

Abstract
The aim of this study was to assess whether hypoxia inhibits endothelial nitric oxide synthase (eNOS) activity and nitric oxide (NO) production, and whether iptakalim may rescue human pulmonary artery endothelial cells (HPAECs) from hypoxia-induced NO system dysfunction. HPAECs were cultured under hypoxic conditions in the absence or presence of 0.1, 10 and 1,000 μM iptakalim or the combination of 10 μM iptakalim and 1, 10 and 100 μM glibenclamide for 24 h, and the eNOS activity and NO levels were measured in the conditioned medium from the HPAEC cultures. The eNOS activity and NO levels were reduced significantly in the conditioned medium from HPAEC cultures under hypoxic conditions. Pre-treatment with 10 μM iptakalim normalized the reduction of the eNOS activity and NO levels caused by hypoxia in the conditioned medium from HPAEC cultures. Iptakalim raised the eNOS activity and NO levels under hypoxic conditions, but was blocked by the KATP channel blocker, glibenclamide. Our results indicate that hypoxia impairs NO system function, whereas the ATP-sensitive K+ channel opener, iptakalim, may rescue HPAECs from hypoxia-induced NO system dysfunction.
Keywords: iptakalim, human pulmonary artery endothelial cells, endothelial nitric oxide synthase, nitric oxide
Introduction
Pulmonary hypertension (PH) is a progressive, often fatal disease that is caused by increased pulmonary vascular resistance (PVR), which is involved in complex processes, including abnormal vascular wall remodeling, vasoconstriction and thrombosis (1,2). According to the 2008 WHO classification, PH may be categorized as pulmonary arterial hypertension (PAH), PH due to left heart disease, PH due to chronic lung disease and/or hypoxia, chronic thromboembolic pulmonary hypertention and miscellaneous forms (3). Among these, hypoxic pulmonary hypertention (HPH) has higher morbidity and mortality. HPH is caused by excessive vasoconstriction and remodeling regulated by endothelial dysfunction. The endothelial cells modulate the activity of smooth muscle cells by producing vasodilators, such as prostacyclin and nitric oxide (NO), and vasoconstrictors, such as thromboxane A2 and endothelin-1 (ET-1). Endothelial dysfunction is a condition in which the physiological balance between vasodilator stimuli and vasoconstrictor substances is shifted towards the latter (4,5); this state has been clearly shown in PH (6). NO has gained attention as a significant mediator of PH by virtue of its ability to produce factors that regulate blood flow and vascular tone. NO is a potent endothelium-derived vasorelaxant substance and an inhibitor of smooth muscle cell growth. The main function of NO is to relax pulmonary vascular smooth muscle cells and inhibit the pulmonary artery smooth muscle cells (PASMCs) proliferation (7). The decrease of NO production and release under hypoxic conditions may promote the development of HPH and pulmonary vascular remodeling. Therefore, to protect endothelial function and promote the production of NO, the therapeutic strategy for HPH is crucial.
The release of NO from pulmonary artery endothelial cells mainly depends on the intercellular concentration of Ca2+, which increases by hyperpolarization. Since endothelial cells do not express voltage-dependent Ca2+ channels, Ca2+ influxes following receptor activation may be facilitated by cell hyperpolarizations mediated by the activation of K+ channels (6). An increasing number of studies have shown that the endothelial cell hyperpolarization is mainly controlled by specific ATP-sensitive K+ (KATP) channels. Consequently, KATP channels may play a key role in generating the electrical activity of endothelial cells and have profound effects in regulating the endothelial function (8). However, it is unclear whether the activation of KATP channels promotes NO release by increasing the intercellular concentration of Ca2+.
Iptakalim, a lipophilic para-amino compound with a low molecular weight, has been demonstrated to be a new selective KATP channel opener via pharmacological, electrophysiological and biochemical studies, and a receptor binding test (9,10). Our previous study revealed that iptakalim can alleviate pulmonary artery remodeling and has the potential to treat pulmonary arterial disorders in PH (11). Moreover, an animal study showed that the activation of KATP channels by iptakalim can enhance NO release in bovine aortic endothelial cells (BAECs) under normoxic conditions (12). However, it is unclear whether iptakalim also protects human pulmonary artery endothelial cells (HPAECs) from hypoxia.
