

Abstract
The Notch signaling pathway plays an important role in the proliferation and differentiation of cells. Although recent studies have shown that Notch plays a role in the mechanisms of cisplatin resistance, the mechanism by which Notch plays roles in intrinsic or acquired cisplatin resistance remains unclear. In the present study, poorly differentiated nasopharyngeal carcinoma cells were treated with a γ-secretase inhibitor (DAPT), which led to a decrease in the Notch intracellular domain and inhibition of Notch signaling. Treatment was not sufficient to induce pronounced apoptosis of CNE-2 cells, but did result in the down-regulation of the P-glycoprotein and ERCC1 protein. In contrast, the combined treatment of DAPT and cisplatin induced substantial cell apoptosis compared to cisplatin treatment alone.
Keywords: Notch signaling, cisplatin resistance, nasopharyngeal carcinoma
Introduction
Nasopharyngeal carcinoma (NPC) is a tumor arising from the epithelial cells that cover the surface and line the nasopharynx. The incidence of NPC is higher in Chinese and Tunisian populations (1). Unfortunately, at diagnosis, 70% of patients have locally advanced, non-metastatic stage III or IV disease (2,3). Despite novel advances in radiotherapy, chemotherapy and gene-targeting agents, the overall survival rate of patients in the aggressive phase remains low (4). Concomitant chemoradiotherapy represents one of the most recent advances in the treatment of NPC patients. A recent meta-analysis confirmed the superiority of concurrent chemoradiotherapy to radiotherapy alone. Cisplatin-based concomitant chemoradiotherapy is now the standard treatment for locally advanced NPC patients (2,5,6).
However, intrinsic or acquired resistance to cisplatin therapy remains a critical problem in the clinical management of NPC patients. Cisplatin resistance in HNSCC may be mediated by a number of different mechanisms, including drug detoxification, up-regulation of DNA repair enzymes or the overexpression of gene products that provide tumor cells with survival advantage relative to normal cells (7,8). Recent studies have shown that Notch may play a role in the mechanisms of cisplatin resistance (9). Moreover, Notch-1 was found to be highly expressed in cisplatin-resistant HNSCC patients (8).
The Notch signaling pathway plays an important role in the proliferation and differentiation of cells. It is an evolutionarily conserved pathway that regulates critical cell fate decisions (10). Notch receptor activation is irreversible as it involves proteolysis-mediated release of the Notch intracellular domain (NICD) which can be interrupted by γ-secretase inhibitor, translocation to the nucleus and transactivation of gene targets associated with the transcription cofactor CBF1, which in turn affect numerous pathways involving cell-fate determination (11–14).
Experimental evidence has also revealed that Notch is involved in anticancer drug resistance, indicating that targeting Notch may be a novel therapeutic approach for the treatment of cancer by overcoming drug resistance of cancer cells (9). Yet, the mechanism of how Notch functions in intrinsic or acquired cisplatin resistance remains unclear, particularly in NPC intrinsic resistance to cisplatin.
In the present study, treatment of poorly differentiated NPC cells with a γ-secretase inhibitor (DAPT), which led to the decline in NICD and inhibition of Notch signaling, was not sufficient to induce pronounced apoptosis of CNE-2 cells, but did result in the down-regulation of the P-glycoprotein and ERCC1 protein. In contrast, combination of DAPT with cisplatin induced substantial cell death compared to cisplatin treatment alone.
Materials and methods
Cell cultures and reagents
The human poorly differentiated nasopharyngeal carcinoma CNE-2 cell line was purchased from the Shanghai Cell Collection (Shanghai, China) and maintained in RPMI-1640 containing 10% fetal bovine serum (Hyclone, UT, USA), 100 U/ml penicillin and 100 mg/ml streptomycin at 37°C in a humidified atmosphere with 5% CO2. The γ-secretase inhibitor (DAPT; N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycinet-butylester, C23H26F2N2O4) was purchased from Sigma-Aldrich Co. LLC and cisplatin were supplied by Qilu Pharmaceutical Co., Ltd.
Cell treatment
Notch signaling pathways were inhibited with the γ-secretase inhibitor (DAPT) at different concentrations. DAPT was dissolved in DMSO to a stock concentration of 10 mM, and was diluted to final concentrations of 25, 50 and 75 μM with conventional culture medium just prior to use. The control group (0 μM DAPT) was mock treated with conventional medium containing the mean volume of DMSO carrier only. Cispatin was dissolved in PBS to a stock concentration of 5 mM and used at a final concentration of 10 μM after a 24-h treatment with various concentrations of DAPT (15).
