

Abstract
Platelet-derived growth factor-BB (PDGF-BB) has been reported to provide tropic support for neurons in the CNS. However, whether PDGF-BB regulates neurogenesis, especially in the context of HIV-associated neurological disorder and drug abuse, remains essentially unknown. In this study, we demonstrate that pretreatment of rat hippocampal neuronal progenitor cells (NPCs) with PDGF-BB restored proliferation that had been impaired by HIV Tat–cocaine via the cognate receptors. We identify the essential role of transient receptor potential canonical (TRPC) channels in PDGF-BB-mediated proliferation. Parallel but distinct ERK/CREB, phosphatidylinositol 3-kinase/Akt signaling pathways with downstream activation of mammalian target of rapamycin (mTOR)/eukaryotic translation initiation factor 4E-binding protein (4E-BP)–p70S6K and nuclear factor-κB were critical for proliferation. Blocking TRPC1 channel suppressed PDGF-mediated proliferation as well as PDGF-BB-induced ERK/CREB and mTOR/4E-BP–p70S6K activation, thereby underscoring its role in this process. In vivo relevance of these findings was further corroborated in Tat transgenic mice wherein hippocampal injection of recombinant AAV2–PDGF-B restored impaired NPC proliferation that was induced by Tat–cocaine. Together, these data underpin the role of TRPC1 channel as a novel target that regulates cell proliferation mediated by PDGF-BB with implications for therapeutic intervention for reversal of impaired neurogenesis inflicted by Tat and cocaine.
Introduction
HIV-associated neurological disorders (HANDs) comprise a range of symptomatology with varying degrees of HIV-related neuropsychiatric impairments. Drugs of abuse have been known to accelerate the incidence and progression of HANDs (Gurwell et al., 2001; Aksenov et al., 2006). Psychostimulant cocaine that is widely abused by HIV-positive individuals has been suggested to worsen HIV progression in the brain. Although the advent of antiretroviral therapy (ART) has decreased the incidence of HANDs, its prevalence is actually on a rise (González-Scarano and Martín-García, 2005). Mounting evidence indicates that brains of patients with HIV-associated CNS disease exhibit not only neuronal damage/loss but also exhibit fewer adult neural stem/progenitor cells (NPCs) in the dentate gyrus of the hippocampus. Such a defect could account for increased prevalence of behavioral deficits observed in patients with HANDs in the post-ART era.
New dentate granule cells are continuously generated from neural progenitor cells and are integrated into the existing hippocampal circuitry in the adult mammalian brain by a process termed as neurogenesis (Venkatesan et al., 2007). Adult hippocampal neurogenesis is regulated by a variety of physiological and pathological stimuli. It has been reported that HIV transactivating protein Tat, which is both released by infected cells and taken up by neighboring cells, is known to impair neurogenesis (Mishra et al., 2010). In addition to this protein, cocaine can also negatively affect the self-renewal capacity of the hippocampus by diminishing the proliferative rate of NPCs (Yamaguchi et al., 2004; Yamaguchi et al., 2005; Hu et al., 2006). These findings raise the possibility that cognitive dysfunction in the setting of HIV infection and drug abuse may, in part, be attributed to impairment of hippocampal neurogenesis.
Neurotrophic family of growth factors plays key roles in maintaining neuronal homeostasis via regulation of neurogenesis (Almeida et al., 2005; Mohapel et al., 2005). Platelet-derived growth factor (PDGF) is composed of a family of five dimeric ligands that act via two receptor tyrosine kinases, PDGF-αR and PDGF-βR (Li et al., 2000; Bergsten et al., 2001; Heldin et al., 2002). PDGF-BB has been implicated as a crucial factor in the developing postnatal rat brains (Smits et al., 1991) and has also been implicated in reversing neuronal toxicity (Yao et al., 2009a). Based on the role of PDGF-BB in the developing CNS and its increased expression in the subventricular zone during peak embryonic proliferative and migration periods (Sasahara et al., 1992), PDGF-BB can be envisioned as a promising candidate for stimulating neurogenesis.
Transient receptor potential canonical (TRPC) channels are Ca2+-permeable, nonselective cation channels formed by homomeric or heteromeric complexes of TRPC protein (Jia et al., 2007). Seven mammalian TRPC proteins (TRPC1–TRPC7) have been identified (Vazquez et al., 2004). Among these subtypes, TRPC1 has been shown to be involved in bFGF-induced self-renewal of embryonic rat NPCs (Fiorio Pla et al., 2005). Whether TRPC functions to modulate PDGF-BB-mediated neurogenesis in NPCs remains unknown.
In the current study, we show direct evidence that PDGF-BB/PDGF-R axis in NPCs may contribute to the NPC proliferation via a previously unidentified role of TRPC1.
Materials and Methods
Reagents.
Recombinant PDGF-BB was purchased from R & D Systems. PDGF-B refers to the RNA expression, whereas PDGF-BB refers to the protein expression of this gene. Tat1–72 was obtained from University of Kentucky College of Medicine (Lexington, KY). The specific phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one], the L-, N-, P-type voltage-dependent calcium channel (VDCC) blocker calcicludine, the phospholipase C (PLC) inhibitor U73122 (1-[6[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione), and the MEK1/2 inhibitor U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene] was purchased from Calbiochem. The tyrosine kinase inhibitor STI-571 (4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide) was obtained from Novartis. Cocaine, the TRPC blocker SKF96365 [1–2-(4-methoxyphenyl)-2-[3-(4-methoxyphenyl)propoxy]ethyl-1H-imidazole], the T-type VDCC blocker NiCl2, and the IκB kinase 2 (IKK2) inhibitor SC514 (3-amino-5-thiophen-3-ylthiophene-2-carboxamide) were purchased from Sigma. H89 (N-[2-(p-bromo-cinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl) and 2-ApB (2-amino-4-phosphonobutanoic acid) were obtained from Tocris Bioscience. Anti-TRPC1, -TRPC4, -TRPC5, and -TRPC6 antibodies were purchased from Alomone Labs. The primary antibodies used included the following: phosphorylated (p)-PDGF-αR–pPDGF-βR/PDGF-αR–PDGF-βR, p-ERK/ERK, p-CREB/CREB, p-Akt/Akt, p65 nuclear factor κB (NF-κB), Histone, p-mammalian target of rapamycin (mTOR)/mTOR, p-p70S6K/p70S6K, p-eukaryotic translation initiation factor 4E-binding protein (4E-BP)/4E-BP, p-S6 ribosomal protein/S6 ribosomal protein (1:200; Cell Signaling Technology), and β-actin (1:4000; Sigma).
Animals.
Pregnant rats were purchased from (Charles River Laboratories). All animals were housed under conditions of constant temperature and humidity on a 12 h light/dark cycle, with lights on at 7:00 A.M. Food and water were available ad libitum. All animal procedures were performed according to the protocols approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center. Tat transgenic mouse is a doxycycline-inducible and brain-specific mouse model developed by Drs. Avindra Nath (NIH, Bethesda, MD) and Kurt Hauser (Virginia Commonwealth University, Richmond, VA). This is a well-characterized rodent model containing a Tat gene driven by GFAP and is induced by doxycylcline.
Isolation, differentiation, and characterization of NPCs.
NPCs derived from the hippocampus of embryonic day 18 fetus were cultured in substrate-free tissue culture T75 flasks as reported previously (Tian et al., 2009). After 4–7 d, NPCs formed neurospheres and were dissociated with trypsin–EDTA for 20 min at 37°C and plated on poly-d-lysine precoated plates and were used if found >90% nestin positive (a marker for progenitor cells). Based on our previous studies, NPCs were treated with varying concentrations of PDGF-BB (20, 50, and 100 ng/ml), HIV-1 Tat (50, 100, and 200 ng/ml), and cocaine (1, 10, and 100 μm). Treatment of NPCs with pharmacological inhibitors [STI-571, 1 μm; SKF96365, 20 μm; EGTA, 2 mm; 2-ApB, 100 μm; Xest-C ([1R-(1R,4aR,11R,12aS,13S,16aS,23R,24aS)]-eicosahydro-5H,17H-1,23:11,13-diethano-2H,14H-[1,11]dioxacycloeicosino[2,3-b:12, 13-b1]dipyridine), 1μm; U73122, 1 μm; U73343 (1-[6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-2,5-pyrrolidine-dione), 1 μm; U0126, 20 μm; LY294002, 20 μm; rapamycin, 1 μm] involved pretreating cells with the respective inhibitors for 1 h, followed by exposure with Tat–cocaine and/or PDGF-BB. Forty-eight hours later, cells were examined for cell proliferation.
Cell proliferation.
Cell proliferation was measured by CyQUANT cell proliferation assay according to the protocol of the manufacturer (Invitrogen). NPCs dissociated from neurosphere were seeded in 96-well plates at a density of 104 cells per well for 2 d and were pretreated with PDGF-BB for 1 h, followed by subsequent treatment with Tat and cocaine for 48 h. Then, 200 μl of the CyQUANT GR dye/cell-lysis buffer was added into each well and incubated in the CO2 incubator for 15 min. Fluorescence intensity of each well was obtained using a Dynatech MR5000 plate counter at excitation and emission wavelengths of 480 and 520 nm, respectively.
MTT assay.
The viability of NPCs treated with cocaine and/or Tat was tested by MTT assay as described in our previous studies (Yao et al., 2009a,b).
Cytotoxicity assay.
To assess the cytotoxicity of cocaine and/or Tat, cells were lysed and assessed for lactate dehydrogenase (LDH) released into the culture medium using a nonradioactive cytotoxicity assay kit (CytoTox 96; Promega) (Peng et al., 2008).