In the present study, to assess whether hypoxia inhibits endothelial nitric oxide synthase (eNOS) activity and NO production, and whether iptakalim can rescue HPAECs from hypoxia-induced NO system dysfunction, HPAECs were cultured under hypoxic conditions in the absence or presence of 0.1, 10 and 1,000 μM iptakalim or the combination of 10 μM iptakalim and 1, 10 and 100 μM glibenclamide for 24 h; the eNOS activity and NO levels were measured in the conditioned medium from the HPAEC cultures.
Materials and methods
Drugs and chemicals
Iptakalim, with a purity of 99.36%, was synthesized and provided by the Institute of Pharmacology and Toxicology, Academy of Military Medical Sciences, China. Glibenclamide was purchased from Sigma-Aldrich Co. (St. Louis, MO, USA).
HPAEC cultures
HPAECs were purchased from ScienCell (San Diego, CA, USA). The cells were routinely maintained in cell culture medium (ScienCell) at 37°C in a humidified atmosphere containing 5% CO2. The third- to sixth-passage cultures were then seeded onto glass-bottom culture dishes (Corning Inc., NY, USA) and allowed to reach subconfluence in 2–3 days.
Hypoxic experiments
For the hypoxic experiments, cell cultures were placed in a modular incubator chamber (Billups-Rothenberg, Del Mar, CA, USA), where the hypoxic gas mixture (95% N2, 5% CO2) was pre-analyzed and infused into airtight incubators with in-flow and out-flow valves at a flow rate of 3 l/min for 15 min to attain a 2% O2 level. The airtight chamber containing the cell cultures was incubated for periods of up to 24 h at 37°C. For the normoxic cultures, the third- to sixth-passage HPAECs were cultured for 24 h in cell culture medium at 37°C in a humidified atmosphere containing 21% O2 and the medium was collected for assay of NO and eNOS. The third- to sixth-passage HPAECs were cultured under hypoxic conditions in the absence or presence of 0.1, 10 and 1,000 μM iptakalim or the combination of 10 μM iptakalim and 1, 10 and 100 μM glibenclamide for 24 h; the eNOS activity and NO levels were measured in the conditioned medium from the HPAEC cultures.
Measurement of NO and eNOS
HPAEC production of NO was determined indirectly in HPAEC supernatants. Due to its instability in physiological solutions, the majority of the NO is rapidly converted to nitrite (NO2−) and further to nitrate (NO3−). Therefore, the levels of NO2−/NO3− in the culture medium were analyzed by a commercially available NO detection kit (Beyotime Institute of Biotechnology, China) according to the manufacturer's instructions. Briefly, nitrate was converted to nitrite with aspergillus nitrite reductase, and the total nitrite was measured with the Griess reagent. The absorbance was determined at 540 nm with a spectrophotometer (13). The eNOS activity was measured using a commercial kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the instructions provided by the manufacturer.
Statistical analysis
Each test was performed and was then repeated six times. Data were expressed as the means ± SD. Comparisons of the measurement data between multiple groups were performed using the one-way ANOVA test. The statistical process was performed with SPSS 12.0 software. Probability values were considered to indicate a statistically significant difference at P<0.05.
Results
Effect of hypoxia on eNOS activity and NO production in HPAECs
To determine whether hypoxia affected eNOS activity and NO production in HPAECs, the cells were cultured for 24 h under hypoxic or normoxic conditions, then the medium was collected to measure NO levels and eNOS activity. The results showed that eNOS activity and NO levels were reduced significantly in the conditioned medium from HPAEC cultures under hypoxic conditions compared to the cultures under normoxic conditions (Fig. 1).
Figure 1.
