Nat Med. 1997;3:879–886. doi: 10.1038/nm0897-879. [DOI] [PubMed] [Google Scholar]
- 76.Gao N, Ding M, Zheng JZ, Zhang Z, Leonard SS, Liu KJ, et al. Vanadate-induced expression of hypoxia-inducible factor 1 alpha and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J Biol Chem. 2002;277:31963–71. doi: 10.1074/jbc.M200082200. [DOI] [PubMed] [Google Scholar]
- 77.Nakagawa T. Uncoupling of VEGF with NO as a mechanism for diabetic nephropathy. Diabetes Res Clin Pract. 2008;82 (Suppl 1):S67–9. doi: 10.1016/j.diabres.2008.09.030. [DOI] [PubMed] [Google Scholar]
- 78.Karalliedde J, Gnudi L. Endothelial factors and diabetic nephropathy. Diabetes Care. 2011;34 (Suppl 2):S291–6. doi: 10.2337/dc11-s241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nakagawa T, Kosugi T, Haneda M, Rivard CJ, Long DA. Abnormal angiogenesis in diabetic nephropathy. Diabetes. 2009;58:1471–8. doi: 10.2337/db09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–50. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 81.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, et al. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17:2664–9. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang CH, Li F, Hiller S, Kim HS, Maeda N, Smithies O, et al. A modest decrease in endothelial NOS in mice comparable to that associated with human NOS3 variants exacerbates diabetic nephropathy. Proc Natl Acad Sci U S A. 2011;108:2070–5. doi: 10.1073/pnas.1018766108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ezzidi I, Mtiraoui N, Mohamed MB, Mahjoub T, Kacem M, Almawi WY. Association of endothelial nitric oxide synthase Glu298Asp, 4b/a, and −786T>C gene variants with diabetic nephropathy. J Diabetes Complications. 2008;22:331–8. doi: 10.1016/j.jdiacomp.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 84.Forbes MS, Thornhill BA, Park MH, Chevalier RL. Lack of endothelial nitric-oxide synthase leads to progressive focal renal injury. Am J Pathol. 2007;170:87–99. doi: 10.2353/ajpath.2007.060610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tan AL, Forbes JM, Cooper ME. AGE, RAGE, and ROS in diabetic nephropathy. Semin Nephrol. 2007;27:130–43. doi: 10.1016/j.semnephrol.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 86.Tsuchida K, Makita Z, Yamagishi S, Atsumi T, Miyoshi H, Obara S, et al. Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia. 1999;42:579–88. doi: 10.1007/s001250051198. [DOI] [PubMed] [Google Scholar]
- 87.Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:1123–37. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, et al. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11:1656–66. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- 89.Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl) 2005;83:876–86. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 90.Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FY, Sourris KC, et al. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes. 2008;57:460–9. doi: 10.2337/db07-1119. [DOI] [PubMed] [Google Scholar]
- 91.Myint KM, Yamamoto Y, Doi T, Kato I, Harashima A, Yonekura H, et al. RAGE control of diabetic nephropathy in a mouse model: effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes. 2006;55:2510–22. doi: 10.2337/db06-0221. [DOI] [PubMed] [Google Scholar]
- 92.Liu H, Luo Y, Zhang T, Zhang Y, Wu Q, Yuan L, et al. Genetic deficiency of aldose reductase counteracts the development of diabetic nephropathy in C57BL/6 mice. Diabetologia. 2011;54:1242–51. doi: 10.1007/s00125-011-2045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fioretto P, Mauer M. Histopathology of diabetic nephropathy. Semin Nephrol. 2007;27:195–207. doi: 10.1016/j.semnephrol.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu L, Derynck R. Essential role of TGF-beta signaling in glucose-induced cell hypertrophy. Dev Cell. 2009;17:35–48. doi: 10.1016/j.devcel.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, Lameire NH. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol. 2001;12:993–1000. doi: 10.1681/ASN.V125993. [DOI] [PubMed] [Google Scholar]
- 96.Flyvbjerg A, Dagnaes-Hansen F, De Vriese AS, Schrijvers BF, Tilton RG, Rasch R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes. 2002;51:3090–4. doi: 10.2337/diabetes.51.10.3090. [DOI] [PubMed] [Google Scholar]
- 97.Veron D, Garcia AM, Reidy K, Tufro A. VEGF-A dysregulation alters the glomerular phenotype of streptozotocin diabetic mice. J Am Soc Nephrol. 19:2008 A. [Google Scholar]
- 98.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta- arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–34. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 99.Suzuki H, Usui I, Kato I, Oya T, Kanatani Y, Yamazaki Y, et al. Deletion of platelet-derived growth factor receptor-3 improves diabetic nephropathy in Ca(2+)/calmodulin-dependent protein kinase II3 (Thr286Asp) transgenic mice. Diabetologia. 2011;54:2953–62. doi: 10.1007/s00125-011-2270-x. [DOI] [PubMed] [Google Scholar]