TUNEL assay.
The in situ cell death detection kit TMR Red (Roche) was used to label apoptotic cells according to the instructions of the manufacturer as described previously (Peng et al., 2008; Yao et al., 2009b).
Measurement of [Ca2+]i.
The changes in [Ca2+]i in NPCs were monitored using Fluo-4/AM as described previously (Yao et al., 2009a,b).
Reverse-transcription PCR.
The primers for rat PDGF-αR and PDGF-βR were obtained from SABiosciences. Total RNA was extracted with Trizol reagent (Invitrogen) according to the instructions of the manufacturer and our previous report for the expression of TRPC channels (Yao et al., 2009a,b).
Western blotting.
Treated cells were lysed using the Mammalian Cell Lysis kit (Sigma) and the NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce). Equal amounts of the proteins were electrophoresed in an SDS-polyacrylamide gel (12%) under reducing conditions, followed by transfer to PVDF membranes. The blots were blocked with 5% nonfat dry milk in PBS as described previously (Yao et al., 2009a,b).
Immunocytochemistry.
For immunocytochemistry, NPCs were plated on coverslips, fixed with 4% paraformaldehyde, and permeabilized with 0.3% Triton X-100 in PBS. For BrdU detection, cells were incubated in medium containing BrdU (10 μm) for 4 h. Fixed cells were incubated in 2N HCl with 0.3% Triton X-100, followed by neutralization with 0.1 m boric acid, pH 8.0. Cells were then incubated with a blocking buffer, followed by incubation with rat anti-nestin (1:100; Millipore), rabbit anti-PDGF-αR and PDGF-βR (1:500; Cell Signaling Technology), and BrdU (1:200; Santa Cruz Biotechnology) antibodies overnight at 4°C. Secondary Alexa Fluor-488 goat anti-rat IgG and Alexa Fluor-569 goat anti-rabbit IgG were added at a 1:500 dilution for 2 h to detect nestin, PDGF-αR, and PDGF-βR, followed by mounting of cells with Vectashield onto glass slides (Vector Laboratories). Fluorescent images were acquired at room temperature on a Carl Zeiss Observer. A Z1 inverted microscope was used; images were processed using AxioVs 40 version 4.8.0.0 software (Carl Zeiss). Photographs were acquired using an AxioCam MRm digital camera.
Adenovirus infection.
NPCs were infected with adenoviral constructs according to previously reported studies (Yao et al., 2009a,b).
Short interfering RNA transfection.
Short interfering RNAs (siRNAs) targeted against PDGF-αR, PDGF-βR, TRPC1, TRPC4, TRPC5, and TRPC6 were obtained from Thermo Scientific Dharmacon RNAi Technologies (Accell SMART Pool) and transfected using the rat stem cell Nucleofector kit (Amaxa) according to the instructions of the manufacturer. The sequences of these siRNAs are listed in Table 1. Briefly, dissociated cells from neurospheres were resuspended in the transfection medium, mixed with respective siRNAs (100 nm), and electroporated, following which cells were quickly centrifuged, resuspended, and plated. The knockdown efficiency was determined by Western blotting (WB) of extracts from transfected cultures 96 h after siRNA delivery. Cells were treated with PDGF-BB for proliferation or WB analyses at 96 h after siRNA delivery.
Table 1.
siRNA sequence of rat TRPC1, TRPC4, TRPC5, and TRPC6
| Items | Sequence |
|---|---|
| Nonspecific siRNA | |
| D-001810-10-20, ON-TARGETplus Non-targeting Pool | UGGUUUACAUGUCGACUAA |
| UGGUUUACAUGUUGUGUGA | |
| UGGUUUACAUGUUUUCUGA | |
| UGGUUUACAUGUUUUCCUA | |
| TRPC1 | |
| L-080128-00-0005, ON-TARGETplus SMARTpool | GAACAUAAAUUGCGUAGAU |
| GGACUACGGUUGUCAGAAA | |
| GAAUUUAAGUCGUCUGAAA | |
| UGAACUUAGUGCUGACUUA | |
| TRPC4 | |
| L-093344-01-0005, ON-TARGETplus SMARTpool | GCCAUUAAGUACCGUCAAA |
| CUUGAAGAUUGUCGCAUUU | |
| AGUCAUUGCCUCCGAGAGA | |
| GUGGAAAUUUGCUCGAACA | |
| TRPC5 | |
| L-094467-01-0005, ON-TARGETplus SMARTpool | CGGCAUGAAUUCACGGAGU |
| GUAUAAUGGUUCUCGUCCA | |
| GCAGAGGCCUGUUCGCAAA | |
| AGAAAGAGUUUGUCGCUCA | |
| TRPC6 | |
| L-095094-00-0005, ON-TARGETplus SMARTpool | GGUCCUCGCUGUCGCCAUU |
| UAAAGGUUAUGUACGGAUU | |
| GGACCAGCAUACAUGUUUA | |
| GAAAUCAGCUGGCACACAA | |
| PDGF-Rα | |
| L-101568-01-0005, ON-TARGETplus SMARTpool | CCAGCAACGUCUCGAAUAU |
| CCAGCGAGUUUAACGUUUA | |
| CUAAGUAGAUGACGAGUUU | |
| GUUUGGAAAUGGACGGACA | |
| PDGF-Rβ | |
| L-091874-01-0005, ON-TARGETplus SMARTpool | GUAGAAAUCCUUGGGUAUA |
| GAGAAAGCCAUCAACGUUA | |
| ACAUCAAAUACGCGGACAU | |
| CAGAUUACCUCUUCGGAGU |
BrdU immunostaining.
The sections were denatured by incubation in 50% formamide in 2× SSC at 65°C. After rinses with PBS, sections were incubated with 2N HCl at 37°C, washed, and treated with 0.1 m boric acid, pH 8.5, followed by incubation with H2O2 for 10 min. After blocking, sections were incubated with mouse anti-BrdU (1:200) overnight at 4°C. Next day, sections were washed, incubated with biotinylated goat anti-mouse IgG (1:200) in immunoblocking buffer at room temperature for 1 h, and incubated with an avidin–biotin–peroxidase kit for 1 h. Horseradish peroxidase reaction product was visualized with nickel-enhanced DAB peroxidase substrate kit. Every sixth section from each animal was used to quantify BrdU-positive cells in the subgranular zone and hillus of the dentate gyrus bilaterally. The number of BrdU-positive cells per section was calculated and multiplied by 6 to obtain the total number of cells per dentate gyrus per mouse as reported previously (Garza et al., 2008).
Construction and transduction of recombinant AAV2-expressing PDGF-B in NPCs.
The rodent GFP PCR product was cloned into rAAV2–MCS–WPRE vector. The vector was packaged in AAV-293 cells (derived from HEK293 cells to produce higher viral titers) by CaPO4 transient transfection of the vector plasmid and virion production plasmids (Stratagene AAV-helperfree kit), followed by lysis of cells to recover replication-deficient virions that were purified by affinity column (ViraTrap AAV purification kit; GeneMega). Recombinant AAV2 (rAAV2) was titered for total particles by serial-dilution transfection in HEK293 and verified by quantitative-competitive PCR assay. rAAV2–GFP was then transduced into NPCs at the multiplicity of infection (2 × 105), followed by monitoring for PDGF-BB expression by imaging and WB analyses.
Microinjection of rAAV2–PDGF-B in vivo.
Ten-week-old HIV Tat transgenic mice were divided into four groups of 24 mice with each group (n = 6; male) treated with the following: (1) saline; (2) doxycycline + cocaine; (3) doxycycline + cocaine + rAAV2–PDGF-B; or (4) doxycycline + cocaine + rAAV2–GFP. In the groups AAV2–PDGF-B and AAV2–GFP, rAAV2 was microinjected into the hippocampus of mice (1 μl of 109 viral genomes/μl) using the microinjection parameters (coordinates: 2.06 mm behind bregma, 2.0 mm lateral from the sagittal midline at a depth of 1.7 mm to skull surface). To reduce the volume of inoculation and increase the precision, a 10 μl Hamilton syringe with a 33 gauge needle was used for intrahippocampal injections as described. One month later, mice were administrated doxycycline feed and cocaine injection daily for an additional 14 d. To evaluate the effect of rAAV2–PDGF-B on cell proliferation, groups of 24 mice were divided as described above, injected with BrdU, and assessed for proliferation.
Statistical analysis.
Data were expressed as mean ± SD. Significance of differences between control and samples treated with various drugs was determined by one-way ANOVA, followed by post hoc least significant difference test. Values of p < 0.05 were taken as statistically significant.
Results
PDGF-BB reverses HIV-1 Tat–cocaine-mediated impairment of proliferation of NPCs
To assess the effect of Tat–cocaine on NPC proliferation, NPCs were exposed to different concentrations of cocaine (1, 10, and 100 μm) or Tat (50,100, and 200 ng/ml) and assessed for cell proliferation. Cocaine concentrations were based on the levels found in the plasma of human volunteers (Van Dyke et al., 1976; Kalasinsky et al., 2000; Stephens et al., 2004). As shown in Figure 1, A and B, there was a concentration-dependent reduction of proliferation in the presence of both Tat–cocaine. For the physiological relevance of cocaine concentration, 10 μm cocaine and 200 ng/ml Tat was therefore chosen for all our additional studies. Treatment with heat-inactivated or mutant Tat had no effect on proliferation.
Figure 1.