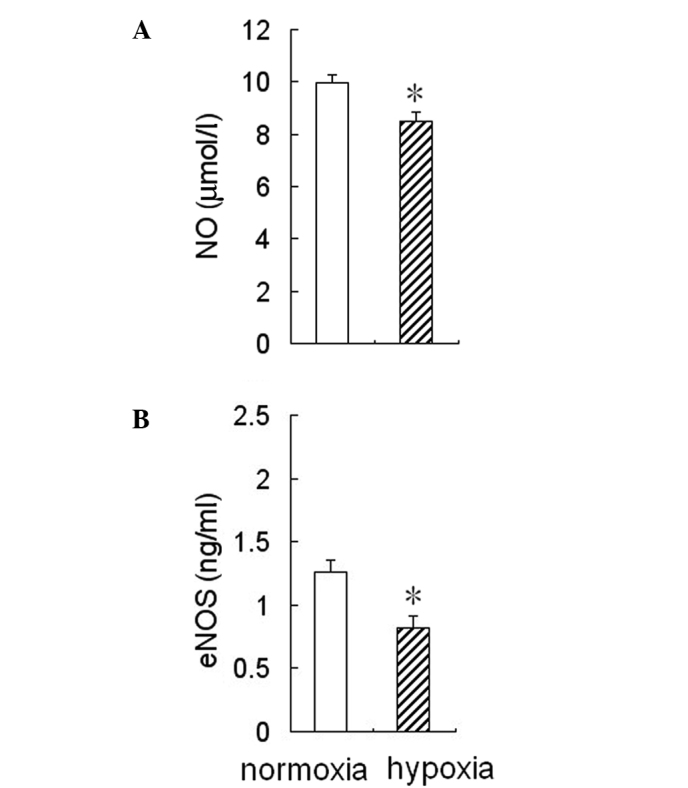
Effect of hypoxia on NO production and eNOS activity in HPAECs. The cultured HPAECs were treated with 2% O2 for 24 h. Hypoxia decreased (A) NO production and (B) eNOS activity in HPAECs. Data are presented as the means ± SD (n=6). *P<0.05 vs. normoxia. NO, nitric oxide; eNOS, endothelial nitric oxide synthase; HPAECs, human pulmonary artery endothelial cells.
Effect of iptakalim on eNOS activity and NO production in HPAECs under hypoxia
To determine whether the treatment with iptakalim antagonized a hypoxia-induced reduction of eNOS activity and NO production in HPAECs, the cells were pre-treated with 0.1 or 10 or 1,000 μM iptakalim for 1 h prior to hypoxia and cultured under hypoxia conditions for 24 h, and eNOS activity and NO levels were measured in the conditioned medium from the HPAEC cultures. The results showed that eNOS activity and NO levels were increased significantly in the conditioned medium from HPAEC cultures pre-treated with 10 or 1,000 μM iptakalim compared to those from hypoxic cultures alone and were the same or even higher than the levels of the normoxic cultures (Fig. 2). However, eNOS activity and NO levels were not raised significantly in the the conditioned medium from HPAEC cultures pre-treated with 0.1 μM iptakalim.
Figure 2.
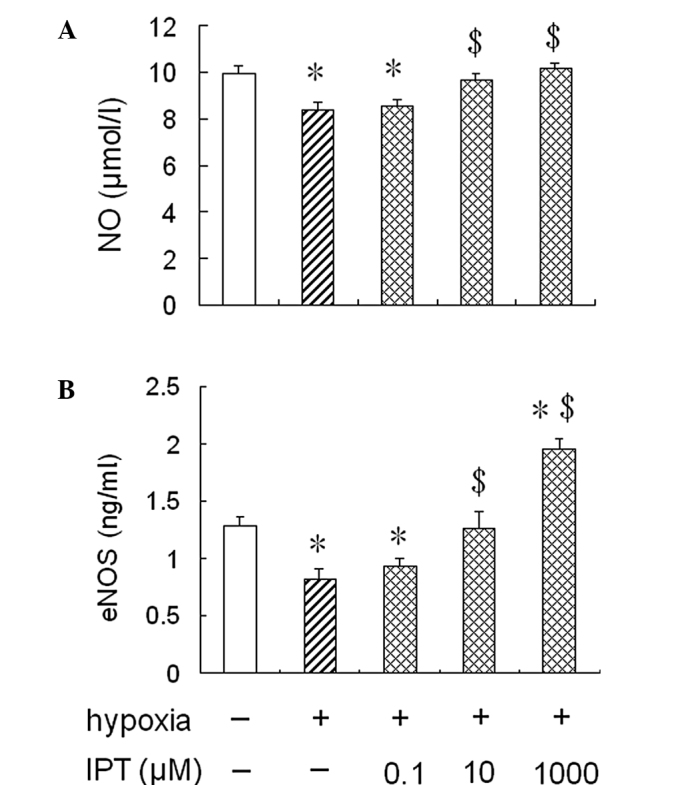
Effect of iptakalim on NO production and eNOS activity in HPAECs under hypoxia. HPAECs were pre-treated with 0.1, 10 or 1,000 μM iptakalim for 1 h prior to hypoxia and were cultured under hypoxic conditions for 24 h. (A) NO levels and (B) eNOS activity were measured in the conditioned medium from HPAEC cultures as described in Materials and methods. Each value is the mean ± SD (n=6). *P<0.05 vs. normoxia; $P<0.05 vs. hypoxia. NO, nitric oxide; eNOS, endothelial nitric oxide synthase; IPT, iptakalim; HPAECs, human pulmonary artery endothelial cells.
