

Abstract
The unfolded protein endoplasmic reticulum stress response has emerged as a cellular physiological target to invoke tumor cell killing due to its homeostatic and cytoprotective functions. In this study, thapsigargin and tunicamycin, two endoplasmic reticulum stress inducers, were investigated for their efficacy on glioblastomas. We demonstrate that clinically relevant concentrations of thapsigargin and tunicamycin eliminate the glioblastoma cell reproductive capacity as a consequence of cell death. The mode of glioblastoma-induced cell death was determined to be via apoptosis as supported by increased C/EBP homologous protein (CHOP) levels and caspase 3 activity, two proteins with established roles in endoplasmic reticulum stress-induced cell death. In conclusion, this study provides evidence that glioblastomas are responsive to endoplasmic reticulum stress induction as a cellular program to eradicate this tumor via programmed cell death.
Keywords: glioblastomas, endoplasmic reticulum stress, C/EBP homologous protein
Introduction
Glioblastomas are among the most chemo-resistant types of human cancers to treat clinically. The refractiveness of glioblastomas to chemotherapy treatment regimens may be attributed in part to the activation of a number of cytoprotective mechanisms in response to chemotherapeutic agents (1,2). One such cytoprotective mechanism that has emerged and received considerable attention as a contributing factor to the therapeutic resistance of a number of types of human cancer in general, and in glioblastomas in particular, is the unfolded protein endoplasmic reticulum stress response (3,4). The unfolded protein endoplasmic reticulum stress response fundamentally functions as an adaptive cellular program essential for normal cellular function and survival, that is triggered by the accumulation of unfolded proteins in the endoplasmic reticulum due to a number of stress inducers, such as hypoxia, oxidative injury, glucose deprivation and aberrant calcium levels. However, if the stress applied to the endoplasmic reticulum is excessive and exceeds its ability to maintain homeostatic control, apoptotic cell death ensues (3,4). Therefore, assessing the efficacy of agents that invoke endoplasmic reticulum stress for their utility as potential chemotherapeutic drugs that promote glioblastoma cell death should be given practical consideration. In support of this, a number of investigational studies have demonstrated the anti-tumorigenic effects of novel endoplasmic reticulum stress inducers in leukemia (5), stomach (6) and prostate cancer (7). Furthermore, recent studies have established that glucose regulated protein 78 (GRP78), an endoplasmic reticulum chaperone with anti-apoptotic properties and a significant role in the unfolded protein endoplasmic reticulum stress response signaling pathway, is overexpressed in malignant gliomas (8–10).
To this end, in the present study, we examined two endoplasmic reticulum stress inducers, thapsigargin and tunicamycin, for their anti-tumor properties on glioblastomas. Thapsigargin, an active component found in root extracts of the umbelliferous plant, Thapsia garganica, acts as a stress inducer by increasing the intracellular calcium concentration via the inhibition of calcium uptake into the endoplasmic reticulum by blocking its ATP-dependent calcium pump (11), while tunicamycin, a nucleoside antibiotic produced by several Streptomyces species, imposes cellular stress by inhibiting protein N-linked glycosylation, the first step in protein glycosylation (12). This study reveals that the endoplasmic reticulum stress inducers, thapsigargin and tunicamycin, promote glioblastoma cell death as a consequence of inducing the pro-apoptotic proteins, C/EBP homologous protein (CHOP) and caspase 3.
Materials and methods
Cells, conditions and reagents
U373 and A172 glioblastoma cells were purchased from the American Type Culture Collection (Manassas, VA, USA). All cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (Invitrogen), 2 mM L-glutamine (Invitrogen), 100 nM MEM non-essential amino acids (Invitrogen) and penicillin-streptomycin (Invitrogen) at 37°C and 5% CO2. Thapsigargin and tunicamycin were purchased from Tocris (Ellisville, MO, USA).
Crystal violet cell proliferation assay
Dose response
Cells were plated in 12-well plates, treated with 1, 5 and 10 μM thapsigargin or tunicamycin and allowed to incubate for 48 h [vehicle controls were treated with dimethyl sulfoxide (DMSO)]. The tissue culture medium was then removed, and the cell monolayer was fixed with 100% methanol for 5 min and stained with 0.5% crystal violet in 25% methanol for 10 min. The cells were then washed three times for 5 min each with distilled water to remove excess dye and allowed to dry overnight at room temperature. The incorporated dye was then solubilized in 0.1 M sodium citrate (Sigma-Aldrich, St. Louis, MO, USA) in 50% ethanol. Subsequently, 100 μl of treated and control samples were transferred to 96-well plates and optical densities were read at 540 nm using an X-mark microplate absorbance spectrophotometer (BioRad, Hercules, CA, USA).