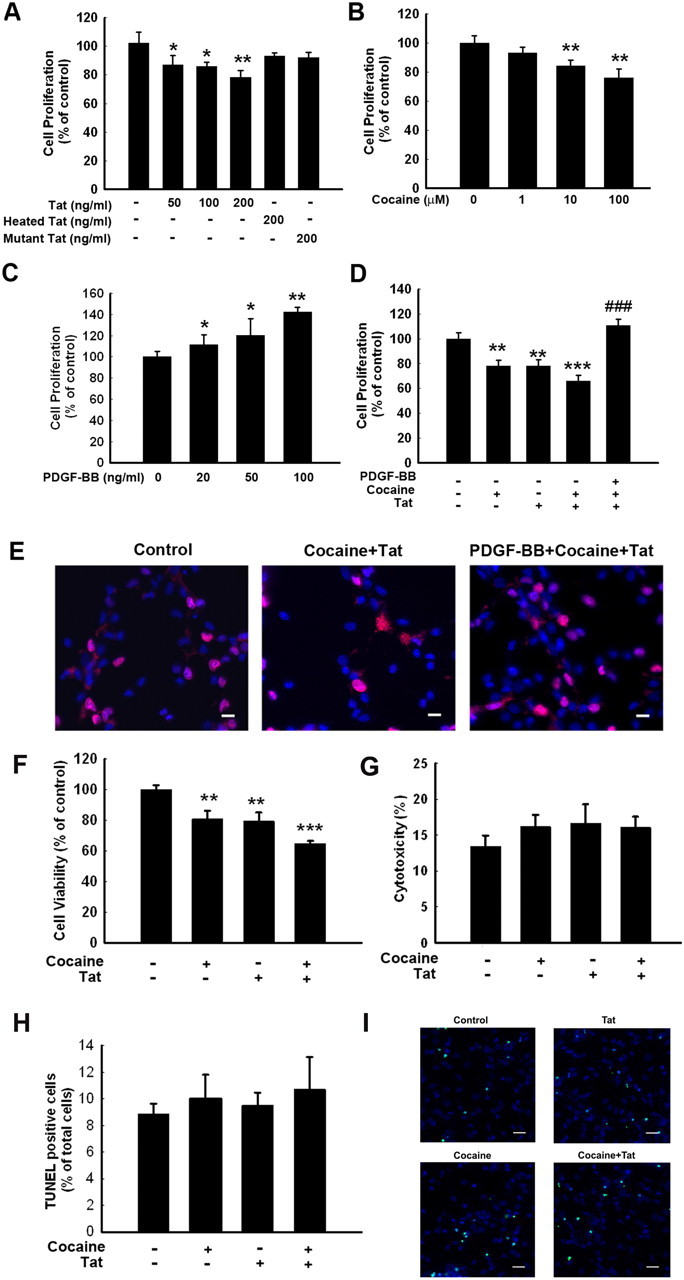
PDGF-BB reverses HIV-1 Tat–cocaine-mediated impairment of proliferation of NPCs. Effect of Tat (A), cocaine (B), and PDGF-BB (C) on NPC proliferation. All the data in these figures are presented as mean ± SD of four individual experiments. *p < 0.05, **p < 0.01 versus control group. D, PDGF-BB restored Tat–cocaine-mediated impairment of NPC proliferation. E, Representative microscopic images showing BrdU labeling in control (left), Tat–cocaine-treated (middle), and PDGF-BB plus Tat–cocaine-treated (right) NPCs. Red, BrdU; blue, DAPI. Scale bars, 10 μm. F, Effect of cocaine and/or Tat on NPC survival as determined by MTT assay. G, Effect of cocaine and/or Tat on the cytotoxicity of NPC using LDH assay. H, Effect of cocaine and/or Tat on the cytotoxicity of NPC using TUNEL assays. I, Representative microscopic images showing TUNEL-positive cells in control, Tat-treated, cocaine-treated, and cocaine–Tat-treated NPCs. Green, TUNEL; blue, DAPI. Scale bars, 20 μm. All the data in these figures are presented as mean ± SD of four individual experiments. **p < 0.01, ***p < 0.001 versus control group; ###p < 0.001 versus Tat–cocaine group.
Next, we assessed the effect of PDGF-BB on proliferation of NPCs. PDGF-BB at all the concentrations tested resulted in significant increase in proliferation of NPCs (Fig. 1C). We next examined the effect of PDGF-BB on Tat–cocaine-mediated inhibition of proliferation. Tat alone decreased NPC proliferation by 20.3%, cocaine alone decreased it by 21.2%, and in conjunction, both caused a statistically significant decrease in proliferation (almost 35%; Fig. 1D). Pretreatment of NPCs with PDGF-BB followed by exposure of cells to Tat–cocaine, however, resulted in restoration of proliferation. These findings were further validated by immunostaining using the anti-BrdU antibody (Fig. 1E).
To test whether cocaine and/or Tat also affect survival of NPCs, MTT assay was used. As shown in Figure 1F, both cocaine and Tat significantly decreased NPC survival. To exclude the possibility of the cytotoxic effects of either cocaine or Tat, exposed cells were assessed for toxicity using the LDH assay. Treatment of NPCs with either agent failed to demonstrate increased cytotoxicity (Fig. 1G). Furthermore, TUNEL analysis as an indicator of cell death revealed that cocaine and Tat did not induce cytoxicity (Fig. 1H,I). Together, these findings suggested that decreased cell proliferation induced by cocaine/Tat was not attributable to the cytotoxic effects.
Engagement of PDGF-αR and PDGF-βR is critical for PDGF-BB-mediated increased proliferation of NPCs
Because PDGF-BB mediates signaling through its receptors PDGF-αR and PDGF-βR, we first sought to examine the expression of these receptors in NPCs. Although NPCs expressed both types of receptors as evidenced by RT-PCR and WB analyses (Fig. 2A,B), there was higher abundance of PDGF-αR, which was confirmed by immunocytochemistry. PDGF-αR and PDGF-βR immunoreactivity colocalized with nestin-positive cells in NPCs (Fig. 2C). Furthermore, exogenous PDGF-BB rapidly phosphorylated PDGF-αR and PDGF-βR in NPCs as evidenced by immunoprecipitation analysis (Fig. 2D). Intriguingly, pretreatment of NPCs with the tyrosine kinase receptor antagonist STI-571 abolished PDGF-BB-mediated increase in proliferation, thus confirming the role of PDGF-BB/PDGF-R axis in this process (Fig. 2E).
Figure 2.
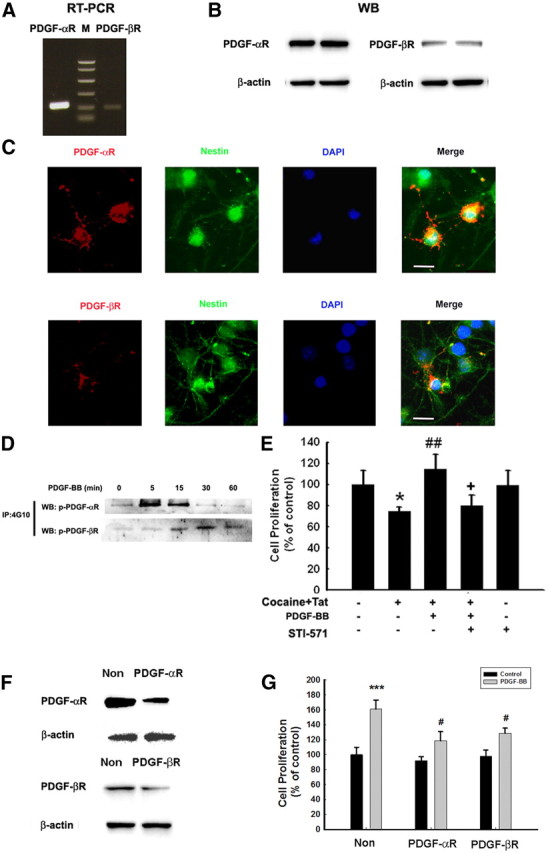
Engagement of PDGF-αR and PDGF-βR is critical for PDGF-BB-mediated increased proliferation of NPCs. RT-PCR (A) and WB (B) of PDGF-αR and PDGF-βR in NPCs; M denotes DNA marker. C, Immunostaining of PDGF-αR and PDGF-βR (red), which colocalized with nestin-positive staining (green) and DAPI (blue). Scale bars, 5 μm. D, PDGF induced PDGF-αR and PDGF-βR phosphorylation as determined by immunoprecipitation. E, STI-571 abolished PDGF-BB-mediated increase in proliferation. The data are presented as mean ± SD of four individual experiments. *p < 0.05 versus control group; ##p < 0.01 versus cocaine–Tat-treated group; +p < 0.05 versus both PDGF-BB-treated and Tat–cocaine-treated groups. F, WB analysis of whole-cell lysates from NPCs transfected with siRNAs PDGF-αR and PDGF-βR or nonsense (Non) siRNA, using antibodies specific for PDGF-αR and PDGF-βR. Data are representative of three independent experiments. G, Transfection of NPCs with siRNAs specific for PDGF-αR/PDGF-βR or nonsense siRNA abolished PDGF-BB-mediated increase of NPC proliferation. The data are presented as mean ± SD of four individual experiments. ***p < 0.001 versus control group; #p < 0.05 versus PDGF-BB-treated cells transfected with nonsense siRNA group.
It is well recognized that STI-571 is not a specific antagonist for either PDGF-αR or PDGF-βR, because it can inhibit other tyrosine kinases as well. As an alternative approach, we thus sought to knock down the respective receptor expression in NPCs using the siRNA strategy. PDGF-αR and PDGF-βR siRNAs abrogated the respective expression of the two receptors in NPCs (Fig. 2F), while also partially abrogating PDGF-BB-mediated increased proliferation of NPCs (Fig. 2G).