Flow cytometric analysis of apoptotic cells using Annexin V-FITC and propidium iodide (PI)
After 72 h of cell treatment as mentioned above, cells were gently trypsinized and washed once with serum-containing media. Each group of cells (1×105 to 2×105) was collected by centrifugation and then resuspended in 500 μl of 1X Annexin-binding buffer. Annexin V-FITC (5 μl) and 10 μl of PI were added, and cells were then incubated at room temperature for 10 min in the dark. Annexin V-FITC binding was analyzed by flow cytometry (Epics Altra, Beckman, USA; Ex=488 nm, Em=530 nm) using FITC signal detector and PI staining by the phycoerythrin emission signal detector.
Cell proliferation and cytotoxicity assay
We dispensed 100 μl of cell suspension (2,000 cells/well) in a 96-well plate. The plate was pre-incubated for 24 h in a humidified incubator. Various concentrations of DAPT (0, 25, 50 and 75 μM) were added to the plate for testing. The plate was incubated for 24, 48 and 72 h in the incubator, and 10 μl of CCK-8 (Dojindo, Japan) solution was added to each well of the plate. The plate was incubated for 1 h in the incubator, and the absorbance at 450 nm was measured using a multilable counter (Victor3 1420; Perkin Elmer).
Flow cytometric analysis of the cell cycle
The cells were treated similarly as for the analysis of cell apoptosis. Then, 1×106 cells of each group were collected and resuspended in 1 ml of 70% ethanol and fixed overnight at 4°C. Cells were collected by centrifugation again, washed with PBS twice and then resuspended in 500 μl of 1X binding buffer containing 0.5% PI. The prepared cells were incubated at 4°C for 20 min in the dark, and then analyzed by flow cytometry.
Western blot analysis
Cells were harvested from the 60 mm cell culture plates, and aliquots of cell extracts were separated on an 8–12% SDS-polyacrylamide gel. Then, proteins were transferred to a polyvinylidene difluoride membrane and incubated overnight at 4°C with the rabbit polyclonal antibody anti-NICD and anti-ERCC1 (Cell Signaling Technology), or mouse monoclonal antibody anti-P-glycoprotein (Abcam), respectively. Membranes were then washed and incubated with HRP-conjugated secondary antibodies in confining liquid for 1 h and the film was developed using an ECL test kit (Beyotime, China). The density of the bands on the film were scanned and analyzed with an image analyzer.
Statistical analysis
Statistical comparison of the data was performed using a Student’s t-test. A P-value of <0.01 was considered to be significant.
Results
Induction of cell death by the combined treatment of DAPT and cisplatin in CNE-2 cells
CNE-2 cells were first treated with increasing concentrations of cisplatin to establish the least effective concentration which substantially affects cell survival. While 5 μM cisplatin for 48 h showed little effect, cell death was observed with 10 μM cisplatin in CNE-2 cells, so this dosage was used for further drug combination studies. The CNE-2 cells were treated with increasing concentrations of DAPT, and there was no obvious effect on cell survival (P>0.01). Cells were initially treated with various concentrations of DAPT, and then combined with 10 μM cisplatin after 24 h. DAPT dose-dependent cell apoptosis was observed after treatment of 10 μM cisplatin for 48 h (Fig. 1).
Figure 1.

(A1–4) CNE-2 cells were treated with different concentrations of DAPT (0, 25, 50 and 75 μM), and the apoptosis of CNE-2 cells was assessed by FACS after 48 h. (B1–4) CNE-2 cells were previously treated with various concentrations of DAPT (0, 25, 50 and 75 μM) before 24 h, and then treated with cisplatin at the same final concentration of 10 μM. The apoptosis of CNE-2 cells was detected by FACS after 48 h of treatment with cisplatin. The control group (0 μM DAPT) was treated with conventional medium containing the mean volume of DMSO. No significant apoptosis was noted only with DAPT treatment when compared to the control group (P>0.01), while pre-treatment with DAPT enhanced the effect of cisplatin in a dose-dependent manner. There was a marked difference compared to the control group treated with cisplatin and DMSO (P<0.01). A1–4 and B1–4 are respresentative of one of three independent experiments that yielded similar results. Values in histograms (A) and (B) are the means ± SD. Proportion of non-apoptotic cells (F3, Annexin V-FITC−/PI−), early apoptotic cells (F4, Annexin V-FITC+/PI−), late apoptotic/necrotic cells (F2, Annexin V-FITC+/PI+) and cell debris or dead cells (F1). *P<0.01 compared to that of 0 μM DAPT group by T-test.