Time course analysis. Cells were plated in 96-well plates for 24 h, treated with 1 μM thapsigargin or tunicamycin and allowed to incubate for 1, 3 and 6 days at 37°C. At the end of each time-point the cells were stained with crystal violet, solubilized with sodium citrate, and the optical densities were read as described above.
Clonogenic survival
Cells were plated for 24 h, treated with 1 μM and 250 nM thapsigargin, tunicamycin or DMSO (vehicle) and allowed to incubate at 37°C for 10–14 days. At the termination of the incubation period, cells were fixed with absolute methanol, stained with 1% crystal violet for 10 min, rinsed in tap water and allowed to dry. Colonies, consisting of ≥50 cells, were then counted to determine the surviving fraction.
Cell motility
Motility assays were conducted according to the manufacturer's instructions (Cell Biolabs Inc., San Diego, CA, USA). A cell suspension containing 0.5–1.0×106 cells/ ml was prepared in serum-free medium with the vehicle (DMSO), 1 μM thapsigargin or 1 μM tunicamycin, while 500 μl of medium containing 10% fetal bovine serum were added to the lower chamber of the migration plate. A total of 300 μl of cell suspension containing vehicle, 1 μM thapsigargin or tunicamycin was then added to the inside of each insert and allowed to incubate for 24 h at 37°C and 5% CO2. Subsequently, non-migratory cells were removed from plate inserts (according to the manufacturer's instructions) and migratory cells were counterstained with cell staining solution (Cell Biolabs Inc.).
Western blotting
Cells were plated in serum-free DMEM for 24 h, treated with 1 μM thapsigargin, 1 μM tunicamycin or the vehicle, and allowed to incubate for 24 and 48 h. The cells were then lysed in lysis buffer (pH 6.8) containing 60 mM Tris and 2% SDS. Protein concentrations were determined using the Bradford method. Subsequently, protein samples were electrophoresed in a 4–12% Tris-HCl polyacrylamide gel, transferred to nitrocellulose membranes and immunoblotted with antibodies against CHOP (Cell Signaling, Danvers, MA, USA). Protein levels were detected using a horseradish peroxidase conjugated secondary antibody and the chemiluminescence detection system (Pierce, Rockford, IL, USA).
Detection of caspase activity
Cells were plated in serum-free DMEM for 24 h, treated with 1 μM thapsigargin, 1 μM tunicamycin or the vehicle and allowed to incubate for 48 h. Cells were lysed in lysis buffer (pH 6.8) containing 60 mM Tris and 2% SDS, and protein concentrations were determined using the Bradford method. Subsequently, caspase 3 activity assays were conducted according to manufacturer's instructions using 30 μg of protein (Promega, Madison, WI, USA).
Results
Endoplasmic reticulum stress inducers impede glioblastoma cell production
Eliciting a hyper-stress response in the endoplasmic reticulum as a means of promoting anti-tumor cell behavior due to the accumulation of unfolded proteins or an unstable physiological cellular environment, such as increased intracellular calcium concentration, has been demonstrated in a variety of types of human cancer (13–17). In this study, we performed a comparative assessment of two endoplasmic reticulum stress inducers, thapsigargin and tunicamycin, with markedly different modes of stressing the endoplasmic reticulum for their anti-tumor properties on glioblastomas. Thapsigargin and tunicamycin were first examined for their effects on glioblastoma cells in dose-response experiments. These experiments revealed a dose-dependent decrease in glioblastoma cell proliferation in A172 and U373 cells exposed to increasing concentrations (1–10 μM) of thapsigargin or tunicamycin, as compared to the vehicle-treated control cells (Fig. 1). Dose response data also revealed that the inhibitory effects on A172 and U373 glioblastoma cell proliferation were more pronounced in the cells treated with 5–10 μM thapsigargin as compared to the cells treated with 5–10 μM tunicamycin. Additionally, A172 cells appeared to be more sensitive to endoplasmic reticulum stress as indicated by a 44±6 and 27±11% reduction in cell proliferation (compared to controls) when treated with 1 μM thapsigargin or tunicamycin, respectively, in comparison to the U373 cells treated with either endoplasmic reticulum stress inducer at the same concentration.
Figure 1.