TRPC channels are critical for PDGF-BB-mediated increased proliferation of NPCs
To study the effects of PDGF-BB on the intracellular Ca2+ level, we measured Ca2+ influx using Fluo-4/AM imaging. Exposure of NPCs to PDGF-BB triggered rapid and sustained intracellular Ca2+ elevation in NPCs (Fig. 3A). This response was inhibited in NPCs pretreated with the pan TRPC inhibitor SKF96365, thus underscoring its role (Fig. 3B).
Figure 3.

TRPC channels contribute to PDGF-BB-increased Ca2+ influx. A, NPCs loaded with Fluo-4 [Ca2+]i-sensitive fluorophores before and after PDGF-BB treatment were recorded within a single field using a Fluoview 300 confocal microscope (right; numbers in the panels indicate time in seconds) and differential interference contrast (left). B, Changes in fluorescence amplitude (Fluo-4/Fura Red) in NPCs exposed to PDGF-BB in the absence or presence of the TRPC blocker SKF96365 (20 μm). C, The TRPC blocker SKF96365 abolished PDGF-BB-mediated increase in proliferation. The data are presented as mean ± SD of four individual experiments. *p < 0.05 versus control group; ##p < 0.01 versus Tat–cocaine-treated group; ++p < 0.01 versus both PDGF-BB-treated and Tat–cocaine-treated groups. D, Cell proliferation of NPCs exposed to Tat–cocaine and/or PDGF-BB in the absence or presence of indicated drugs (EGTA, 2 mm; 2-ApB, 100 μm; Xest-C, 1 μm; U73122, 1 μm; U73343, 1 μm). All the data in these figures are presented as mean ± SD of four individual experiments. *p < 0.05 versus control group; ###p < 0.001 versus Tat-treated group; +++p < 0.001 versus both PDGF-BB-treated and Tat–cocaine-treated group. E, Expression of TRPC subtypes in NPCs as detected by RT-PCR. M denotes DNA marker. F, WB analysis of whole-cell lysates from NPCs transfected with TRPC1, TRPC4, TRPC5, TRC6 or nonsense (Non) siRNAs using antibodies specific for TRPC1, TRPC4, TRPC5, or TRPC6. Data are representative of three independent experiments. G, NPCs transfected with TRPC1, TRPC4, TRPC5, or TRPC6 siRNAs were monitored for PDGF-BB-mediated proliferation. H, NPCs were knocked down for three TRPC channels with the expression of only one channel and were then monitored for PDGF-BB-mediated proliferation. The data are presented as mean ± SD of four individual experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus control group; ##p < 0.01 versus PDGF-BB-treated cells transfected with nonsense siRNA group.
Based on the premise that Ca2+ mediates proliferation and that TRPC functions as Ca2+ influx channels, it was rationalized that Ca2+ influx through TRPC channels is a prerequisite for increased NPC proliferation mediated by PDGF-BB. NPCs were pretreated with the TRPC blocker and PDGF-BB and subsequently assessed for proliferation in the presence of Tat–cocaine. Pretreatment of NPCs with SKF96365 resulted in failure of PDGF-BB to rescue Tat–cocaine-mediated inhibition of proliferation (Fig. 3C). Additional validation of the role of extracellular Ca2+ influx in PDGF-BB mediated proliferation of NPCs was supported by pretreating the cells with EGTA, which resulted in suppression of NPC proliferation mediated by PDGF-BB (Fig. 3D).
PDGF-BB, on activating its receptor, is known to stimulate PLC, resulting in inositol triphosphate (IP3)-dependent release of Ca2+ from intracellular stores as well from extracellular sources. Interference of the PLC–IP3 pathway thus ought to suppress the increased NPC proliferation exerted by PDGF-BB. As expected, pretreatment of NPCs with the PLC inhibitor U73122 but not its inactive analog U73343 abolished PDGF-mediated increase in proliferation in Tat and cocaine-treated NPCs. The effect of PDGF-BB on proliferation was also dependent on IP3 receptor activation, because exposure of NPCs to antagonists specific for IP3 receptor, 2-ApB and Xest-C, resulted in suppression of PDGF-BB-mediated increase in proliferation (Fig. 3D). All of inhibitors alone failed to inhibit cell proliferation significantly.
Because TRPC family comprises of seven subtypes, it was important to first investigate the expression pattern of these subtypes in NPCs. NPCs expressed TRPC1, TRPC4, TRPC5, and TRPC6 but not TRPC2, TRPC3, or TRPC7 (Fig. 3E). To ascertain the TRPC subtype(s) critical for PDGF-BB-mediated increased NPC proliferation, each of the subtypes was individually downregulated using the specific siRNAs, followed by assessment of proliferation. siRNA against TRPC1, TRPC4, TRPC5, and TRPC6 suppressed the expression of the respective TRPC subtype as expected (Fig. 3F); however, TRPC1, but not any of the other TRPC siRNAs, alleviated PDGF-BB-mediated proliferation of NPCs (Fig. 3G). To specifically confirm the role of TRPC1 in PDGF-BB-mediated cell proliferation, we took the approach of knocking down the three TRPC channels together while leaving only one TRPC effective. As shown in Figure 3H, only expression of TRPC1 but not TRPC4, TRPC5, and TRPC6 was essential for PDGF-BB-mediated cell proliferation.
Involvement of PI3K in TRPC-mediated calcium influx
The activation of TRPC channels can be regulated via multiple mechanisms, including hydrolysis of phosphoinositide (Kwon et al., 2007). A recent study provides evidence for regulation of TRPC-dependent Ca2+ influx by phosphatidylinositol (3,4,5)-triphosphate (PIP3). Consistent with previous findings (Henle et al., 2011), pretreatment of NPCs with the PI3K inhibitor LY294002 significantly attenuated Ca2+ entry induced by PDGF-BB (Fig. 4A). To assess the role of PI3K in specifically regulating TRPC-dependent Ca2+ influx, NPCs were also pretreated with the other VDCC blockers [calcicludine (200 nm, an L-, N-, P-type VDCC blocker) and NiCl2 (50 μm, a T-type VDCC); Sigma-Aldrich] to specifically isolate TRPC-dependent Ca2+ influx from voltage-dependent Ca2+ influx, followed by treatment of cells with the PI3K inhibitor. As shown in Figure 4B, VDCC blocker decreased PDGF-BB-induced Ca2+ influx, and this was further decreased in cells treated with LY294002, thereby underpinning the role of PI3K in TRPC-dependent Ca2+ influx.
Figure 4.
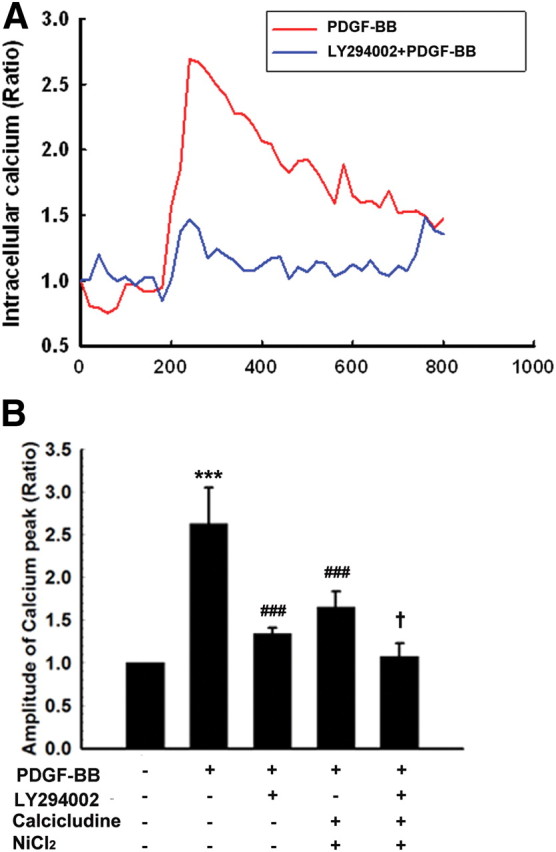
Involvement of PI3K in TRPC-mediated Ca2+ influx. A, Changes in fluorescence amplitude (Fluo-4/Fura Red) in NPCs exposed to PDGF-BB in the absence or presence of LY294002 (1 μm). B, Changes in fluorescence amplitude (Fluo-4/Fura Red) in NPCs exposed to PDGF-BB in the absence or presence of the indicated drugs (LY294002, 1 μm; calcicludine, 200 nm[scap]; NiCl2, 50 μm). Voltage-dependent channel blockers include calcicludine (L-, N-, P-type VDCC) and NiCl2 (T-type VDCC). All the data are presented as mean ± SD of four individual experiments. ***p < 0.001 versus control group; ###p < 0.001 versus PDGF-treated group; +p < 0.05 versus both PDGF-BB-treated and voltage-dependent channel blocker (calcicludine and NiCl2)-treated groups.