- 100.Hanahan D. Signaling vascular morphogenesis and maintenance. Science. 1997;277:48–50. doi: 10.1126/science.277.5322.48. [DOI] [PubMed] [Google Scholar]
- 101.Tapia R, Guan F, Gershin I, Teichman J, Villegas G, Tufro A. Semaphorin3a disrupts podocyte foot processes causing acute proteinuria. Kidney Int. 2008;73:733–40. doi: 10.1038/sj.ki.5002726. [DOI] [PubMed] [Google Scholar]
- 102.Reidy KJ, Villegas G, Veron D, Jimenez J, Thomas D, Tufro A. Semaphorin3a regulates endothelial cell number and podocyte differentiation during glomerular development. Development. 2009;136:3979–89. doi: 10.1242/dev.037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jeansson M, Gawlik A, Anderson G, Li C, Kerjaschki D, Henkelman M, et al. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. 2011;121:2278–89. doi: 10.1172/JCI46322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sung SH, Ziyadeh FN, Wang A, Pyagay PE, Kanwar YS, Chen S. Blockade of vascular endothelial growth factor signaling ameliorates diabetic albuminuria in mice. J Am Soc Nephrol. 2006;17:3093–104. doi: 10.1681/ASN.2006010064. [DOI] [PubMed] [Google Scholar]
- 105.Zhang SX, Wang JJ, Lu K, Mott R, Longeras R, Ma JX. Therapeutic potential of angiostatin in diabetic nephropathy. J Am Soc Nephrol. 2006;17:475–86. doi: 10.1681/ASN.2005020217. [DOI] [PubMed] [Google Scholar]
- 106.Yamamoto Y, Maeshima Y, Kitayama H, Kitamura S, Takazawa Y, Sugiyama H, et al. Tumstatin peptide, an inhibitor of angiogenesis, prevents glomerular hypertrophy in the early stage of diabetic nephropathy. Diabetes. 2004;53:1831–40. doi: 10.2337/diabetes.53.7.1831. [DOI] [PubMed] [Google Scholar]
- 107.Ichinose K, Maeshima Y, Yamamoto Y, Kitayama H, Takazawa Y, Hirokoshi K, et al. Antiangiogenic endostatin peptide ameliorates renal alterations in the early stage of a type 1 diabetic nephropathy model. Diabetes. 2005;54:2891–903. doi: 10.2337/diabetes.54.10.2891. [DOI] [PubMed] [Google Scholar]
- 108.Rask-Madsen C, King GL. Diabetes: Podocytes lose their footing. Nature. 2010;468:42–4. doi: 10.1038/468042a. [DOI] [PubMed] [Google Scholar]
- 109.Thomas S, Vanuystel J, Gruden G, Rodríguez V, Burt D, Gnudi L, et al. Vascular endothelial growth factor receptors in human mesangium in vitro and in glomerular disease. J Am Soc Nephrol. 2000;11:1236–43. doi: 10.1681/ASN.V1171236. [DOI] [PubMed] [Google Scholar]
- 110.Iglesias-de la Cruz MC, Ziyadeh FN, Isono M, Kouahou M, Han DC, Kalluri R. Effects of high glucose and TGF-beta1 on the expression of collagen IV and vascular endothelial growth factor in mouse podocytes. Kidney Int. 2002;62:901–13. doi: 10.1046/j.1523-1755.2002.00528.x. [DOI] [PubMed] [Google Scholar]
- 111.Wang Y, Fei D, Vanderlaan M, Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7:335–45. doi: 10.1007/s10456-004-8272-2. [DOI] [PubMed] [Google Scholar]
- 112.Tahara A, Tsukada J, Tomura Y, Yatsu T, Shibasaki M. Vasopressin regulates rat mesangial cell growth by inducing autocrine secretion of vascular endothelial growth factor. J Physiol Sci. 2011;61:115–22. doi: 10.1007/s12576-010-0128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rooney B, O’Donovan H, Gaffney A, Browne M, Faherty N, Curran SP, et al. CTGF/CCN2 activates canonical Wnt signalling in mesangial cells through LRP6: implications for the pathogenesis of diabetic nephropathy. FEBS Lett. 2011;585:531–8. doi: 10.1016/j.febslet.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 115.Suzuma K, Naruse K, Suzuma I, Takahara N, Ueki K, Aiello LP, King GL. Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, Flt1, and phosphatidylinositol 3-kinase-akt-dependent pathways in retinal vascular cells. J Biol Chem. 2000;275:40725–31. doi: 10.1074/jbc.M006509200. [DOI] [PubMed] [Google Scholar]
- 116.Inoki I, Shiomi T, Hashimoto G, Enomoto H, Nakamura H, Makino K, et al. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2002;16:219–21. doi: 10.1096/fj.01-0332fje. [DOI] [PubMed] [Google Scholar]
- 117.Hashimoto G, Inoki I, Fujii Y, Aoki T, Ikeda E, Okada Y. Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J Biol Chem. 2002;277:36288–95. doi: 10.1074/jbc.M201674200. [DOI] [PubMed] [Google Scholar]
- 118.Keil A, Blom IE, Goldschmeding R, Rupprecht HD. Nitric oxide down-regulates connective tissue growth factor in rat mesangial cells. Kidney Int. 2002;62:401–11. doi: 10.1046/j.1523-1755.2002.00462.x. [DOI] [PubMed] [Google Scholar]
- 119.Hakroush S, Moeller MJ, Theilig F, Kaissling B, Sijmonsma TP, Jugold M, et al. Effects of increased renal tubular vascular endothelial growth factor (VEGF) on fibrosis, cyst formation, and glomerular disease. Am J Pathol. 2009;175:1883–95. doi: 10.2353/ajpath.2009.080792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329–40. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang Y, Heilig K, Saunders T, Minto A, Deb DK, Chang A, et al. Transgenic overexpression of GLUT1 in mouse glomeruli produces renal disease resembling diabetic glomerulosclerosis. Am J Physiol Renal Physiol. 2010;299:F99–F111. doi: 10.1152/ajprenal.00466.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]