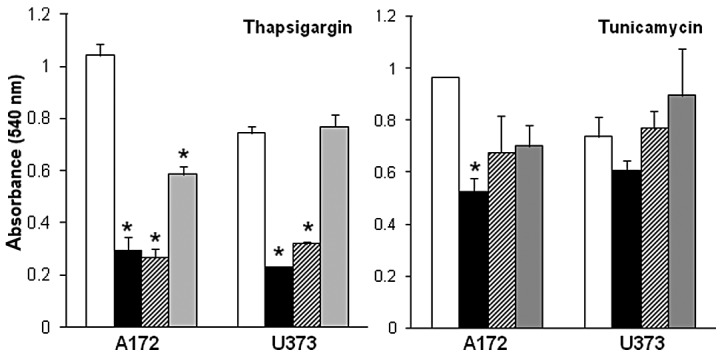
Dose response of thapsigargin and tunicamycin on A172 and U373 glioblastoma cells. Vehicle-treated (DMSO) control cells (white bars); 10 μM (solid black bars); 5 μM (hatched bars); 1 μM (graybars). Data shown are representative of 3 independent experiments (means ± SE) performed in duplicate showing similar results (*p<0.05, Student's t-test). DMSO, dimethyl sulfoxide; SE, standard error.
To further examine the inhibitory effects of thapsigargin and tunicamycin on glioblastoma cell production, clonogenic survival assays that measure cellular reproductive capacity at low cell plating densities were performed by treating A172 and U373 cells with 1 μM thapsigargin or tunicamycin. This concentration was selected for its non-lethal effects on U373 cells, and showed an inhibitory concentration greater than 50 (>IC50) in A172 cells, as shown in the dose-response experiments. Clonogenic survival experiments revealed that in comparison to the vehicle-treated control cells, thapsigargin or tunicamycin completely abrogated colony formation of A172 (Fig. 2) and U373 (Fig. 3) cells.
Figure 2.
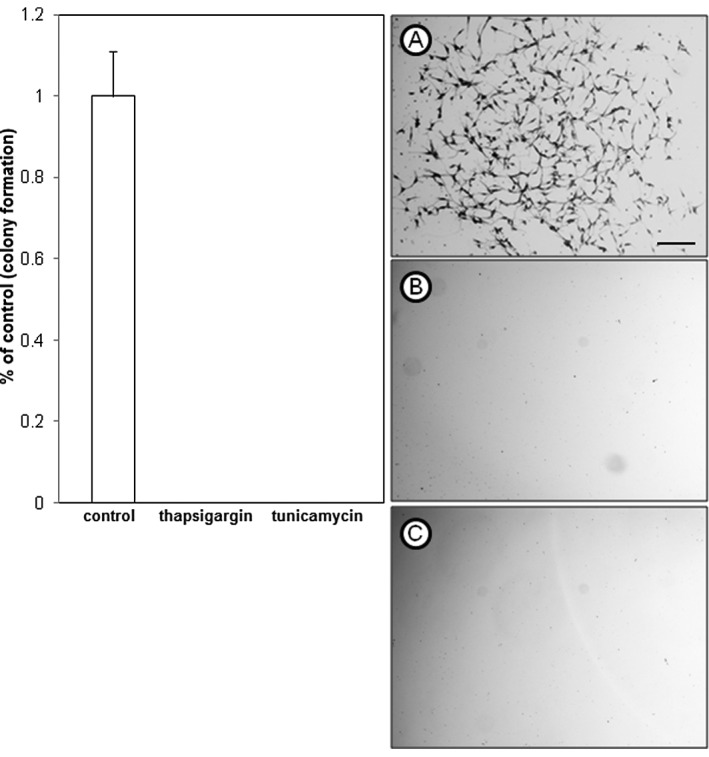
Clonogenic survival of A172 glioblastoma cells treated with endoplasmic reticulum stress inducers. (A) Vehicle control (DMSO); (B) thapsigargin (1 μM); (C) tunicamycin (1 μM). Cells shown are of a single colony stained with crystal violet from a clonogenic survival experiment. Data shown are representative of at least 3 independent experiments (means ± SE) performed in duplicate with comparable results (scale bar = 200 μm). DMSO, dimethyl sulfoxide; SE, standard error.
Figure 3.