PDGF-BB-mediated activation of TRPC channels involves activation of ERK/CREB signal
The MAPK pathway has been demonstrated to play a crucial role in NPC proliferation (Learish et al., 2000). It was therefore of interest to examine the effect of PDGF-BB on ERK and its downstream transcription factor CREB in NPCs. Exposure of NPCs to PDGF-BB resulted in sustained and time-dependent activation of ERK/CREB (Figs. 5A, 6A). Having determined the role of TRPC channels in PDGF-BB-mediated cell proliferation, we next wanted to examine the role of these channels in PDGF-BB-mediated activation of ERK/CREB. Pretreatment of NPCs with SKF96365 significantly attenuated PDGF-BB-induced ERK/CREB phosphorylation, thereby suggesting that PDGF-BB-mediated activation of ERK/CREB involved TRPC (Figs. 5B, 6B). The functional role of PDGF-BB-induced ERK/CREB activation was further corroborated using proliferation assays, wherein PDGF-BB failed to exert proliferative effect in cells pretreated with ERK inhibitor (Fig. 5C), dominant-negative (DN) MEK (Fig. 5D), or PKA inhibitor (Fig. 6C). Similar to the role of TRPC1 in PDGF-BB-mediated proliferation, transfection of NPCs with siRNA for TRPC1 resulted in abrogation of PDGF-mediated activation of ERK/CREB (Figs. 5E, 6D). To specifically confirm the role of TRPC in PDGF-BB-mediated ERK phosphorylation, we again sought the approach of knocking down the three TRPC channels together while leaving only one TRPC effective. Expression of TRPC1, but not TRPC4, TRPC5, and TRPC6, was essential for PDGF-BB-mediated phosphorylation of ERK (Fig. 5F) and CREB (Fig. 6E).
Figure 5.

TRPC channels are involved in PDGF-BB-induced activation of the ERK pathway. A, PDGF-BB induced time-dependent phosphorylation of ERK. B, Representative immunoblot of NPCs exposed to PDGF-BB in the presence of the TRPC blocker SKF96365 (20 μm) was monitored for ERK activation. Densitometric analysis of p-ERK/ERK from four individual experiments. **p < 0.01 versus control group; ##p < 0.01 versus PDGF-BB group. C, Pretreatment of NPCs with the MEK inhibitor U0126 (10 μm) for 1 h significantly attenuated the effect of PDGF-BB on the restoration of Tat–cocaine-mediated impairment of proliferation. Data are presented as mean ± SD of four individual experiments. **p < 0.01 versus control group; ##p < 0.01 versus Tat–cocaine-treated group; +p < 0.05 versus both PDGF-BB-treated and Tat–cocaine-treated groups. D, PDGF-BB failed to restore impaired proliferation induced by Tat–cocaine in the presence of DN MEK but not in the wild-type group. Data are presented as mean ± SD of four individual experiments. ***p < 0.001 versus control group; ##p < 0.01 versus Tat–cocaine-treated group. E, Representative immunoblot of PDGF-BB-mediated ERK phosphorylation in NPCs transfected with TRPC1, TRPC4, TRPC5, or TRPC6 siRNAs (top), followed by densitometric analyses from three individual experiments (bottom). F, NPCs were knocked down for three TRPC channels with the expression of only one channel and were then monitored for PDGF-BB-mediated ERK phosphorylation (top), followed by densitometric analyses from three individual experiments (bottom). *p < 0.05, **p < 0.01, ***p < 0.001 versus control group; #p < 0.01 versus PDGF-BB-treated cells transfected with nonsense (Non) siRNA group.
Figure 6.

TRPC channels are involved in PDGF-BB-induced activation of the CREB pathway. A, PDGF-BB induced time-dependent phosphorylation of CREB. B, Representative immunoblot of NPCs exposed to PDGF-BB in the presence of the TRPC blocker SKF96365 (20 μm) was monitored for CREB activation. Densitometric analysis of p-CREB/CREB from four individual experiments. *p < 0.05, versus control group; #p < 0.05, versus PDGF-BB group. C, Pretreatment of NPCs with the PKA inhibitor H89 (10 μm) for 1 h significantly attenuated the effect of PDGF-BB on the restoration of Tat–cocaine-mediated impaired proliferation. Data are presented as mean ± SD of four individual experiments. **p < 0.01 versus control group; ##p < 0.01 versus Tat–cocaine-treated group; ++p < 0.01 versus both PDGF-BB-treated and Tat–cocaine-treated groups. D, Representative immunoblot of PDGF-BB-mediated CREB phosphorylation in NPCs transfected with TRPC1, TRPC4, TRPC5, or TRPC6 siRNAs (top), followed by densitometric analyses from three individual experiments (bottom). E, NPCs were knocked down for three TRPC channels with the expression of only one channel and were then monitored for PDGF-BB-mediated CREB phosphorylation (top), followed by densitometric analyses from three individual experiments (bottom). **p < 0.01, ***p < 0.001 versus control group; #p < 0.01 versus PDGF-BB-treated cells transfected with nonsense (Non) siRNA group.
TRPC channels are not required for PDGF-BB-induced Akt/NF-κB activation
In addition to the activation of the MEK/ERK pathway, Akt pathway also is known to play a critical role in proliferation (Peltier et al., 2007). To examine the role of this pathway in PDGF-BB-mediated proliferation, cell lysates were examined for phosphorylation of Akt. After exposure of NPCs to PDGF-BB, there was an enhanced and sustained activation of Akt and its downstream mediator NF-κB (Figs. 7A, 8A). Intriguingly, pretreatment of NPCs with SKF96365 did not attenuate PDGF-BB-induced Akt/NF-κB activation (Figs. 7B, 8B). The functional role of PDGF-BB-induced Akt/NF-κB activation in mediating cell proliferation was corroborated in cells pretreated with either PI3K (Fig. 7C) or IKK2 (Fig. 8C) inhibitors. The role of Akt and NF-κB in PDGF-BB-mediated cell proliferation was further validated in cells transduced with either a DN Akt (Fig. 7D) or mutant NF-κB (Fig. 8D) construct. Unlike the involvement of TRPC1 in the activation of the ERK/CREB pathway, PDGF-BB-mediated activation of Akt/NF-κB, although critical for NPC proliferation, did not involve TRPC1, as evidenced by the failure of siRNA to suppress activation of the latter pathway (Figs. 7E, 8E). To specifically confirm the role of TRPC in PDGF-BB-mediated activation of Akt/NF-κB, cells were simultaneously knocked down for the three TRPC channels together while leaving only one TRPC effective. None of the combinations of TRPC knockdown had any effect on PDGF-BB-mediated activation of Akt (Fig. 7F) or NF-κB (Fig. 8F). These findings thus lead us to speculate that PDGF-BB-mediated activation of Akt/NF-κB pathway was independent of TRPC activation. Furthermore, treatment of NPCs with LY294002 attenuated ERK phosphorylation induced by PDGF-BB, but reciprocal inhibition of MEK by U0126 failed to exert any effect on Akt phosphorylation induced by PDGF-BB (Fig. 7G).
Figure 7.

TRPC channels are not involved in PDGF-BB-induced activation of the Akt pathway. A, PDGF-BB induced time-dependent activation of Akt. B, Representative immunoblot of NPCs exposed to PDGF-BB in the presence of the TRPC blocker SKF96365 (20 μm) was monitored for Akt activation. Densitometric analysis of p-Akt/Akt from four individual experiments. **p < 0.01 versus control group. C, Pretreatment of NPCs with the PI3K inhibitor LY294002 (1 μm) for 1 h significantly attenuated the effect of PDGF-BB on the impaired proliferation induced by Tat–cocaine. Data are presented as mean ± SD of four individual experiments. *p < 0.05 versus control group; #p < 0.05 versus Tat–cocaine-treated group; +p < 0.05 versus both PDGF-BB-treated and Tat–cocaine-treated group. D, PDGF-BB failed to restore impaired proliferation induced by Tat–cocaine in the presence of DN Akt but not in the wild-type (WT) group. Data are presented as mean ± SD of four individual experiments. ***p < 0.001 versus control group; #p < 0.05 versus Tat–cocaine-treated group. E, Representative immunoblot of PDGF-BB-mediated Akt activation in NPCs transfected with TRPC1, TRPC4, TRPC5, or TRPC6 siRNAs (top), followed by densitometric analyses from three individual experiments (bottom). F, NPCs were knocked down for three TRPC channels with the expression of only one channel and were then monitored for PDGF-BB-mediated Akt phosphorylation (top), followed by densitometric analyses from three individual experiments (bottom). *p < 0.05, **p < 0.01, ***p < 0.001 versus control group. G, Effect of LY294002 or U0126 on ERK and Akt phosphorylation induced by PDGF-BB (left). Densitometric analyses of PDGF-mediated phosphorylation of ERK and Akt in NPCs pretreated with LY294002 (1 and 10 μm) or U0126 (10 μm) from a representative immunoblot from three individual experiments (right). ***p < 0.001 versus control group; ##p < 0.01, ###p < 0.001 versus PDGF-BB-treated group. Non, Nonsense.
Figure 8.

TRPC channels are not involved in PDGF-BB-induced activation of the NF-κB pathway. A, PDGF-BB induced time-dependent activation of NF-κB. B, Representative immunoblot of NPCs exposed to PDGF-BB in the presence of the TRPC blocker SKF96365 (20 μm) was monitored for NF-κB activation. Densitometric analysis of NF-κB/Histone from four individual experiments. *p < 0.05, versus control group. C, Pretreatment of NPCs with the IκB inhibitor SC514 (5 μm) for 1 h significantly attenuated the effect of PDGF-BB on impaired proliferation induced by Tat–cocaine. Data are presented as mean ± SD of 4 individual experiments. **p < 0.01 versus control group; ##p < 0.01 versus Tat–cocaine-treated group; +p < 0.05 versus both PDGF-BB-treated and Tat–cocaine-treated groups. D, PDGF-BB failed to restore impaired proliferation induced by Tat–cocaine in the presence of mutant p65 but not in the wild-type (WT) group. Data are presented as mean ± SD of four individual experiments. ***p < 0.001 versus control group; #p < 0.05 versus Tat–cocaine-treated group. E, Representative immunoblot of PDGF-BB-mediated NF-κB activation in NPCs transfected with TRPC1, TRPC4, TRPC5, or TRPC6 siRNAs (top), followed by densitometric from three individual experiments (bottom). F, NPCs were knocked down for three TRPC channels with the expression of only one channel and were then monitored for PDGF-BB-mediated activation of NF-κB (top), followed by densitometric analyses from three individual experiments (bottom). **p < 0.01, ***p < 0.001 versus control group. Non, Nonsense.