Effect of glibenclamide on eNOS activity and NO production in HPAECs pre-treated with iptakalim under hypoxia
To further assess whether iptakalim increases eNOS activity and NO levels through the activation of the KATP channel, the cells were pre-treated with 1.0 or 10 or 100 μM glibenclamide, a KATP channel blocker, for 1 h prior to the addition of 10 μM iptakalim, and were cultured under hypoxia conditions for 24 h; eNOS activity and NO levels were measured in the conditioned medium from the HPAEC cultures. The treatment of glibenclamide alone did not alter eNOS activity and NO levels in the conditioned medium from the HPAEC cultures under normoxic or hypoxic conditions (data not shown). However, the increased eNOS activity and NO levels induced by the pre-treatment with 10 μM iptakalim in the conditioned medium from the HPAEC cultures were blocked completely by the pre-treatment of 10 or 100 μM glibenclamide. However, eNOS activity and NO levels were not blocked completely in the conditioned medium from the HPAEC cultures pre-treated with 1.0 μM glibenclamide (Fig. 3).
Figure 3.
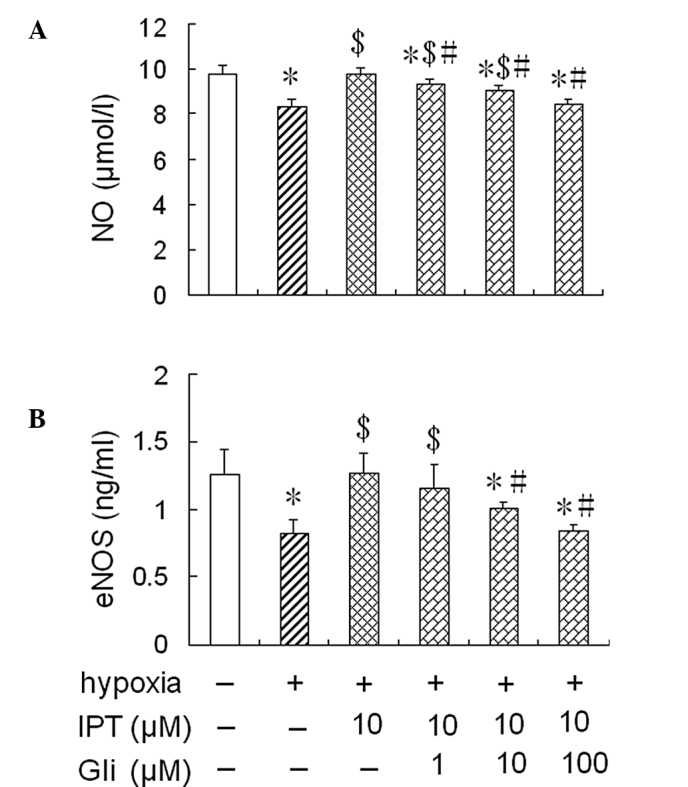
Effect of glibenclamide on NO production and eNOS activity in HPAECs pre-treated with iptakalim under hypoxic conditions. HPAECs were pre-treated with 1.0, 10 or 100 μM glibenclamide, a KATP channel blocker, for 1 h prior to the addition of 10 μM iptakalim and were cultured under hypoxic conditions for 24 h. (A) NO levels and (B) eNOS activity were measured in the conditioned medium from HPAEC cultures as described in Materials and methods. Each value is the mean ± SD (n=6). *P<0.05 vs. normoxia; $P<0.05 vs. hypoxia; #P<0.05 vs. iptkalim 10 μM + hypoxia. NO, nitric oxide; eNOS, endothelial nitric oxide synthase; IPT, iptakalim; Gli, glibenclamide; HPAECs, human pulmonary artery endothelial cells.