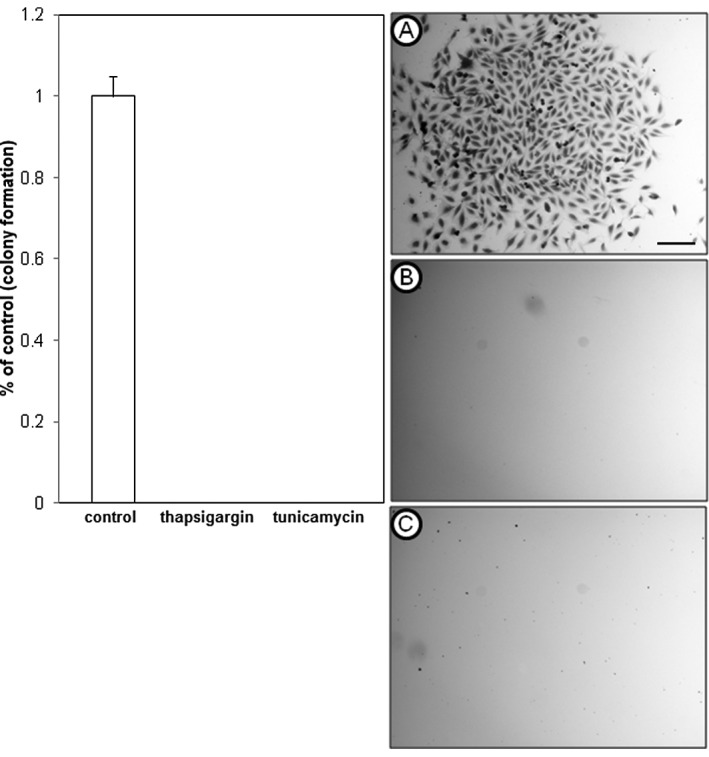
Clonogenic survival of U373 glioblastoma cells treated with endoplasmic reticulum stress inducers. (A) Vehicle control (DMSO); (B) thapsigargin (1 μM); (C) tunicamycin (1 μM). Cells shown are of a single colony stained with crystal violet from a clonogenic survival experiment. Data shown are representative of at least 3 independent experiments (means ± SE) performed in duplicate with comparable results (scale bar = 200 μm). DMSO, dimethyl sulfoxide; SE, standard error.
In addition to clonogenic survival experiments, time-course experiments were performed as a means to determine the cellular behavior underlying the inhibition of glioblastoma cell proliferation in response to thapsigargin or tunicamycin exposure (Fig. 4). Analysis of glioblastoma cell proliferation was conducted over a 6-day period post-exposure to the vehicle, thapsigargin or tunicamycin. The most demonstrative effects of thapsigargin or tunicamycin on A172 and U373 cell production were observed on day 6, with a statistically significant (p<0.05) decrease in glioblastoma cell proliferation as compared to the vehicle-treated control cells examined at the same time-point (Fig. 4). Time course data also displayed a 28±4 and 52±4% reduction in A172 cell proliferation 6 days post-treatment with thapsigargin or tunicamycin, respectively, in comparison to A172 cell proliferation observed on day 0 (Fig. 4). A comparative assessment between these same time-points in U373 cells also revealed a decrease in cell proliferation 6 days post-treatment with either thapsigargin or tunicamycin when compared to U373 cell proliferation on day 0 (Fig. 4). Taken together, the time course data presented here suggest that the overall inhibitory effect of thapsigargin and tunicamycin was due to glioblastoma cell death. These results are similar to those from other studies on several types of human cancer that also demonstrated that thapsigargin (18–21) and tunicamycin (22–24) promoted tumor cell death.
Figure 4.

Time course analysis of glioblastoma cell proliferation post-exposure to thapsigargin or tunicamycin. Cells were exposed on day 0 to solvent (DMSO), thapsigargin (1 μM) or tunicamycin (1 μM) and monitored for a period of 6 days. The proliferation curves displayed show that, when compared to the controls, glioblastoma cell proliferation is significantly reduced when treated with thapsigargin or tunicamycin. Data shown are representative of 3 independent experiments (means ± SE) performed in duplicate displaying similar results (*p<0.05, Student's t-test). DMSO, dimethyl sulfoxide; SE, standard error.
ER stress inducers increase CHOP expression and caspase 3 activity
It is well established that the inability of the endoplasmic reticulum to activate compensatory mechanisms essential for cellular survival in response to stress will subsequently lead to apoptotic cell death (3,4). CHOP, a basic-leucine zipper (bZIP) transcription factor, and caspase 3, a cysteine protease, are pro-apoptotic proteins known to play prominent roles in endoplasmic reticulum stress-induced cell death (25,26). We therefore evaluated the protein expression levels of CHOP and caspase 3 in thapsigargin- and tunicamycin-treated glioblastoma cells. A temporal analysis using immunoblotting procedures revealed an up-regulation of CHOP protein levels at 24 and 48 h in A172 and U373 cells treated with 1 μM thapsigargin or tunicamycin (Fig. 5). Caspase 3 activity assessment displayed a 2.6- and 1.43-fold increase in caspase 3 activity at 48 h in A172 cells treated with thapsigargin or tunicamycin, respectively, while a 1.32- and 1.30-fold increase in caspase 3 activity was observed at the same time-point in U373 cells treated with thapsigargin or tunicamycin (Fig. 6). Although our observations are consistent with previous ones on colon cancer, leukemia and neuroblastomas, which also demonstrated that thapsigargin or tunicamycin invoked CHOP expression or caspase 3 activity (24,27–30), few studies have shown the induction of both of these pro-apoptotic proteins in the same tumor type in response to single exposures of endoplasmic reticulum stress inducers with different modes of action. This suggests that glioblastomas are susceptible to endoplasmic reticulum stress-induced cell death by diverse physiological stressors.