PDGF-BB-mediated activation of TRPC channels involves the mTOR/4E-BP and p70S6K pathway
Intracellular Ca2+ elevations have been implicated in the activation of ERK and mTOR pathways (Xu et al., 2011). Based on the activation of the ERK pathway by PDGF-BB, we next sought to examine whether the mTOR/4E-BP and p70S6K pathways played a role in this process. mTOR and its downstream mediators 4E-BP and p70S6K/S6 ribosomal protein (S6) were phosphorylated time dependently in the presence of PDGF-BB (Fig. 9A). The functional implication of PDGF-BB-induced mTOR activation in NPC proliferation was further corroborated using proliferation assays, wherein PDGF-BB failed to induce proliferation in NPCs pretreated with the mTOR inhibitor rapamycin (Fig. 9B). These findings underpinned the role of mTOR in PDGF-BB-mediated NPC proliferation.
Figure 9.

TRPC channels are involved in PDGF-BB-induced activation of mTOR/4E-BP–p70S6K pathways. A, PDGF-BB induced time-dependent phosphorylation of mTOR/4E-BP–p70S6K/S6. B, Pretreatment of NPCs with the mTOR inhibitor rapamycin (1 μm) for 1 h significantly attenuated the effect of PDGF-BB in restoring impaired proliferation induced by Tat–cocaine. Data are presented as mean ± SD of four individual experiments. **p < 0.01 versus control group; ##p < 0.01 versus Tat–cocaine-treated group; +p < 0.05 versus both PDGF-BB-treated and Tat–cocaine-treated groups. C, Representative immunoblot of NPCs exposed to PDGF-BB in the presence of the TRPC blocker SKF96365 (20 μm) or the MEK inhibitor U0126 (10 μm) monitored for mTOR/4E-BP–p70S6K activation (left). Densitometric analysis of mTOR/4E-BP and p70S6K from four individual experiments (right). **p < 0.01, ***p < 0.001 versus control group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus PDGF-BB group. Representative immunoblot of PDGF-mediated mTOR (D), 4E-BP (E), and p70S6K (F) phosphorylation in NPCs transfected with TRPC1, TRPC4, TRPC5, or TRPC6 siRNAs (top). Densitometric analysis of PDGF-BB-mediated phosphorylation of mTOR/4E-BP–p70S6K6 in NPCs transfected with TRPC1, TRPC4, TRPC5, or TRPC6 from three individual experiments (bottom). *p < 0.05, **p < 0.01, ***p < 0.001 versus control group; #p < 0.05, ###p < 0.001 versus PDGF-BB-treated cells transfected with nonsense (Non) siRNA group.
To further unravel the role of TRPC channels in PDGF-BB-mediated activation of the mTOR/4E-BP and p70S6K pathways, NPCs were pretreated with the TRPC blocker and assessed for expression of the activation mediators. Pretreatment of cells with SKF96365 markedly attenuated PDGF-BB-induced phosphorylation of mTOR/4E-BP and p70S6K (Fig. 9C).
Because TRPC was involved in both ERK and mTOR activation, we next wanted to assess the relationship between ERK and mTOR. Intriguingly, pharmacological blocking of ERK activation by U0126 inhibited PDGF-BB-mediated mTOR/4E-BP–p70S6K phosphorylation. In agreement with the reported finding by Bodine et al. (2001), our results also confirmed the link between Akt and mTOR/4E-BP–p70S6K phosphorylation, as evidenced by the fact that PI3K inhibitor significantly inhibited mTOR/4E-BP–p70S6K phosphorylation induced by PDGF-BB (data not shown). To confirm the role of specific TRPC channels in the mTOR pathway, NPCs were transfected with the respective TRPC siRNAs and assessed for expression of the activated signaling mediators. NPCs transfected with TRPC1 siRNA exhibited attenuation of mTOR/4E-BP–p70S6K phosphorylation induced by PDGF-BB (Fig. 9D–F). These results implicate the role of TRPC1 channel in PDGF-BB-mediated activation of mTOR/p-P70S6/4E-BP.
In vivo restoration of Tat–cocaine-mediated impairment of neurogenesis by PDGF-BB
To examine the relevance of PDGF-BB-mediated proliferation of NPCs in vivo, we assessed the proliferative potential of microinjected rAAV2–PDGF-B in the hippocampi of Tat transgenic mice that were administered cocaine for 14 d. Having determined the successful expression of the rAAV2–PDGF-B constructs (Fig. 10A–C), the next step was to test the efficacy of rAAV2–GFP/PDGF-B transduction in vivo. Expression of GFP was primarily restricted within the hippocampus (Fig. 10D), and, as expected, there was enhanced expression of PDGF-BB in the hippocampi of rAAV2–PDGF-B-injected mice compared with mice injected with AAV2–GFP (Fig. 10E). Four weeks after adenoviral construct injections (to ensure optimal expression of the transgene; our unpublished observations), mice were put on a doxycycline feed (for induction of Tat in the CNS) and also administered cocaine daily for 14 d, followed by assessment of BrdU-positive cells in the hippocampi. There was decreased proliferation in doxycycline-fed, cocaine-injected mice as evidenced by decreased BrdU-positive cells compared with controls (saline-injected and normal-fed diet) (Fig. 10F). Decreased proliferation induced by Tat and cocaine was ameliorated in mice pretreated with AAV2–PDGF-B (quantified in Fig. 10G), thereby suggesting the role of PDGF-BB in restoring impaired NPC proliferation.
Figure 10.
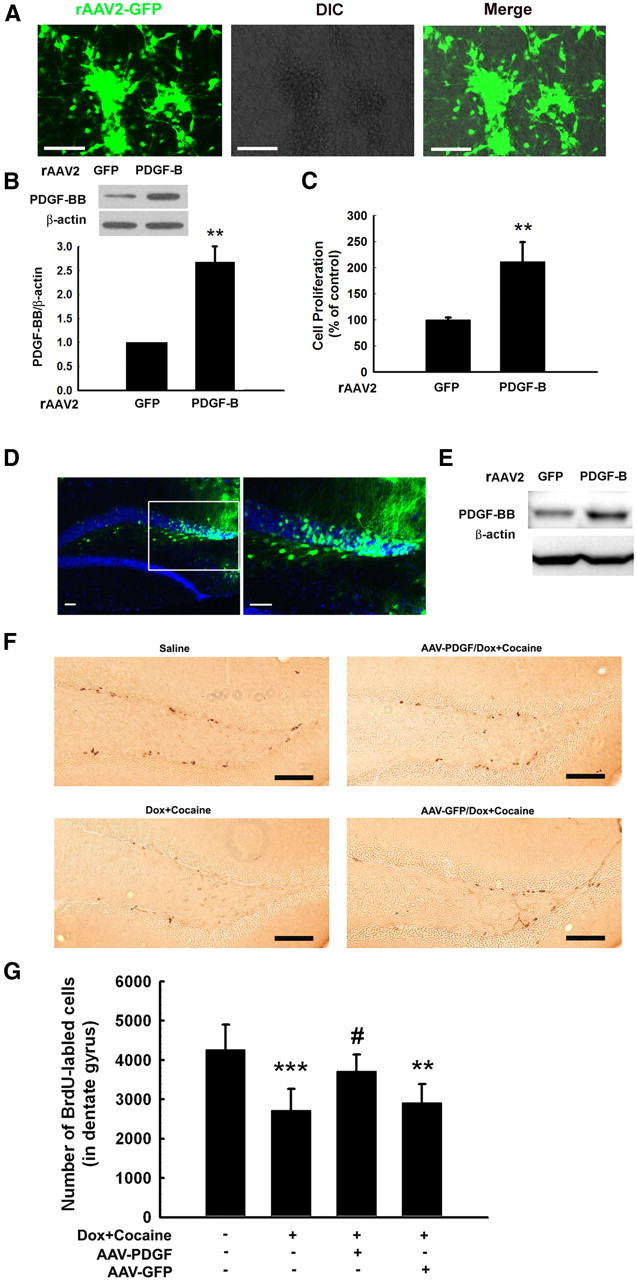
PDGF-BB ameliorates Tat–cocaine-mediated impairment of neurogenesis in vivo. A, GFP protein expression in NPCs transduced with rAAV2–GFP. Representative pictures were taken 4 d after transduction. Scale bars, 80 μm. B, PDGF-BB expression in NPCs transduced with rAAV2–GFP or rAAV2–PDGF-B. C, Effect of rAAV2–PDGF-B on proliferation of NPCs from four individual experiments. **p < 0.01 versus rAAV2–GFP-transduced NPC group. D, Microinjection of rAAV2–GFP into the hippocampus. rAAV2–GFP expression in the hippocampus of mice 1 week after microinjection. Slides were costained with DAPI. Right is a higher magnification of the inset in the left. Scale bars, 40 μm. E, PDGF-BB expression was increased in hippocampus microinjected with rAAV2–PDGF-B compared with rAAV2–GFP. F, Representative hippocampal sections from four different groups of mice administrated doxycycline feed and cocaine in the presence or absence of microinjected rAAV2–PDGF-B or rAAV2–GFP were examined for BrdU staining. Scale bars, 100 μm. G, Quantification of BrdU-positive cells in the hippocampus of doxycycline (Dox)–cocaine-treated group of mice compared with the saline controls. Microinjection with rAAV2–PDGF-B, but not rAAV2–GFP, resulted in restoration of NPC proliferation in the hippocampus. **p < 0.01, ***p < 0.001 versus saline group; #p < 0.05 versus Tat–cocaine-treated group.