Discussion
In the present study, we found that the eNOS activity and NO levels were reduced significantly in the conditioned medium from the HPAEC cultures under hypoxic conditions. Previous studies have shown that the release of NO is decreased in bovine aortic and pulmonary endothelial cells and human umbilical vein endothelial cells (HUVECs) by hypoxia (13,14). It has also been shown that eNOS activities decrease in bovine and human endothelial cells exposed to chronic hypoxia (15–18). However, hypoxia has been found to increase the formation of NO and its products in cultured coronary endothelial cells (19–22). The differences in the NO release under hypoxic conditions may be due to the endothelial cell type and duration of hypoxic exposure. Our results indicate that hypoxia may impair NO system function in HPAECs.
NO is an endogenous vasoactive compound that contributes to pulmonary vascular homeostasis and is produced by three NOS isoforms: Neuronal NOS (nNOS), inducible NOS (iNOS) and eNOS. Though all these NOS isoforms are present in the lung, it was thought that eNOS-derived NO plays a significant role in modulating pulmonary vascular tone and attenuating PH (23). The vasorelaxation of smooth muscle cells may be achieved by the release of NO from endothelial cells in response to various stimuli (24), and the ability of the endothelium to produce NO is essential for the maintenance of vascular homeostasis. Reduced endothelium-derived NO production in pulmonary arterial vessels has been implicated in the pathophysiology of PH. It has been confirmed that NO synthase expression is reduced in pulmonary endothelial cells from patients suffering from PAH (25).
It has been shown that the synthesis of NO is Ca2+-dependent (constitutive form). The Ca2+ influx in endothelial cells is controlled by the membrane potential (26). KATP channels are present in endothelial cells of the vascular system (6,27) and are responsible for maintaining the resting potential of endothelial cells and modulating the release of vasoactive compounds. Thus, KATP channels may play a key role in generating the electrical activity of endothelial cells and have profound effects on endothelial function. In fact, pinacidil, a KATP channel opener, has been shown to cause an increase of Ca2+ influx in rat aorta and brain microvascular endothelial cells (6). Iptakalim, a new compound of the potassium channel opener class, is a promising drug undergoing Phase II clinical trials to treat pulmonary hypertension (28). By opening the KATP channels in vascular smooth muscle cells, iptakalim induces membrane hyperpolarization, relaxing the vessels and reducing blood pressure.
We previously found that iptakalim antagonized the vascular contraction evoked by ET-1 in isolated rat aorta rings (29,30). It was also found that iptakalim increased the intercellular concentration of Ca2+ and promoted the NO production in BAECs under normoxic conditions. Since the decreased eNOS/NO activities have been implicated in the vascular remodeling and endothelial dysfunction observed in the hypertensive models, opening the endothelial KATP channels may have protective effects on endothelial functions under hypoxic conditions and have certain therapeutic functions in HPH. Indeed, in the present study, we found that iptakalim increased the NO production and eNOS activity in HPAECs under hypoxia conditions. Glibenclamide, a KATP channel blocker, blocked the increased NO levels and eNOS activity caused by iptakalim under hypoxic conditions. These results demonstrate that the effect of iptakalim on NO production and eNOS activity in HPAECs under hypoxic conditions occurs through the activation of the KATP channel.
In conclusion, our results indicate that hypoxia impairs the NO system function, whereas the ATP-sensitive K channel opener, iptakalim, rescues HPAECs from hypoxia-induced NO system dysfunction. Combined with previous findings where iptakalim not only reduced the blood pressure indefinitely, but also antagonized the proliferation of human PASMCs induced by ET-1 through the activation of KATP channels. Our results show that iptakalim may be effective in the treatment of PAH by reversing human PASMCs remodeling and protecting HPAEC functions. Perhaps it should be considered as a promising drug for the treatment of PH.