Figure 5.

Western blot analysis of CHOP protein levels in glioblastoma cells treated with thapsigargin (1 μM) or tunicamyicin (1 μM). A172 cells (lanes 1–5); U373 cells (lanes 6–10). Vehicle-treated control cells (DMSO) (lanes 1 and 6); thapsigargin treatment for 24 h (lanes 2 and 7); thapsigargin treatment for 48 h (lanes 3 and 8); tunicamycin treatment for 24 h (lanes 4 and 9); tunicamycin treatment for 48 h (lanes 5 and 10). The images shown are representative of 3 independent experiments performed in duplicate that displayed similar results. CHOP, C/EBP homologous protein; DMSO, dimethyl sulfoxide.
Figure 6.
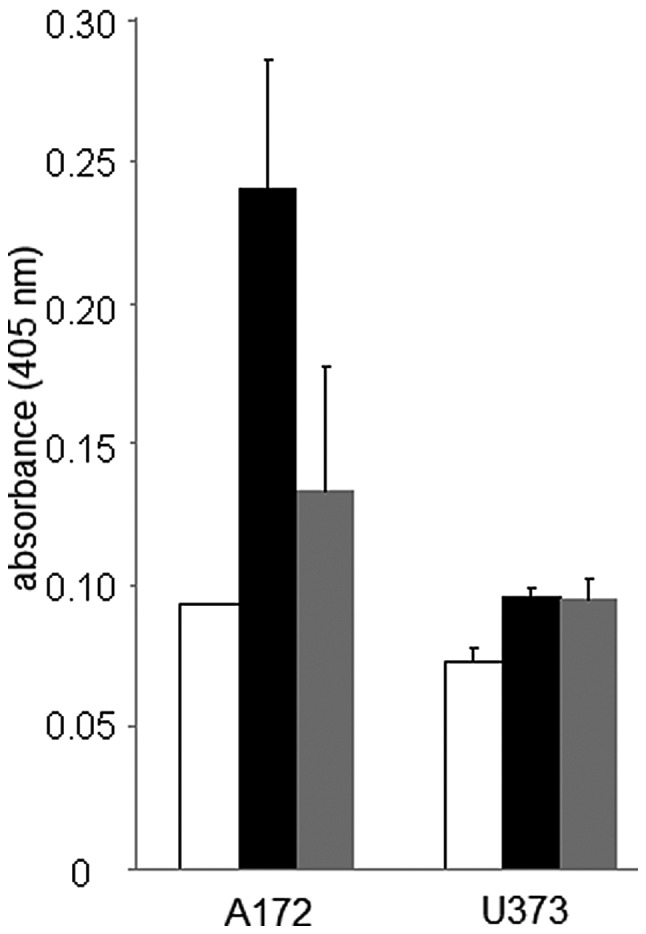
Assessment of caspase 3 activity in A172 and U373 glioblastoma cells in response to thapsigargin (1 μM) and tunicamycin (1 μM) exposure. Caspase protein activity was measured 48 h post-exposure to thapsigargin or tunicamycin (vehicle control, white bars; thapsigargin, black bars; tunicamycin, gray bars). Data are represented as an average of 3 separate independent experiments (means ± SE). SE, standard error.
Effects of ER stress on glioblastoma cell motility
Glioblastomas are notoriously invasive tumors with high rates of recurrence, which are major factors contributing to their therapeutic refractiveness during clinical treatment (surgery, chemo- and radiotherapy) regimens. Few studies to date have investigated the role of the unfolded protein endoplasmic reticulum stress response in tumor cell motility and invasion. However, a study by Chiu et al (31) on head and neck cancers revealed that silencing the function of the endoplasmic reticulum chaperone protein, GRP78, using siRNA reduced the metastatic potential of these cancers; thus, providing evidence for a role of the unfolded protein endoplasmic reticulum stress response in the metastatic invasion of tumor cells. Therefore, in this study, we examined the effects of thapsigargin and tunicamycin on glioblastoma cell motility, a prerequisite cellular program for invasive tumor cells. In contrast to the findings by Chiu et al (31), we did not observe antagonistic effects of thapsigargin or tunicamycin on glioblastoma cell motility (Fig. 7), which is likely due to variations in the experimental approach and the differential mechanisms targeted.
Figure 7.