Discussion
It is well recognized that new dentate granule cells are continuously generated from neural progenitor cells and are integrated into the existing hippocampal circuitry in the adult mammalian brain through an orchestrated process termed adult neurogenesis (Venkatesan et al., 2007). Neurogenesis is regulated by a variety of physiological and pathological stimuli. Both viral protein and drugs of abuse, such as cocaine, can negatively affect the self-renewal capacity of the hippocampus by diminishing the proliferative rate of NPCs (Yamaguchi et al., 2004, 2005; Hu et al., 2006). These findings thus raise the possibility that cognitive impairment attributable to comorbid conditions, such as HIV infection and drug abuse, may in part be attributed to impaired hippocampal neurogenesis. Intriguingly, patients with HANDs manifest fewer adult NPCs in the dentate gyrus compared with either the non-infected or HIV-positive subjects without dementia as reported by Krathwohl et al. (2004).
PDGF is composed of a family of five dimeric ligands assembled from four gene products (PDGF-A–PDGF-D) that act via two receptor tyrosine kinases, PDGF-αR and PDGF-βR (Li et al., 2000; Bergsten et al., 2001; Heldin et al., 2002). Intriguingly, PDGF-BB has been implicated as a crucial factor in the developing postnatal rat brains (Smits et al., 1991). Furthermore, PDGF-BB pretreatment is known to induce striatal neurogenesis in a parkinsonian rat model of 6-hydroxydopamine lesions (Mohapel et al., 2005). The novel finding of this report is that PDGF-BB via binding to its cognate receptors was able to rescue Tat–cocaine-mediated impairment of NPC proliferation both in vitro and in vivo.
Another novel finding of this study is the role of TRPC 1 in PDGF-BB-mediated increased proliferation against Tat–cocaine, thereby lending credence to previous reports indicating the involvement of TRPC signaling in neurogenesis (Fiorio Pla et al., 2005; Weick et al., 2009). Consistent with the previous reports demonstrating the colocalization of TRPC (TRPC1, TRPC4, TRPC5, and TRPC6) with the progenitor cells in hippocampus (De March et al., 2006; Martorana et al., 2006), our findings also provide evidence that all these subtypes were expressed in NPCs. The present study identified a novel molecular target, TRPC1, underlying the restoration of PDGF-BB-mediated neurogenesis. These findings are in agreement with a previous study indicating that bFGF-mediated Ca2+ increase and cell proliferation were significantly reduced in TRPC1 knock-out animals (Fiorio Pla et al., 2005).
PDGF is known to exerts its action by triggering [Ca2+]i transients in neuronal precursor cells (Cuddon et al., 2008), but the mechanism of action remains less clear. Herein, we report that PDGF-BB induced [Ca2+]i elevations through activation of TRPC1. The mammalian TRPC channels can be activated by G-protein-coupled receptors and RTKs (receptor tyrosine kinases) (Clapham, 2003). PDGF-R belongs to the receptor tyrosine kinase family and is known to activate PLC, leading to hydrolysis of phosphatidylinositol (4,5)-bisphosphate into membrane-bound diacyglycerol and soluble IP3. Generation of IP3 results in IP3 receptor-mediated release of Ca2+ from intracellular stores and Ca2+ influx extracellularly (White et al., 2005). In addition to the role of IP3 in regulating activation of TRPC, Lu et al. (1998) have also shown that activation of PI3K leads to generation of PIP3-mediated Ca2+ influx via the TRPC channels in platelets and Jurkat and mast cells (Ching et al., 2001). A key finding here is that PDGF-BB-induced influx of Ca2+ via TRPC activation involved PI3K. As was evident from our findings, PDGF-BB-mediated activation of PI3K involved PIP3-dependent activation of TRPC1, resulting in influx of Ca2+ and ensuing NPC proliferation.
Using both pharmacological and genetic approaches, we demonstrated activation of parallel but distinct cell signaling pathways, including ERK/CREB, Akt/mTOR/4E-BP–p70S6K, and Akt/NF-κB in PDGF-BB-mediated proliferation. Interestingly, blocking TRPC1 resulted in suppression of PDGF-BB-induced ERK/CREB and mTOR/4E-BP–p70S6K activation but had no impact on Akt/NF-κB activation. The findings reported here on PDGF-BB-mediated activation of the ERK/CREB pathway are consistent with several lines of published reports, specifically, the study wherein activation of TRPC3 and TRPC6 by the growth factor BDNF resulted in stimulation of two signaling pathways: Ca2+/Ras/MEK/ERK and Ca2+/CaM/CaMK, intersecting at CREB (Jia et al., 2007; Yao et al., 2009a,b).
In addition to activation of the mTOR/4E-BP–p70S6K signaling pathway by PI3K, our findings also underpin the role of TRPC1 channel in activating this pathway. Our data, however, are in disagreement with a report describing increased mTOR phosphorylation in lysates of heart tissue isolated from TRPC1 knock-out mice after the transverse aortic constriction operation compared with the wild-type controls (Seth et al., 2009). This inconsistency could be attributable to a compensatory response in the TRPC1 knock-out mouse model because mTOR may positively affect the protein translation of other TRPC channels or proteins involved in the TRPC/PI3K network (Vollenbröker et al., 2009). Our results suggest that the ERK pathway, which lies downstream of TRPC, could be involved in the regulation of mTOR. We demonstrate a crosstalk between MEK/ERK and mTOR pathways as evidenced by the fact that MEK inhibitor was capable of inhibiting PDGF-BB-induced phosphorylation of mTOR/4E-BP–p70S6K. This is in agreement with a previous report indicating ERK pathway-mediated regulation of mTOR activation through TCS2 phosphorylation (Ma et al., 2005).
Another interesting finding herein was the observation that inhibition of PI3K/Akt resulted in reversal of PDGF-BB-mediated NPC proliferation, thereby highlighting the potential role of this pathway. The role of the Akt pathway was further confirmed using a loss-of-function approach by transfecting NPCs with a DN construct of Akt. Our results are in agreement with previous reports that PI3K/Akt transduces intracellular signals that regulate NPC proliferation (Peltier et al., 2007). Interestingly, unlike the TRPC-mediated activation of ERK/CREB, PDGF-BB-mediated activation of Akt and its downstream NF-κB was independent of TRPC1 channel. Activation of NF-κB plays a key role in enhancing NPC proliferation after exposure to a wide array of stimuli (Piotrowska et al., 2006; Kaus et al., 2010). Our findings are consistent with a report indicating Akt-mediated transduction of signal via activation of NF-κB in toll-like receptor-mediated modulation of adult hippocampal neurogenesis (Rolls et al., 2007).
To investigate the relevance of PDGF-BB-mediated cell proliferation in vivo, a genetic approach using the inducible Tat transgenic mice was used. Microinjection of rAAV2–PDGF-B into the hippocampi of mice that were induced to express Tat in the CNS and that were also administered cocaine for 14 d resulted in restoration of impaired NPC proliferation, thus confirming the role of PDGF-BB in NPC proliferation. These findings are in agreement with the previous reports describing the role of PDGF-BB in induction of striatal neurogenesis in adult rats with 6-hydroxydopamine lesions (Mohapel et al., 2005).
In summary, activation of the PDGF-BB/PDGF-R axis resulted in stimulation of PI3K and PLC/IP3 receptor pathways, leading to activation of TRPC channels, which in turn resulted in Ca2+ influx, culminating in activation of ERK/CREB and mTOR/4E-BP–p70S6K (Fig. 11). Together, our findings suggest that, although the two pathways involved in PDGF-BB-mediated restoration of impaired proliferation of NPCs operate independent of each other, their combined actions are necessary for the observed proliferation potential of PDGF-BB. A better understanding of these molecular pathways could be critical for the development of therapeutic interventions aimed at targeting cognitive impairment observed in cocaine-abusing HIV-infected individuals.
Figure 11.

Schematic illustration demonstrating putative signaling pathways involved in PDGF-BB-mediated increase in proliferation of NPCs. PDGF-BB-mediated engagement of the PDGF-αR and PDGF-βR stimulated the PLC/IP3 receptor and PI3K pathways, which in turn, activated TRPC1 channels, resulting in Ca2+ influx transients, culminating in activation of ERK/CREB and mTOR/4E-BP–p70S6K6 but not the Akt/NF-κB pathway. PIP2, Phosphatidylinositol (4,5)-bisphosphate.
Footnotes
This work was supported by National Institutes of Health Grants DA024442 (S.B.), DA030285 (H.Y.), DA 033614 (S.B.), and DA 033150 (S.B. and H.Y.).
The authors declare no competing financial interests.