Acknowledgments
The authors wish to thank Dr H. Wang (Institute of Pharmacology and Toxicology, Academy of Military Medical Sciences, Beijing, China) for the excellent technical assistance. This study was supported by the National Natural Science Foundation of China (No. 30971319), the ‘Six Talent Peak’ project of Jiangsu Province (No. 08-B), and the grant from Open Project Program of the key discipline of Public Health Department of Jiangsu Province (No. XK13_200902).
References
- 1.Zhu Y, Zhang S, Xie W, Li Q, Zhou Y, Wang H. Iptakalim inhibited endothelin-1-induced proliferation of human pulmonary arterial smooth muscle cells through the activation of KATP channel. Vascul Pharmacol. 2008;48:92–99. doi: 10.1016/j.vph.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Klok FA, Huisman MV. Epidemiology and management of chronic thromboembolic pulmonary hypertension. Neth J Med. 2010;68:347–351. [PubMed] [Google Scholar]
- 3.Hoeper MM. Definition, classification, and epidemiology of pulmonary arterial hypertension. Semin Respir Crit Care Med. 2009;30:369–375. doi: 10.1055/s-0029-1233306. [DOI] [PubMed] [Google Scholar]
- 4.Alan B, Nalbantgil S. Genetic, cellular and molecular mechanisms of pulmonary arterial hypertension. Anadolu Kardiyol Derg. 2010;10(Suppl 1):9–13. doi: 10.5152/akd.2010.114. [DOI] [PubMed] [Google Scholar]
- 5.Bauersachs J, Widder JD. Endothelial dysfunction in heart failure. Pharmacol Rep. 2008;60:119–126. [PubMed] [Google Scholar]
- 6.Janigro D, West GA, Gordon EL, Winn HR. ATP-sensitive K+ channels in rat aorta and brain microvascular endothelial cells. Am J Physiol. 1993;265:C812–C821. doi: 10.1152/ajpcell.1993.265.3.C812. [DOI] [PubMed] [Google Scholar]
- 7.Shirai M, Pearson JT, Shimouchi A, et al. Changes in functional and histological distributions of nitric oxide synthase caused by chronic hypoxia in rat small pulmonary arteries. Br J Pharmacol. 2003;139:899–910. doi: 10.1038/sj.bjp.0705312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatterjee S, Al-Mehdi AB, Levitan I, Stevens T, Fisher AB. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am J Physiol Cell Physiol. 2003;285:C959–C967. doi: 10.1152/ajpcell.00511.2002. [DOI] [PubMed] [Google Scholar]
- 9.Hu LF, Wang S, Shi XR, et al. ATP-sensitive potassium channel opener iptakalim protected against the cytotoxicity of MPP+ on SH-SY5Y cells by decreasing extracellular glutamate level. J Neurochem. 2005;94:1570–1579. doi: 10.1111/j.1471-4159.2005.03306.x. [DOI] [PubMed] [Google Scholar]
- 10.Misaki N, Mao X, Lin YF, et al. Iptakalim, a vascular ATP-sensitive potassium (KATP) channel opener, closes rat pancreatic beta-cell KATP channels and increases insulin release. J Pharmacol Exp Ther. 2007;322:871–878. doi: 10.1124/jpet.107.121129. [DOI] [PubMed] [Google Scholar]
- 11.Xie W, Wang H, Wang H, Hu G. Effects of iptakalim hydrochloride, a novel KATP channel opener, on pulmonary vascular remodeling in hypoxic rats. Life Sci. 2004;75:2065–2076. doi: 10.1016/j.lfs.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Long C, Duan Z, Shi C, Jia G, Zhang Y. A new ATP-sensitive potassium channel opener protects endothelial function in cultured aortic endothelial cells. Cardiovasc Res. 2007;73:497–503. doi: 10.1016/j.cardiores.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Whorton AR, Simonds DB, Piantadosi CA. Regulation of nitric oxide synthesis by oxygen in vascular endothelial cells. Am J Physiol. 1997;272:1161–1166. doi: 10.1152/ajplung.1997.272.6.L1161. [DOI] [PubMed] [Google Scholar]