Motility of A172 (left) and U373 (right) glioblastoma cells treated with the endoplasmic reticulum stress inducers, thapsigargin and tunicamycin. Motility was determined with boyden chamber assays and the results are shown as micrographs of glioblastoma cells stained with crystal violet. (A and D) Vehicle (DMSO)-treated cells; (B and E) cells treated with 1 μM thapsigargin; (C and F) cells treated with 1 μM tunicamycin. The images shown are representative of 3 independent experiments performed in duplicate that displayed similar results (scale bar = 100 μm). DMSO, dimethyl sulfoxide.
Discussion
Targeting the unfolded protein endoplasmic reticulum stress response is a relatively avant-garde yet practical therapeutic approach for the treatment of human cancers. Human tumors and their microenvironment are in a continuous flux of imbalance due to the presence of abnormally folded proteins and physiological instability, relating to fluctuations in pH and ion concentration, all of which invoke stress on tumor cells. As a survival mechanism, tumor cells respond via the activation of stress responders, such as GRP78/binding immunoglobulin protein (BiP), inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6) that collectively underlie the unfolded protein endoplasmic reticulum stress response and confer tumor cell cytoprotection (3,4). The involvement of cytoprotective stress responders in glioblastomas has been demonstrated in a number of recent investigations that together establish that high levels of GRP78 expression correlate with increased survival and drug resistance (8–10) in these tumors. However, studies to date have failed to demonstrate that antagonizing stress responder function, particularly GRP78, promotes glioblastoma cell death.
The dichotomy of the unfolded protein endoplasmic reticulum stress response is that stress exceeding the endoplasmic reticulum's capacity to promote cell survival will consequently result in cell death. To this end, in this study, we demonstrate that two endoplasmic reticulum stress inducers, thapsigargin and tunicamycin, promote glioblastoma cell death. Our findings are consistent with those from previous studies on several human cancers that also showed that thapsigargin (prostate, breast, leukemia, and melanoma) (18–20,30,32) and tunicamycin (neuroblastoma and melanoma) induced tumor cell death (24,33).
It is well established that the mechanism affiliated with endoplasmic reticulum stress-induced cell death is the activation of the transcription factor, CHOP, a downstream pro-apoptotic component of the IRE1α, PERK and ATF6 stress responder pathways (25). Concomitantly with glioblastoma cell death, we observed a significant increase in CHOP expression in response to endoplasmic recticulum stress inducers. These findings parallel those observed by Rosati et al (22) and Oda et al (24) who also detected increased CHOP expression levels in leukemia and neuroblastoma cells, respectively, in response to thapsigargin and tunicamycin. Although effector molecules of CHOP still remain somewhat elusive, its overexpression has been shown to lead to a decrease in the pro-survival protein, Bcl-2 (34), providing evidence that the pro-apoptotic functions of CHOP are associated with mitochondria-dependent mechanisms of cell death. Our data support this premise and were substantiated by observations of increased caspase 3 activity in glioblastoma cells treated with thapsigargin or tunicamycin.
In spite of the pro-apoptotic effects of endoplasmic reticulum stress inducers on glioblastoma cell production observed in this study, thapsigargin and tunicamycin failed to impair glioblastoma cell motility. This is in contrast to experimental data by Chiu et al who demonstrated that silencing GRP78 function reduced cell motility and prevented tumor cell invasion of head and neck cancers (31). The differential cell motility responses observed in this study and by Chiu et al are likely attributed to divergent cellular and physiological mechanisms targeted in the unfolded protein endoplasmic reticulum stress response between our two studies, and further emphasizes the pleiotropic effects of the endoplasmic reticulum stress response on cell behavior.
Taken together, we provided evidence that hyper-stressing the endoplasmic reticulum, by using endoplasmic reticulum stress inducers with markedly different modes of action, is a viable approach for the promotion of killing glioblastomas. Furthermore, our study suggests that endoplasmic reticulum stress inducers exert their anti-tumorigenic effects on proliferating cells, a selective advantage for treating clinical glioblastomas which typically reside in regions of the human brain that contain dormant cells.
Acknowledgments
This work was supported by grant number P20MD002731 from the National Institute on Minority Health and Health Disparities (MOF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Minority Health and Health Disparities or the National Institutes of Health.