References
- Aksenov MY, Aksenova MV, Nath A, Ray PD, Mactutus CF, Booze RM. Cocaine-mediated enhancement of Tat toxicity in rat hippocampal cell cultures: the role of oxidative stress and D1 dopamine receptor. Neurotoxicology. 2006;27:217–228. doi: 10.1016/j.neuro.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Grãos MM, Carvalho RF, Carvalho AP, Duarte CB. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005;12:1329–1343. doi: 10.1038/sj.cdd.4401662. [DOI] [PubMed] [Google Scholar]
- Bergsten E, Uutela M, Li X, Pietras K, Ostman A, Heldin CH, Alitalo K, Eriksson U. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat Cell Biol. 2001;3:512–516. doi: 10.1038/35074588. [DOI] [PubMed] [Google Scholar]
- Ching TT, Hsu AL, Johnson AJ, Chen CS. Phosphoinositide 3-kinase facilitates antigen-stimulated Ca2+ influx in RBL-2H3 mast cells via a phosphatidylinositol 3,4,5-trisphosphate-sensitive Ca2+ entry mechanism. J Biol Chem. 2001;276:14814–14820. doi: 10.1074/jbc.M009851200. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Cuddon P, Bootman MD, Richards GR, Smith AJ, Simpson PB, Roderick HL. Methacholine and PDGF activate store-operated calcium entry in neuronal precursor cells via distinct calcium entry channels. Biol Res. 2008;41:183–195. doi: 10.4067/S0716-97602008000200008. [DOI] [PubMed] [Google Scholar]
- De March Z, Giamp à C, Patassini S, Bernardi G, Fusco FR. Cellular localization of TRPC5 in the substantia nigra of rat. Neurosci Lett. 2006;402:35–39. doi: 10.1016/j.neulet.2006.03.061. [DOI] [PubMed] [Google Scholar]
- Fiorio Pla A, Maric D, Brazer SC, Giacobini P, Liu X, Chang YH, Ambudkar IS, Barker JL. Canonical transient receptor potential 1 plays a role in basic fibroblast growth factor (bFGF)/FGF receptor-1-induced Ca2+ entry and embryonic rat neural stem cell proliferation. J Neurosci. 2005;25:2687–2701. doi: 10.1523/JNEUROSCI.0951-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Scarano F, Martín-García J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience. 2001;102:555–563. doi: 10.1016/s0306-4522(00)00461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH, Eriksson U, Ostman A. New members of the platelet-derived growth factor family of mitogens. Arch Biochem Biophys. 2002;398:284–290. doi: 10.1006/abbi.2001.2707. [DOI] [PubMed] [Google Scholar]
- Henle SJ, Wang G, Liang E, Wu M, Poo MM, Henley JR. Asymmetric PI(3,4,5)P3 and Akt signaling mediates chemotaxis of axonal growth cones. J Neurosci. 2011;31:7016–7027. doi: 10.1523/JNEUROSCI.0216-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Cheeran MC, Sheng WS, Ni HT, Lokensgard JR, Peterson PK. Cocaine alters proliferation, migration, and differentiation of human fetal brain-derived neural precursor cells. J Pharmacol Exp Ther. 2006;318:1280–1286. doi: 10.1124/jpet.106.103853. [DOI] [PubMed] [Google Scholar]
- Jia Y, Zhou J, Tai Y, Wang Y. TRPC channels promote cerebellar granule neuron survival. Nat Neurosci. 2007;10:559–567. doi: 10.1038/nn1870. [DOI] [PubMed] [Google Scholar]
- Kalasinsky KS, Bosy TZ, Schmunk GA, Ang L, Adams V, Gore SB, Smialek J, Furukawa Y, Guttman M, Kish SJ. Regional distribution of cocaine in postmortem brain of chronic human cocaine users. J Forensic Sci. 2000;45:1041–1048. [PubMed] [Google Scholar]
- Kaus A, Widera D, Kassmer S, Peter J, Zaenker K, Kaltschmidt C, Kaltschmidt B. Neural stem cells adopt tumorigenic properties by constitutively activated NF-kappaB and subsequent VEGF up-regulation. Stem Cells Dev. 2010;19:999–1015. doi: 10.1089/scd.2009.0416. [DOI] [PubMed] [Google Scholar]
- Kwon Y, Hofmann T, Montell C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol Cell. 2007;25:491–503. doi: 10.1016/j.molcel.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Learish RD, Bruss MD, Haak-Frendscho M. Inhibition of mitogen-activated protein kinase kinase blocks proliferation of neural progenitor cells. Brain Res Dev Brain Res. 2000;122:97–109. doi: 10.1016/s0165-3806(00)00064-x. [DOI] [PubMed] [Google Scholar]
- Li X, Pontén A, Aase K, Karlsson L, Abramsson A, Uutela M, Bäckström G, Hellström M, Boström H, Li H, Soriano P, Betsholtz C, Heldin CH, Alitalo K, Ostman A, Eriksson U. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat Cell Biol. 2000;2:302–309. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Martorana A, Giamp à C, DeMarch Z, Viscomi MT, Patassini S, Sancesario G, Bernardi G, Fusco FR. Distribution of TRPC1 receptors in dendrites of rat substantia nigra: a confocal and electron microscopy study. Eur J Neurosci. 2006;24:732–738. doi: 10.1111/j.1460-9568.2006.04932.x. [DOI] [PubMed] [Google Scholar]
- Mishra M, Taneja M, Malik S, Khalique H, Seth P. Human immunodeficiency virus type 1 Tat modulates proliferation and differentiation of human neural precursor cells: implication in NeuroAIDS. J Neurovirol. 2010;16:355–367. doi: 10.3109/13550284.2010.513028. [DOI] [PubMed] [Google Scholar]
- Mohapel P, Frielingsdorf H, Häggblad J, Zachrisson O, Brundin P. Platelet-derived growth factor (PDGF-BB) and brain-derived neurotrophic factor (BDNF) induce striatal neurogenesis in adult rats with 6-hydroxydopamine lesions. Neuroscience. 2005;132:767–776. doi: 10.1016/j.neuroscience.2004.11.056. [DOI] [PubMed] [Google Scholar]
- Peltier J, O'Neill A, Schaffer DV. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol. 2007;67:1348–1361. doi: 10.1002/dneu.20506. [DOI] [PubMed] [Google Scholar]
- Peng F, Dhillon N, Callen S, Yao H, Bokhari S, Zhu X, Baydoun HH, Buch S. Platelet-derived growth factor protects neurons against gp120-mediated toxicity. J Neurovirol. 2008;14:62–72. doi: 10.1080/13550280701809084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowska MJ, Widera D, Kaltschmidt B, an der Heiden U, Kaltschmidt C. Mathematical model for NF-kappaB-driven proliferation of adult neural stem cells. Cell Prolif. 2006;39:441–455. doi: 10.1111/j.1365-2184.2006.00403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- Sasahara A, Kott JN, Sasahara M, Raines EW, Ross R, Westrum LE. Platelet-derived growth factor B-chain-like immunoreactivity in the developing and adult rat brain. Brain Res Dev Brain Res. 1992;68:41–53. doi: 10.1016/0165-3806(92)90246-s. [DOI] [PubMed] [Google Scholar]
- Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, Tsiokas L, Winn M, Abramowitz J, Rockman HA, Birnbaumer L, Rosenberg P. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res. 2009;105:1023–1030. doi: 10.1161/CIRCRESAHA.109.206581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits A, Kato M, Westermark B, Nistér M, Heldin CH, Funa K. Neurotrophic activity of platelet-derived growth factor (PDGF): rat neuronal cells possess functional PDGF beta-type receptors and respond to PDGF. Proc Natl Acad Sci U S A. 1991;88:8159–8163. doi: 10.1073/pnas.88.18.8159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens BG, Jentzen JM, Karch S, Mash DC, Wetli CV. Criteria for the interpretation of cocaine levels in human biological samples and their relation to the cause of death. Am J Forensic Med Pathol. 2004;25:1–10. doi: 10.1097/01.paf.0000118960.58334.a9. [DOI] [PubMed] [Google Scholar]
- Van Dyke C, Barash PG, Jatlow P, Byck R. Cocaine: plasma concentrations after intranasal application in man. Science. 1976;191:859–861. doi: 10.1126/science.56036. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW., Jr The mammalian TRPC cation channels. Biochim Biophys Acta. 2004;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Venkatesan A, Nath A, Ming GL, Song H. Adult hippocampal neurogenesis: regulation by HIV and drugs of abuse. Cell Mol Life Sci. 2007;64:2120–2132. doi: 10.1007/s00018-007-7063-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollenbröker B, George B, Wolfgart M, Saleem MA, Pavenstädt H, Weide T. mTOR regulates expression of slit diaphragm proteins and cytoskeleton structure in podocytes. Am J Physiol Renal Physiol. 2009;296:F418–F426. doi: 10.1152/ajprenal.90319.2008. [DOI] [PubMed] [Google Scholar]
- Weick JP, Austin Johnson M, Zhang SC. Developmental regulation of human embryonic stem cell-derived neurons by calcium entry via transient receptor potential channels. Stem Cells. 2009;27:2906–2916. doi: 10.1002/stem.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Steflik J, Cortright DN, Lamotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005;102:14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Chen S, Luo Y, Chen Z, Liu L, Zhou H, Chen W, Shen T, Han X, Chen L, Huang S. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS One. 2011;6:e19052. doi: 10.1371/journal.pone.0019052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Suzuki T, Seki T, Namba T, Juan R, Arai H, Hori T, Asada T. Repetitive cocaine administration decreases neurogenesis in adult rat hippocampus. Ann N Y Acad Sci. 2004;1025:351–362. doi: 10.1196/annals.1316.043. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Suzuki T, Seki T, Namba T, Liu J, Arai H, Hori T, Shiga T. Decreased cell proliferation in the dentate gyrus of rats after repeated administration of cocaine. Synapse. 2005;58:63–71. doi: 10.1002/syn.20182. [DOI] [PubMed] [Google Scholar]
- Yao H, Peng F, Fan Y, Zhu X, Hu G, Buch SJ. TRPC channel-mediated neuroprotection by PDGF involves Pyk2/ERK/CREB pathway. Cell Death Differ. 2009a;16:1681–1693. doi: 10.1038/cdd.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Peng F, Dhillon N, Callen S, Bokhari S, Stehno-Bittel L, Ahmad SO, Wang JQ, Buch S. Involvement of TRPC channels in CCL2-mediated neuroprotection against tat toxicity. J Neurosci. 2009b;29:1657–1669. doi: 10.1523/JNEUROSCI.2781-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]