Induction of cell cycle arrest and cell proliferation is suppressed by DAPT
Although the application of the maximum dose of DAPT at 75 μM had no obvious effect on cell survival, it substantially retarded CNE-2 cell proliferation in a dose-dependent manner (Fig. 2). To assess whether DAPT-mediated Notch 1 inhibition is associated with changes in the cell cycle progression of CNE-2 cells, flow cytometry was employed to detect their allocation at different cell cycle phases in the presence of various concentrations of DAPT at 48 h. Notch 1 inhibition increased the percentage of cells in the G0/G1 phase at the expense of S-phase (Fig. 3).
Figure 2.
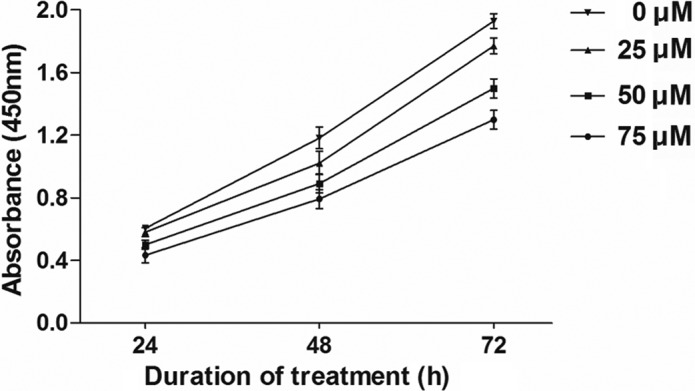
Cells were treated as described in Materials and methods, and cell proliferation and cytotoxicity assays were carried out. After 48 h, there was a notable difference in the absorbance between the DAPT-treated group and the control group (P<0.01), which indicated that the γ-secretase inhibitor retarded CNE-2 cell proliferation in a dose-dependent manner.
Figure 3.

(C1–4) Cell cycle of CNE-2 cells treated with different concentrations of DAPT. DAPT treatment had an impact on the cell cycle of long-term self-renewing CNE-2 cells. The histogram shows the relative distribution of CNE-2 cells across the different cell cycle phases under self-renewing conditions in the presence or absence of DAPT (P<0.01).
Mechanism of enhanced apoptosis is induced by the combined treatment of DAPT and cisplatin in CNE-2 cells
CNE-2 cells were treated with increasing concentrations of DAPT, and the γ-secretase-generated Notch 1 fragment Val1744-NICD was decreased after 48 h in a dose-dependent manner (P<0.01). The activation of γ-secretase was almost completely inhibited by DAPT at the concentration of 50 μM. Meanwhile, the expression of P-gp and ERCC1 protein was detected by western blotting. There was a significant decrease in the expression of P-gp and ERCC1 protein in a dose-dependent manner (P<0.01; Fig. 4).
Figure 4.

Western blotting of Val1744-NICD, P-gp and ERCC1 expression of CNE-2 cells treated with different concentrations of DAPT. At 48 h, lysates from CNE-2 cells were subjected to western blot analysis as described in Materials and methods. The images are from one of three independent experiments that yielded similar results.
Discussion
Chemotherapy fails to eliminate all tumor cells due to intrinsic or acquired drug resistance, which is the most common cause of tumor recurrence. The mechanisms responsible for drug resistance are complex and remain poorly understood. Resistance may be due to either the specific nature and genetic background of the cancer cell itself or the genetic changes that follow toxic chemotherapy (9,16,17).
Intrinsic or acquired resistance to cisplatin therapy remains a critical problem in the clinical management of NPC patients. Cisplatin resistance in HNSCC may be mediated by a number of different mechanisms, including drug detoxification, up-regulation of DNA repair enzymes or overexpression of gene products that provide tumor cells with survival advantage relative to normal cells. Studies have demonstrated that Notch regulates the formation of cancer stem cells and contributes to the acquisition of the epithelial-mesenchymal transition phenotype, which are also critically associated with drug resistance (18,19).
There is little evidence indicating that Notch signaling pathways are involved in cisplatin resistance through regulating drug detoxification or up-regulating DNA repair enzymes. In our study, we found that treatment with DAPT (γ-secretase inhibitor) markedly down-regulated P-gp and ERCC1 protein expression in CNE-2 cells, and the combination of DAPT and cisplatin in CNE-2 cells enhanced cisplatin sensitivity, eliminated the drug resistance and induced greater cell apoptosis compared to cisplatin treatment alone.