References
- 1.Krakstad C, Chekenya M. Survival signaling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol Cancer. 2010;9:135. doi: 10.1186/1476-4598-9-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kögel D, Fulda S, Mittelbronn M. Therapeutic exploitation of apoptosis and autophagy for glioblastoma. Anticancer Agents Med Chem. 2010;10:438–449. doi: 10.2174/1871520611009060438. [DOI] [PubMed] [Google Scholar]
- 3.Wang G, Yang Z, Zhang K. Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. Am J Transl Res. 2010;2:65–74. [PMC free article] [PubMed] [Google Scholar]
- 4.Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol. 2011;2:143–149. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lust S, Vanhoecke B, Van Gele M, Boelens J, Van Melckebeke H, Kaileh M, Vanden Berghe W, Haegeman G, Philippé J, Bracke M, Offner F. Xanthohumol activates the proapoptotic arm of the unfolded protein response in chronic lymphocytic leukemia. Anticancer Res. 2009;10:3797–3805. [PMC free article] [PubMed] [Google Scholar]
- 6.Huang X, Zhang Z, Jia L, Zhao Y, Zhang X, Wu K. Endoplasmic reticulum stress contributes to vitamin E succinate-induced apoptosis in human gastric cancer SGC-7901 cells. Cancer Lett. 2010;296:123–131. doi: 10.1016/j.canlet.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Ravanan P, Sano R, Priti T, Ogasawara S, Matsuzawa SI, Cuddy M, Singh SK, Rao GS, Kondaiah P, Reed JC. Synthetic triterpenoid cyano enone of methyl boswellate (CEMB) activates intrinsic, extrinsic and endoplasmic reticulum stress cell death pathways in tumor cell lines. Mol Cancer Ther. 2011;10:1635–1643. doi: 10.1158/1535-7163.MCT-10-0887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pyrko P, Schönthal AH, Hofman F, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–9816. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 9.Virrey JJ, Dong D, Stiles C, Patterson JB, Pen L, Ni M, Schonthal AH, Chen TC, Hofman FM, Lee AS. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol Cancer Res. 2008;6:1268–1275. doi: 10.1158/1541-7786.MCR-08-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee HK, Xiang C, Cazacu S, Finniss S, Kazimirsky G, Lemke N, Lehman NL, Rempel SA, Mikkelsen T, Brodie C. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol. 2008;10:236–243. doi: 10.1215/15228517-2008-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabała P, Czarny M, Woronczak JP, Barańska J. Thapsigargin: potent inhibitor of Ca2+ transport ATP-ases of endoplasmic and sarcoplasmic reticulum. Acta Biochim Pol. 1993;40:309–319. [PubMed] [Google Scholar]
- 12.Isono K. Nucleoside antibiotics: structure, biological activity, and biosynthesis. J Antibiot (Tokyo) 1988;41:1711–1739. doi: 10.7164/antibiotics.41.1711. [DOI] [PubMed] [Google Scholar]
- 13.Hancock CN, Stockwin LH, Han B, Divelbiss RD, Jun JH, Malhotra SV, Hollingshead MG, Newton DL. A copper chelate of thiosemicarbazone NSC 689534 induces oxidative/ER stress and inhibits tumor growth in vitro and in vivo. Free Radic Biol Med. 2011;50:110–121. doi: 10.1016/j.freeradbiomed.2010.10.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Min SK, Lee SK, Park JS, Lee J, Paeng JY, Lee SI, Lee HJ, Kim Y, Pae HO, Lee SK, Kim EC. Endoplasmic reticulum stress is involved in hydrogen peroxide induced apoptosis in immortalized and malignant human oral keratinocytes. J Oral Pathol Med. 2008;37:490–498. doi: 10.1111/j.1600-0714.2008.00679.x. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe Y, Tsuchiya H, Sakabe T, Matsuoka S, Akechi Y, Fujimoto Y, Yamane K, Ikeda R, Nishio R, Terabayashi K, et al. CD437 induces apoptosis in ovarian adenocarcinoma cells via ER stress signaling. Biochem Biophys Res Commun. 2008;366:840–847. doi: 10.1016/j.bbrc.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 16.Joo JH, Liao G, Collins JB, Grissom SF, Jetten AM. Farnesol-induced apoptosis in human lung carcinoma cells is coupled to the endoplasmic reticulum stress response. Cancer Res. 2007;67:7929–7936. doi: 10.1158/0008-5472.CAN-07-0931. [DOI] [PubMed] [Google Scholar]