- 14.Liao JK, Zulueta JJ, Yu FS, Peng HB, Cote CG, Hassoun PM. Regulation of bovine endothelial constitutive nitric oxide synthase by oxygen. J Clin Invest. 1995;96:2661–2666. doi: 10.1172/JCI118332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnet UA, McMillan A, Dinerman JL, Ballermann B, Lowenstein CJ. Regulation of endothelial nitric-oxide synthase during hypoxia. J Biol Chem. 1996;271:15069–15073. doi: 10.1074/jbc.271.25.15069. [DOI] [PubMed] [Google Scholar]
- 16.Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 17.McQuillan LP, Leung GK, Marsden PA, Kostyk SK, Kourembanas S. Hypoxia inhibits expression of eNOS via transcriptional and posttranscriptional mechanisms. Am J Physiol. 1994;267:1921–1927. doi: 10.1152/ajpheart.1994.267.5.H1921. [DOI] [PubMed] [Google Scholar]
- 18.Phelan MW, Faller DV. Hypoxia decreases constitutive nitric oxide synthase transcript and protein in cultured endothelial cells. J Cell Physiol. 1996;167:469–476. doi: 10.1002/(SICI)1097-4652(199606)167:3<469::AID-JCP11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 19.Chen JX, Meyrick B. Hypoxia increases Hsp90 binding to eNOS via PI3K-Akt in porcine coronary artery endothelium. Lab Invest. 2004;84:182–190. doi: 10.1038/labinvest.3700027. [DOI] [PubMed] [Google Scholar]
- 20.Justice JM, Tanner MA, Myers PR. Endothelial cell regulation of nitric oxide production during hypoxia in coronary microvessels and epicardial arteries. J Cell Physiol. 2000;182:359–365. doi: 10.1002/(SICI)1097-4652(200003)182:3<359::AID-JCP6>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 21.Sohn HY, Krotz F, Gloe T, et al. Differential regulation of xanthine and NAD(P)H oxidase by hypoxia in human umbilical vein endothelial cells. Role of nitric oxide and adenosine Cardiovasc Res. 2003;58:638–646. doi: 10.1016/s0008-6363(03)00262-1. [DOI] [PubMed] [Google Scholar]
- 22.Xu XP, Pollock JS, Tanner MA, Myers PR. Hypoxia activates nitric oxide synthase and stimulates nitric oxide production in porcine coronary resistance arteriolar endothelial cells. Cardiovasc Res. 1995;30:841–847. [PubMed] [Google Scholar]
- 23.Ostergaard L, Stankevicius E, Andersen MR, et al. Diminished NO release in chronic hypoxic human endothelial cells. Am J Physiol Heart Circ Physiol. 2007;293:2894–2903. doi: 10.1152/ajpheart.01230.2006. [DOI] [PubMed] [Google Scholar]
- 24.Taddei S, Virdis A, Ghiadoni L, Sudano I, Salvetti A. Effects of antihypertensive drugs on endothelial dysfunction: clinical implications. Drugs. 2002;62:265–284. doi: 10.2165/00003495-200262020-00003. [DOI] [PubMed] [Google Scholar]
- 25.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–221. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 26.Kuo L, Chancellor JD. Adenosine potentiates flow-induced dilation of coronary arterioles by activating KATP channels in endothelium. Am J Physiol. 1995;69:541–549. doi: 10.1152/ajpheart.1995.269.2.H541. [DOI] [PubMed] [Google Scholar]
- 27.Mederos y Schnitzler M, Derst C, Daut J, Preisig-Müller R. ATP-sensitive potassium channels in capillaries isolated from guinea-pig heart. J Physiol. 2000;525:307–317. doi: 10.1111/j.1469-7793.2000.t01-1-00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H. Pharmacological characteristics of the novel antihypertensive drug, iptakalim hydrochloride, and its molecular mechanisms. Drug Dev Res. 2003;58:65–68. [Google Scholar]
- 29.Xue H, Zhang YL, Liu GS, Wang H. A new ATP-sensitive potassium channel opener protects the kidney from hypertensive damage in spontaneously hypertensive rats. J Pharmacol Exp Ther. 2005;315:501–509. doi: 10.1124/jpet.105.089722. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Zhang YL, Tang XC, Feng HS, Hu G. Targeting ischemic stroke with a novel opener of ATP-sensitive potassium channels in the brain. Mol Pharmacol. 2004;66:1160–1168. doi: 10.1124/mol.104.003178. [DOI] [PubMed] [Google Scholar]