The majority of published data suggest that P-gp acts as a transmembrane pump which removes drugs from the cell membrane and cytoplasm (20). Overexpression of P-gp, the ATP-binding cassette drug transporter, confers an intrinsic or acquired multidrug resistance (MDR) due to its capability of transporting a broad range of chemically diverse anticancer drugs. Occurrence of MDR prevents efficient killing of cancer cells, leading to chemotherapeutic treatment failure (21). As a non-specific drug transporter, the P-gp also removes the γ-secretase inhibitor from the cytoplasm in theory. In fact, in this study, the concentration of DAPT applied in CNE-2 cells was higher compared to other studies (22,23), which indicated more P-gp pumps in the membrane of drug-resistant cancer cells. The nearly saturated concentration of DAPT guarantees the inhibition of γ-secretase activity and the interruption of NICD release, which in turn affects the transactivation of the MDR1 gene or other target genes encoding some enzymes or proteins, such as PKC, which activates the P-gp protein (24).
The expression of ERCC1 was also decreased after DAPT treatment. ERCC1 activity is involved in resistance to platinum chemotherapy drugs. Nucleotide excision repair (NER) is the primary DNA repair mechanism that removes the therapeutic platinum-DNA adducts from the tumor DNA. ERCC1 activity levels, being an important part of the NER common final pathway, may serve as a marker of general NER throughput. One study reported that ERCC1-positive NSCLC tumors do not benefit from adjuvant platinum chemotherapy. Thus, down-regulation of ERCC1 enhances the cancer cell sensitivity to platinum chemotherapy drugs, and ERCC1 may be involved in the mechanism of Notch-regulated cancer drug resistance (25–28).
In this study, we also found that application of γ-secretase inhibitor had no detectable effect on CNE-2 cell survival, which was not in accord with previous reports (29,30), but it substantially retarded CNE-2 cell proliferation in a dose-dependent relationship. Flow cytometry was employed to detect the allocation to different cell cycle phases in the presence of various concentrations of DAPT at 48 h. Notch 1 inhibition increased the percentage of cells in G0/G1 phase at the expense of S-phase. Borghese et al found similar results which showed that inhibition of Notch signaling in human embryonic stem cell-derived neural stem cells delayed G1/S phase transition and accelerated neuronal differentiation (23). Huang et al (22) also reported that the γ-secretase inhibitor DAPT induced adipogenesis of adipose-derived stem cells. Application of γ-secretase inhibitor may alter the differentiation state of CNE-2 cells and induce CNE-2 cells toward a well-differentiated state. Whether this change is involved in drug resistance of CNE-2 cells, requires future research.
In conclusion, inhibition of the Notch signaling pathway by a γ-secretase inhibitor markedly enhanced the cisplatin sensitivity of CNE-2 cells, which may be achieved through down-regulation of P-gp and ERCC1 protein. The combination of GSI with platinum compounds may provide an option to improve treatment for a subset of nasopharyngeal carcinoma patients.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No. 30872851 and 30901662), and the Science and Technology Department of Hubei Province (No. 2007AA302B08). This study also received assistance from Ms. Qing He, the Director of the Central Laboratory of Renmin Hospital.