- 17.Yacoub A, Hamed HA, Allegood J, Mitchell C, Spiegel S, Lesniak MS, Ogretmen B, Dash R, Sarkar D, Broaddus WC, et al. PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res. 2010;70:1120–1129. doi: 10.1158/0008-5472.CAN-09-4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackisch C, Hahm HA, Tombal B, McCloskey D, Butash K, Davidson NE, Denmeade SR. Delayed micromolar elevation in intracellular calcium precedes induction of apoptosis in thapsigargin-treated breast cancer cells. Clin Cancer Res. 2000;6:2844–2850. [PubMed] [Google Scholar]
- 19.Huang JK, Chou CT, Chang HT, Shu SS, Kuo CC, Tsai JY, Liao WC, Wang JL, Lin KL, Lu YC, et al. Effect of thapsigargin on Ca2+ fluxes and viability in human prostate cancer cells. J Recept Signal Transduct Res. 2011;31:247–255. doi: 10.3109/10799893.2011.563311. [DOI] [PubMed] [Google Scholar]
- 20.Vander Griend DJ, Antony L, Dalrymple SL, Xu Y, Christensen SB, Denmeade SR, Isaacs JT. Amino acid containing thapsigargin analogues deplete androgen receptor protein via synthesis inhibition and induce the death of prostate cancer cells. Mol Cancer Ther. 2009;8:1340–1349. doi: 10.1158/1535-7163.MCT-08-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaneko Y, Tsukamoto A. Thapsigargin-induced persistent intracellular calcium pool depletion and apoptosis in human hepatoma cells. Cancer Lett. 1994;79:147–155. doi: 10.1016/0304-3835(94)90253-4. [DOI] [PubMed] [Google Scholar]
- 22.Rosati E, Sabatini R, Rampino G, De Falco F, Di Ianni M, Falzetti F, Fettucciari K, Bartoli A, Screpanti I, Marconi P. Novel targets for endoplasmic reticulum stress-induced apoptosis in B-CLL. Blood. 2010;116:2713–2723. doi: 10.1182/blood-2010-03-275628. [DOI] [PubMed] [Google Scholar]
- 23.Girnita L, Wang M, Xie Y, Nilsson G, Dricu A, Wejde J, Larsson O. Inhibition of N-linked glycosylation down-regulates insulin-like growth factor-1 receptor at the cell surface and kills Ewing's sarcoma cells: therapeutic implications. Anticancer Drug Des. 2000;15:67–72. [PubMed] [Google Scholar]
- 24.Oda T, Kosuge Y, Arakawa M, Ishige K, Ito Y. Distinct mechanism of cell death is responsible for tunicamycin-induced ER stress in SK-N-SH and SH-SY5Y cells. Neurosci Res. 2008;60:29–39. doi: 10.1016/j.neures.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 25.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 26.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi H, Bhalla K, Wang HG. Bax plays a pivotal role in thapsigargin-induced apoptosis of human colon cancer HCT116 cells by controlling Smac/Diablo and Omi/HtrA2 release from mitochondria. Cancer Res. 2003;63:1483–1489. [PubMed] [Google Scholar]
- 28.Kitamura Y, Miyamura A, Takata K, Inden M, Tsuchiya D, Nakamura K, Taniguchi T. Possible involvement of both endoplasmic reticulum-and mitochondria-dependent pathways in thapsigargin-induced apoptosis in human neuroblastoma SH-SY5Y cells. J Pharmacol Sci. 2003;92:228–236. doi: 10.1254/jphs.92.228. [DOI] [PubMed] [Google Scholar]
- 29.Dahmer MK. Caspases-2, -3, and -7 are involved in thapsigargin-induced apoptosis of SH-SY5Y neuroblastoma cells. J Neurosci Res. 2005;80:576–583. doi: 10.1002/jnr.20471. [DOI] [PubMed] [Google Scholar]
- 30.Feng XQ, You Y, Xiao J, Zou P. Thapsigargin-induced apoptosis of K562 cells and its mechanism. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2006;14:25–30. [PubMed] [Google Scholar]
- 31.Chiu CC, Lin CY, Lee LY, Chen YJ, Kuo TF, Chang JT, Liao CT, Wang HM, Yen TC, Shen CR, Liao SK, Cheng AJ. Glucose-regulated protein 78 regulates multiple malignant phenotypes in head and neck cancer and may serve as a molecular target of therapeutic intervention. Mol Cancer Ther. 2008;7:2788–2797. doi: 10.1158/1535-7163.MCT-08-0172. [DOI] [PubMed] [Google Scholar]
- 32.Chen LH, Jiang CC, Kiejda KA, Wang YF, Thorne RF, Zhang XD, Hersey P. Thapsigargin sensitizes human melanoma cells to TRAIL-induced apoptosis by up-regulation of TRAIL-R2 through the unfolded protein response. Carcinogenesis. 2007;28:2328–2336. doi: 10.1093/carcin/bgm173. [DOI] [PubMed] [Google Scholar]
- 33.Dricu A, Carlberg M, Wang M, Larsson O. Inhibition of N-linked glycosylation using tunicamycin causes cell death in malignant cells: role of down-regulation of the insulin-like growth factor 1 receptor in induction of apoptosis. Cancer Res. 1997;57:543–548. [PubMed] [Google Scholar]
- 34.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]