References
- 1.Brennan B. Nasopharyngeal carcinoma. Orphanet J Rare Dis. 2006;1:23. doi: 10.1186/1750-1172-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afqir S, Ismaili N, Errihani H, et al. Concurrent chemoradiotherapy in the management of advanced nasopharyngeal carcinoma: current status. J Can Res Ther. 2009;5:3–7. doi: 10.4103/0973-1482.48763. [DOI] [PubMed] [Google Scholar]
- 3.Albiruni RA, Razak, Lillian L, Liu FF, Ito E, O’Sullivan B, Chan K. Nasopharyngeal carcinoma: the next challenges. Eur J Cancer. 2010;46:1967–1978. doi: 10.1016/j.ejca.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 4.Chen SM, Liu JP, Zhou JX, et al. Suppression of the notch signaling pathway by γ-secretase inhibitor GSI inhibits human nasopharyngeal carcinoma cell proliferation. Cancer Lett. 2011;306:76–84. doi: 10.1016/j.canlet.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 5.Guigay J. Advances in nasopharyngeal carcinoma. Curr Opin Oncol. 2008;20:264–269. doi: 10.1097/CCO.0b013e3282fad846. [DOI] [PubMed] [Google Scholar]
- 6.Lua HM, Pang LX, Yuan XB, et al. Concurrent chemoradiotherapy in locally advanced nasopharyngeal carcinoma: a treatment paradigm also applicable to patients in Southeast Asia. Cancer Treat Rev. 2009;35:345–353. doi: 10.1016/j.ctrv.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Kartalou M, Essigmann JM. Mechanisms of resistance to Cisplatin. Mutat Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 8.Gu F, Ma Y, Zhang Z, Zhao J, Kobayashi H, Zhang L, Fu L. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol Rep. 2010;23:671–676. doi: 10.3892/or_00000683. [DOI] [PubMed] [Google Scholar]
- 9.Wang ZW, Li YW, Ahmad A, et al. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim Biophys Acta. 2010;18:258–267. doi: 10.1016/j.bbcan.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 11.Kadesch T. Notch signaling: the demise of elegant simplicity. Curr Opin Genet. 2004;14:506–512. doi: 10.1016/j.gde.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 12.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih IM, Wang TL. Notch signaling, γ-secretase inhibitors, and cancer therapy. Cancer Res. 2007;67:1879–1882. doi: 10.1158/0008-5472.CAN-06-3958. [DOI] [PubMed] [Google Scholar]
- 14.Sarah J. Notch signaling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–687. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 15.Aleksic T, Stephan M. Gamma-secretase inhibition combined with platinum compounds enhances cell death in a large subset of colorectal cancer cells. Cell Commun Signal. 2008;6:1–13. doi: 10.1186/1478-811X-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 17.Gottesman MM. Mechanisms of cancer drug resistance. Ann Rev Med. 2002;5:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Li Y, Kong D, Ahmad A, Banerjee S, Sarkar FH. Cross-talk between miRNA and Notch signaling pathways in tumor development and progression. Cancer Lett. 2010;292:141–148. doi: 10.1016/j.canlet.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009;279:8–12. doi: 10.1016/j.canlet.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su LL, Yan-Cheng C, Dolores D. Drug transporter, P-glycoprotein (MDR1), is an integrated component of the mammalian blood-testis barrier. Int J Biochem Cell Biol. 2009;41:2578–2587. doi: 10.1016/j.biocel.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang XB. A molecular understanding of ATP-dependent solute transport by multidrug resistance-associated protein MRP1. Cancer Metastasis Rev. 2007;26:15–37. doi: 10.1007/s10555-007-9041-7. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y, Yang X, Wu Y, et al. γ-secretase inhibitor induces adipogenesis of adipose-derived stem cells by regulation of Notch and PPAR-γ. Cell Prolif. 2010;43:147–156. doi: 10.1111/j.1365-2184.2009.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borghese L, Dolezalova D, Opitz T, et al. Inhibition of Notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells. 2010;28:955–964. doi: 10.1002/stem.408. [DOI] [PubMed] [Google Scholar]
- 24.Fine RL, Chambers TC, Sachs CW. P-glycoprotein, multidrug resistance and protein kinase C. Stem Cells. 1996;14:47–55. doi: 10.1002/stem.140047. [DOI] [PubMed] [Google Scholar]
- 25.Kirschner K, Melton DW. Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res. 2010;30:3223–3232. [PubMed] [Google Scholar]
- 26.Bellmunt J, Paz-Ares L, Cuello M, et al. Gene expression of ERCC1 as a novel prognostic marker in advanced bladder cancer patients receiving cisplatin-based chemotherapy. Ann Oncol. 2007;18:522–528. doi: 10.1093/annonc/mdl435. [DOI] [PubMed] [Google Scholar]
- 27.Olaussen KA, Dunant A, Fouret P, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355:983–991. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 28.Wang QE, Milum K, Han CH, et al. Differential contributory roles of nucleotide excision and homologous recombination repair for enhancing cisplatin sensitivity in human ovarian cancer cells. Mol Cancer. 2011;10:24. doi: 10.1186/1476-4598-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nefedova Y, Daniel M, Sophia CB, William SD, Dmitry IG. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. 2008;111:2220–2228. doi: 10.1182/blood-2007-07-102632. [DOI] [PubMed] [Google Scholar]
- 30.Koduru S, Kumar R, Srinivasan S, et al. Notch-1 inhibition by Withaferin-A: a therapeutic target against colon carcinogenesis. Mol Cancer Ther. 2010;9:202–210. doi: 10.1158/1535-7163.MCT-09-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]